

ABSTRACT
Mutations in SH2D1A gene that encodes SAP (SLAM-associated protein) result in X-linked lymphoproliferative disease (XLP), a rare primary immunodeficiency disease defined by exquisite sensitivity to the B-lymphotropic Epstein–Barr virus (EBV) and B cell lymphomas. However, the precise mechanism of how the loss of SAP function contributes to extreme vulnerability to EBV and the development of B cell lymphomas remains unclear. Here, we investigate the hypothesis that SAP is critical for CD8+ T cell immune surveillance of antigen (Ag)-expressing B cells or B lymphoma cells under conditions of defined T cell receptor (TCR) signaling. Sh2d1a−/− CD8+ T cells exhibited greatly diminished proliferation relative to wild type when Ag-presenting-B cells or -B lymphoma cells served as the primary Ag-presenting cell (APC). By contrast, Sh2d1a−/− CD8+ T cells responded equivalently to wild-type CD8+ T cells when B cell-depleted splenocytes, melanoma cells or breast carcinoma cells performed Ag presentation. Through application of signaling lymphocyte activation molecule (SLAM) family receptor blocking antibodies or SLAM family receptor-deficient CD8+ T cells and APCs, we found that CD48 engagement on the B cell surface by 2B4 is crucial for initiating SAP-dependent signaling required for the Ag-driven CD8+ T cell proliferation and differentiation. Altogether, a pivotal role for SAP in promoting the expansion and differentiation of B cell-primed viral-specific naive CD8+ T cells may explain the selective immune deficiency of XLP patients to EBV and B cell lymphomas.
KEYWORDS: Antigen-presenting cells, B cells, B cell lymphoma, CD8+ T cells, Epstein–Barr virus, immune deficiency, SLAM-associated protein, SLAM family receptors, T cell receptor, X-linked proliferative disease
Introduction
Epstein–Barr virus (EBV) is a ubiquitous γ-herpesvirus that has infected about 90% of the worldwide adult population. Virus acquisition is initiated following infection of oropharyngeal epithelial cells, resulting in lytic replication and production of infectious virions, and subsequent contagion spread to nearby B cells. Upon resolution of acute infection, EBV establishes a lifelong viral reservoir within a small fraction of circulating B cells containing viral genomes.1 In EBV-infected B cells, the viral life cycle may shunt between a lytic (replicative) phase and various forms of latency to avoid T cell surveillance by expressing restricted profiles of viral proteins. Although early life viral infection is predominately asymptomatic, EBV often becomes pathogenic during adolescence, when primary infection results in infectious mononucleosis (IM), or in adulthood, with latent infection promoting development of EBV-positive lymphoid and epithelial malignancies. In addition, individuals with compromised T or NK cell immunity are especially vulnerable to virus-driven B cell lymphoproliferative disorders and B cell lymphomas. Currently, the immune correlates associated with protection are unclear and no vaccines are effective in preventing EBV infection.
X-linked lymphoproliferative disease (XLP) is a rare primary immunodeficiency disease, resulting from inactivating mutations within the SH2D1A gene encoding SLAM-associated protein (SAP),2-4 whose hallmark is defined by exquisite sensitivity to EBV.5-7 In contrast to many primary immunodeficiencies,8,9 SAP-deficient patients do not exhibit similar vulnerabilities to other pathogens, including other Herpesviridae family members such as cytomegalovirus, herpes simplex virus and varicella zoster. EBV infection of XLP patients results in life-threatening IM that is associated with uncontrolled expansions of virally infected B cells and sometimes, B cell lymphomas.5,6 However, the heightened susceptibility of XLP patients to B cell lymphomas is independent of infection by EBV.10,11 Importantly, the control of EBV-infected B cells seems to be a key determinant in driving fulminant IM in XLP patients given that B cell-depletion therapy with rituximab resolves symptoms and reduces viral DNA among circulating lymphocytes.12,13 Together, these findings support the hypothesis that SAP-dependent immunity is essential for the surveillance of infected and malignant B cells.
SAP functions as an intracellular adaptor protein that utilizes its SH2 domain to associate with immunoreceptor tyrosine-based switch motifs (ITSM: TxYxxI/V in which “x” denotes any amino acid) present in all cell surface SLAM family receptors except CD48.5–7 The SLAM family receptors—SLAM (SLAMF1), CD48 (SLAMF2), LY9 (SLAMF3), 2B4 (SLAMF4), CD84 (SLAMF5), NTB-A/Ly108 (SLAMF6) and CRACC (SLAMF7)—share homologous immunoglobulin-like extracellular domains and are principally expressed by haematopoietic cells. Most SLAM family receptors are self-ligands (i.e., LY9 binds LY9) with the one exception being 2B4's recognition of CD48. Consequently, SLAM receptors are capable of regulating either homotypic– or heterotypic–cell/cell interactions between immune cells. Through investigations of XLP patients and gene-targeted mice, a common theme has emerged for SAP in regulating lymphocyte–lymphocyte contact, communicating signals necessary for lymphocyte differentiation and executing effector function: CD4+ T cell–B cell interactions in generating TFH cells, germinal centers, B cell isotype-switching and B cell memory;14-17 thymocyte–thymocyte interactions instructing the development of NKT cells;18-20 NK cell–haematopoietic target interactions controlling cytotoxicity21-23 and effector CD8 T cell–B cell interactions modulating CD8+ T cell killing.24-28 Although multiple immune defects have been attributed to SAP deficiency,5-7 it remains unclear how SAP facilitates control of EBV infection and whether dysfunction of one or more immune cell types underlies the vulnerability of XLP patients to EBV and B cell malignancies.
B cells likely function as the critical antigen (Ag)-presenting cell (APC) during EBV infection as the virus selectively infects B cells and B cells may present viral Ags not expressed by other infected host cells. Consequently, we hypothesized that extreme vulnerability of XLP patients to EBV and B cell malignancies may be related to the crucial roles that SAP and SLAM family receptors play in the priming of naive CD8+ T cells by B cells. Here, we show that SAP expression in naive CD8+ T cells is essential for Ag-driven proliferation and differentiation when B cells or B lymphoma cells act as APCs. By contrast, SAP appears to be dispensable when naive CD8+ T cells are primed by B cell-depleted splenocytes or tumor cell lines that lack expression of SLAM family receptors. Furthermore, the engagement of 2B4 on naive CD8+ T cells by CD48 on the surface of B cells or B lymphoma cells was found to be required for initiating SAP-dependent signaling necessary for the Ag-driven CD8+ T cell differentiation. Altogether, our findings indicate that SLAM family receptors and SAP provide critical co-stimulatory signals necessary for CD8+ T cell immune surveillance of transformed B cells, and suggest why XLP patients are especially prone to EBV and B cell lymphomas.
Results
SAP is critical for naive CD8+ T cell differentiation upon stimulation with antigen-presenting B cells
Previous studies have found that Sh2d1a−/− CD8+ T cells do not exhibit inherent proliferative defects,29-32 however, we speculated that the susceptibility of XLP patients to the B-lymphotropic EBV results from a critical role of SAP in the differentiation of naive CD8+ T cells upon stimulation by B cell APCs. Consequently, we tested this hypothesis by activating WT and Sh2d1a−/− OT-I TCR CD8+ T cells with cognate Ag along with either purified splenic B cell APCs, B cell-depleted APCs (anti-Ig-depleted splenocytes) or whole splenocytes. Strikingly, Sh2d1a−/− CD8+ T cells displayed greatly diminished proliferation relative to WT CD8+ T cells when Ag-priming was performed by B cells, particularly at low Ag concentrations (Fig. 1A: 9.0-fold at 10−10 M, p < 0.0001; 5.8-fold at 10−9 OVA, p < 0.0001; 5.1-fold at 10−8 M, p < 0.001). By contrast, both WT and Sh2d1a−/− CD8+ T cells responded robustly upon stimulation with B cell-depleted splenocyte APCs (Fig. 1B) or whole splenocyte APCs (data not shown). These findings indicate that SAP is important for naive CD8+ T cell differentiation following antigen priming by B cells.
Figure 1.
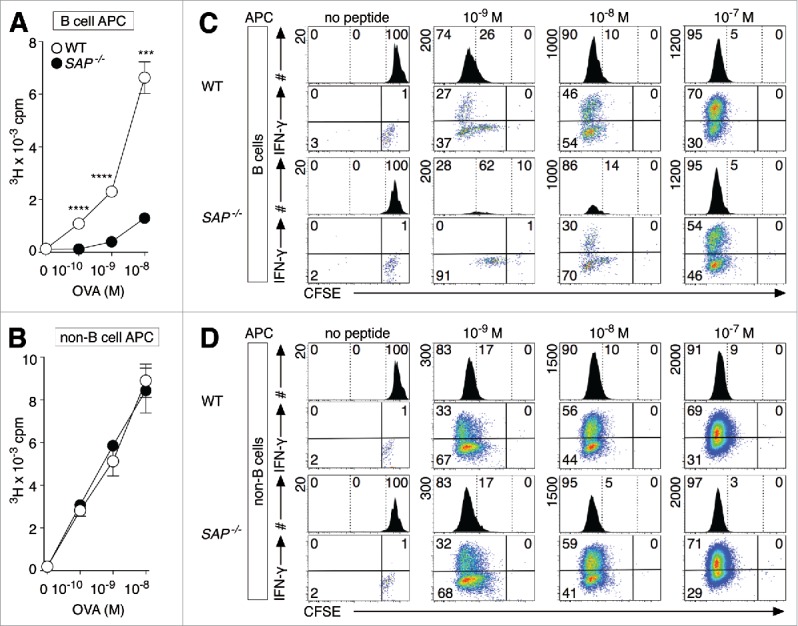
SAP is critical for naive CD8+ T cell differentiation upon stimulation with antigen-presenting B cells. (A, B) WT and Sh2d1a−/− (SAP−/−) OT-I CD8+ T cells were stimulated with OVA and highly purified B cells (A) or B cell-depleted splenocytes (B) for 3 d and proliferation measured by [3H]-thymidine incorporation. (C, D) CFSE-labeled WT and SAP−/− OT-I CD8+ T cells were activated with OVA and purified B cell APCs (C) or B cell-depleted splenic APCs (D) and proliferation tracked after 4 d of culture. At day 4 post-activation, cells were re-stimulated before measuring cytokine production. Samples were acquired for 1 min intervals and the maximum scale (y-axis) is shown vertically at the top left for each histogram. Error bars represent SD and statistical significance was calculated using unpaired, two-tailed Student's t-tests. Triple (***) and quadruple (****) indicate p values of less than 0.001 and 0.0001, respectively. The data are representative of at least three independent experiments.
We next investigated how the type of APC used for stimulation affects SAP's influence on CD8+ T cell division and differentiation using the mitotic tracker CFSE and performing stimulations as described above. At 4 d post-activation, cultures were either directly stained to discriminate OT-I CD8+ T cells or re-stimulated for 4 h with PMA and ionomycin to assess capacity to secrete IFNγ. During flow acquisition, samples were collected for the same length of time (1 min) so that OT-I CD8+ T cell numbers would correlate with burst size (larger scale on y-axis plotted on histograms), reflecting total cell numbers rather than just CFSE cell division history. As expected, WT CD8+ T cells underwent proliferation and differentiation regardless of APC used and higher doses of Ag resulted in larger cell recoveries (greater maximum value for y-axis) and an increased frequency of IFNγ producers (Figs. 1C and D). By contrast, Sh2d1a−/− CD8+ T cells exhibited a considerably smaller burst size relative to WT CD8+ T cells exclusively when activated with Ag-presenting B cells and secreted less IFN-γ (Fig. 1C; 10−9 M: 0% vs. 27%; 10−8 M: 30% vs. 46% and 10−7 M: 54% vs. 70%), suggesting that SAP is critical for naive CD8+ T cell growth and differentiation when B cells act as APCs.
SAP-deficient CD8+ T cells secrete decreased IL-2 when activated by B cells and exogenous IL-2 rescues their defective proliferation
We next determined whether defective proliferation of Sh2d1a−/− CD8+ T cells was associated with alterations in IL-2 secretion or CD25 (IL-2Rα receptor) upregulation following Ag priming by B cells. Strikingly, Sh2d1a−/− CD8+ T cells produced less IL-2 than WT CD8+ T cells when activated by Ag-presenting B cells but not when stimulated with B cell-depleted splenocytes (Fig. 2A, 2.1-fold less IL-2+ cells at 2 h, p < 0.001; 2.0-fold less at 6 h, p < 0.01). In addition, Sh2d1a−/− CD8+ T cells displayed a modest delay in CD25 induction relative to WT CD8+ T cells when stimulated by B cells (Fig. 2B, 3.0-fold decrease at 4 h, p < 0.01; 1.2-fold decrease at 6 h, p < 0.01). Furthermore, the addition of exogenous IL-2 was able to rescue the defective proliferation and differentiation of B cell-primed Sh2d1a−/− CD8+ T cells (Figs. 2C and D). Altogether, these findings indicate that SAP promotes CD8+ T cell responses against Ag-presenting B cells by regulating IL-2 production.
Figure 2.

SAP-deficient CD8+ T cells secrete decreased IL-2 when activated by B cells and exogenous IL-2 rescues their defective proliferation. (A) Intracellular IL- 2 and (B) surface CD25 expression was monitored on WT and Sh2d1a−/− (SAP−/−) OT-I CD8+ T cells upon activation with OVA (10−8 M) and B cell- or B cell-depleted splenocyte-APCs for the indicated time. (C) WT and SAP−/− OT-I CD8+ T cells were stimulated with Ag, B cell APCs and exogenous IL-2 and proliferation measured by [3H]-thymidine incorporation assays. (D) WT and SAP−/− OT-I CD8+ T cells were stimulated as described in (C) except proliferation quantitated through CFSE dilution 4 d post-activation. Samples were collected for 1 min intervals and the maximum scale (y-axis) for each histogram is shown vertically at the top left of histograms. Error bars indicate ± SD and significance calculated using unpaired, two-tailed Student's t-tests with double (**) and triple asterisks (***) indicating p values of less than 0.01 and 0.001, respectively. The data are representative of three independent experiments.
2B4. is required for CD8+ T cell responses against antigen-presenting B cells
To establish which SLAM family receptors that SAP may act through to regulate antigen priming of CD8+ T cells by B cells, we pre-treated Ag-presenting B cells with individual SLAM family receptor antibodies to determine whether any could inhibit the proliferation of WT CD8+ T cells. Interestingly, only anti-CD48 antibodies were found to block WT CD8+ T cell proliferation (Fig. S1A). Furthermore, experiments using Ly108−/− B cell APCs showed that LY108 expression did not affect responses by WT OT-I CD8+ T cells (data not shown). Given that CD48's ligand is 2B45, our results suggest that CD48 on B cells provides co-stimulatory signals through 2B4 on naive CD8+ T cells. Consequently, we next investigated whether naive OT-I CD8+ T cells upregulate 2B4 upon activation because they lack appreciable levels of 2B4 directly ex vivo. Definitively, WT and Sh2d1a−/− CD8+ T cells strongly induced 2B4 upon stimulation with Ag-presenting B cells but not B cell-depleted APCs (Figs. 3A and B), demonstrating that naive CD8+ T cells rapidly express 2B4 specifically upon B cell priming and that its induction is SAP-independent.
Figure 3.

2B4–CD48 interactions promote naive CD8+ T cell responses toward Ag-presenting B cells. (A, B) Surface 2B4 levels were monitored on WT and Sh2d1a−/− (SAP−/−) OT-I CD8+ T cells at the indicated times (h) post-stimulation with OVA (10−8 M) and purified B cells or B cell-depleted splenocytes acting as APCs. Shaded histograms represent background fluorescence-minus-one (FMO) control staining. (C, D) WT and 2B4−/− OT-I CD8+ T cells were activated with Ag-presenting B cells and proliferation quantitated by (C) [3H]-thymidine DNA incorporation assays or (D) CFSE-dilution analyses. (E, F) WT OT-I CD8+ T cells were stimulated with OVA and WT or CD48−/− B cells acting as APCs and proliferation measured by (E) [3H]-thymidine incorporation or (F) CFSE dilution assays. Samples were acquired for 1 min intervals and the maximum scale (y-axis) is shown vertically at the top left of each histogram. For cytokine detection, samples were re-stimulated before staining for intracellular IFNγ. Error bars represent the SD and statistical significance determined using unpaired, two-tailed Student's t tests. Triple (***) and quadruple (****) indicate p values of less than 0.001 and 0.0001, respectively. The data shown are representative from at least three independent experiments.
To address whether 2B4 regulates B cell-primed CD8+ T cell responses, WT and 2B4−/− OT-I CD8+ T cells were setup in proliferation assays as described above. Strikingly, 2B4−/− CD8+ T cells exhibited dramatically reduced proliferation upon Ag stimulation with B cells relative to WT CD8+ T cells (Fig. 3C: 4.4-fold at 10−10 M, p < 0.0001; 5.8-fold at Ag 10−9 M, p < 0.0001; 6.5-fold at 10−8 M, p < 0.0001). Furthermore, B cell-primed 2B4−/− CD8+ T cells exhibited impaired expansion and decreased IFNγ production compared with WT CD8+ T cells (Fig. 3D). By contrast, 2B4−/− CD8+ T cells proliferated robustly and differentiated into IFNγ-producing effectors when B cell-depleted splenocytes acted as the APC (Figs. 1SB, 1SC). Together, these results indicate that 2B4 plays a selective role in priming of naive CD8+ T cells by Ag-presenting B cells.
CD48 expression by B cells provides critical co-stimulation for CD8+ T cells
CD48 is a glycophosphatidylinositol (GPI)-linked protein expressed by most haematopoietic cells that has been associated with immune cell regulation through interactions with its ligands 2B4 and CD2.33 We next sought to determine whether CD48 on B cells is also critical for this immune reaction by Ag priming WT and Sh2d1a−/− CD8+ T cells with WT or CD48−/− B cells. WT CD8+ T cells displayed greatly reduced proliferation when activated by CD48−/− B cells compared with WT B cells (Fig. 3E, 7.5-fold at 10−10 M, p < 0.0001; 4.3-fold at 10−9 M, p < 0.0001; 2.5-fold at Ag 10−8 M, p < 0.001). Furthermore, WT CD8+ T cells stimulated with CD48-deficient B cells exhibited reduced proportions of IFNγ-secreting effectors versus those activated by WT B cells (Fig. 3F). However, the anemic response of Sh2d1a−/− CD8+ T cells toward cognate Ag-presenting B cells was not impacted by CD48 expression (data not shown). In contrast to its vital role on B cells, CD48 expression on non-B cell APCs appears dispensable for inducing the proliferation and differentiation of WT CD8+ T cells (Figs. S1D and E). Together, these data indicate that CD48 plays a selective role on B cells critical for the priming of naive CD8+ T cells.
SAP is essential for anti-B cell lymphoma CD8+ T cell responses in vitro
XLP patients are also susceptible to B cell lymphomas independent of EBV infection status,10,11 suggesting that SAP is required for immune surveillance of malignant B cells. To test whether SAP is important for CD8+ T cell responses directed against B lymphoma cells, CFSE-labeled WT and Sh2d1a−/− OT-I CD8+ T cells were stimulated with previously described Ag-negative (B-GFP) or Ag-expressing Eμ-Myc B lymphoma cells (B-OVA).34,35 After 4 d culture, WT CD8+ T cells activated with B-OVA had proliferated vigorously with most (71%) of cells capable of secreting IFNγ (Fig. 4A). By contrast, Sh2d1a−/− CD8+ T cells expanded poorly (54% undivided), resulting in a poor cell recovery, and most cells (96%) lacked IFNγ expression. Furthermore, the provision of exogenous IL-2 was found to rescue Sh2d1a−/− CD8+ T cell responses against B-OVA cells. To determine whether SAP-deficiency could be overcome by increasing Ag concentrations, WT and Sh2d1a−/− OT-I CD8+ T cells were stimulated with varying amounts of OVA and B-GFP cells (Fig. 4B). However, Sh2d1a−/− OT-I CD8+ T cells exhibited lower cell yields and increased proportions remaining undivided relative to WT CD8+ T cells regardless of Ag concentration.
Figure 4.

SAP regulates CD8+ T cell responses toward antigen-presenting B lymphoma cells. (A) WT and Sh2d1a−/− (SAP−/−) OT-I CD8+ T cells were stimulated with GFP- (B-GFP) or OVA-expressing B lymphoma cells (B-OVA) in the absence (left) or presence of exogenous IL-2 (right) and proliferation measured by CFSE dilution after 4 d. (B) WT and SAP−/− OT-I CD8+ T cells were activated with indicated concentrations of OVA and B-GFP cells for 4 d before flow cytometric analyses. (C) WT and SAP−/− OT-I CD8+ T cells were incubated with OVA-expressing B16 melanoma cells (B16-OVA) or breast carcinoma cells (NOP12) and proliferation history tracked using CFSE dilution after 4 d. Cultures were re-stimulated for 4 h before intracellular detection of IFNγ. Samples were acquired for 1 min intervals so that the acquisition events would be reflective of CD8+ T cell numbers present within cultures. The maximum scale (y-axis) is shown vertically at the top left of each histogram. The data are representative of at least four independent experiments.
To assess whether SAP is important for naive CD8+ T cell responses against tumor cell APCs lacking SLAM family receptors, CFSE-labeled WT and Sh2d1a−/− OT-I CD8+ T cells were stimulated with OVA-expressing B16-OVA melanoma36 or NOP12 breast carcinoma cells.37 After 4 d culture, WT and Sh2d1a−/− CD8+ T cells exhibited comparable levels of proliferation and differentiation into secreting IFNγ effectors (Fig. 4C). These observations suggest that SAP is dispensable for naive CD8+ T cell responses when tumor cell APCs lack SLAM family receptors.
2B4–CD48. interactions regulate CD8+ T cell responses against B lymphoma cells
To investigate whether the SLAM family receptors 2B4/CD48 also participate in CD8+ T cell responses against B lymphoma cells, CFSE-labeled WT and 2B4−/− OT-I CD8+ T cells were stimulated with B-OVA lymphoma cells. 2B4−/− CD8+ T cells exhibit dramatically decreased proliferation (69% remain CFSE high vs. 0%) and differentiation (2% vs. 85% secrete IFNγ) relative to WT CD8+ T cells (Fig. 5A). By contrast, 2B4−/− CD8+ T cells responded similarly to WT CD8+ T cells when activated by SLAM family receptor-negative carcinoma NOP12 cells. Collectively, these findings indicate that 2B4 is critical for naive CD8+ T cell responses against B lymphoma APCs.
Figure 5.

2B4–CD48 interactions are required for efficient priming of naive CD8+ T cells by antigen-presenting B lymphoma cells. (A) CFSE-labeled WT and 2B4−/− OT-I CD8+ T cells were stimulated with B-GFP, B-OVA or OVA-expressing NOP12 carcinoma cells and proliferation measured 4 d later. (B) CFSE-labeled WT and Sh2d1a−/− (SAP−/−) OT-I CD8+ T cells were incubated with B-OVA cells that had been left untreated (No Tx) or pre-coated with anti-CD48 (+ α-CD48) or isotype-matched control antibodies (+ control Ab) for 4 d. CD8+ T cell proliferation and cytokine production was assessed after a brief 4 h re-stimulation. Samples were acquired for 1 min intervals and the maximum scale (y-axis) is shown vertically at the top left for each histogram. The data are representative from three independent experiments.
Next, to examine whether CD48 on B lymphoma cells is required for naive CD8+ T cell proliferation, CFSE-labeled WT and Sh2d1a−/− OT-I CD8+ T cells were activated with B-OVA cells that were untreated or pre-coated with anti-CD48 or isotype-control antibodies (Fig. 5B). WT CD8+ T cells stimulated with B-OVA that had been left untreated or coated with control antibody responded vigorously whereas those activated by α-CD48 antibody-treated B-OVA cells exhibited severely diminished proliferation and differentiation (4% vs. 82% IFNγ+ for α-CD48 vs. control Ab). In addition, Sh2d1a−/− CD8+ T cell response was modestly decreased when B-OVA cells were treated with anti-CD48 relative to control antibody, perhaps a consequence of blocking CD2–CD48 interactions. These results demonstrate that CD48 on B lymphoma APCs is important for co-stimulating naive CD8+ T cell responses.
SAP is critical for anti-B cell lymphoma immunity mediated by naive CD8+ T cells
To determine if SAP is important for antigen-stimulated CD8+ T cell responses in vivo, a 1:1 ratio of CFSE-labeled WT (Thy1.1+CD45.2+) and Sh2d1a−/− (Thy1.2+CD45.2+) OT-I CD8+ T cells were co-transferred into recipients (Thy1.2+CD45.1+) one day before infection with recombinant Listeria monocytogenes expressing OVA (Fig. 6A). At one week post-infection, both donor WT and Sh2d1a−/− OT-I CD8+ T cells completely lost their CFSE fluorescence and their relative cell proportions to one another was almost equal (50:50). As Listeria predominately infects phagocytic cells and DCs are key APCs in priming T cells,38,39 these results suggest that SAP is dispensable for CD8+ T cell immunity when DCs are primarily responsible for antigen priming.
Figure 6.

SAP is critical for anti-B cell lymphoma immunity mediated by naive CD8+ Tcells. (A) CFSE-labeled WT (Thy1.1+) and Sh2d1a−/− (SAP−/−, Thy1.2+) OT-I CD8+ T cells were co-transferred at a 1:1 ratio into congenic (CD45.1+) recipients (n = 3) and infected with recombinant L. monocytogenes expressing OVA (+ LM-OVA) one day later. CFSE histograms and distributions of donor WT vs. SAP−/− CD8+ T cells recovered from spleens of LM-OVA infected mice (black histograms) are shown. Gray histograms represent CFSE profiles for donor WT and SAP−/− CD8+ T cells recovered from spleens of naive (uninfected) mice. (B, C) CFSE-labeled WT or SAP−/− OT-I CD8+ T cells (Thy1.1+ CD45.2+) were adoptively transferred into CD45.1+ hosts (n = 6 per group) one day before infusion with B-OVA cells (CD45.2+). (B) Representative donor WT and SAP−/− OT-I CD8+ T cell proliferation and absolute cell number recovery (right) within host spleens at day 9 post-challenge are shown. (C) Splenocytes were left untreated (unstimulated) or re-stimulated (+ PMA/Iono) for detection of surface CD107a and intracellular IFNγ to assess donor CD8+ T cell effector functions. The proportions of donor WT and SAP−/− CD8+ T cells that co-express CD107a and IFNγ upon re-stimulation are presented (bar graph, right). Error bars represent the SD and statistical significance was calculated using unpaired, two-tailed Student's t tests. Triple asterisks (***) indicate p values of less than 0.001. The data in panels B and C are representative of three independent experiments.
To investigate whether our in vitro lymphoma findings could be translated in vivo, CFSE-labeled WT or Sh2d1a−/− OT-I CD8+ T cells (Thy1.1+CD45.2+) were adoptive transferred before infusing recipients (CD45.1+) with B-OVA (CD45.2+) cells. Despite the likely presentation of lymphoma Ags on the surface of B-OVA cells34,35 and DCs via cross-presentation, Sh2d1a−/− CD8+ T cells displayed a 5.0-fold reduction in total cell numbers relative to WT CD8+ T cells at 9 d post-challenge (Fig. 6B, p < 0.001). Furthermore, recovered Sh2d1a−/− CD8+ T cells also exhibited decreased effector functioning compared with WT CD8+ T cells, showing a 4.9-fold reduction in the frequency of cells double positive for IFNγ and CD107a, a marker of degranulating cytotoxic T cells (Fig. 6C, p < 0.001). However, splenic lymphoma burdens between the two cohorts of mice were very similar despite the increased proliferation and effector function by WT CD8+ T cells relative to Sh2d1a−/− CD8+ T cells (Recipients with WT vs. Sh2d1a−/− OT-I CD8+ T cells: 1.5 ± 2.1 × 107 vs. 1.7 ± 3.3 × 107 lymphoma cells, p = 0.6457). The lack of protection afforded by WT OT-I T cells may be related to observations demonstrating that these B lymphoma cells are resistant to OT-I CD8+ T cell effectors by mediating their functional inactivation.40 Collectively, these results indicate that SAP is critical for driving the expansion and effector functions of lymphoma-specific CD8+ T cells in vivo.
Discussion
SAP function and EBV immunity may intersect because of SAP's regulation of physical interactions between haematopoietic cells and EBV's tendency to latently infect B lymphocytes. During primary infection, EBV morphs its viral gene expression from a complex profile responsible for lytic viral replication to a simpler, more restricted viral protein subset responsible for the latent cycle that initiates host cellular proliferation as the virus transits from oropharyngeal epithelial cells to B cells.1 Consequently, EBV-infected B cells, rather than oral epithelial cells or cross-priming DCs, may function as critical APCs necessary for the differentiation of viral-specific naive T cells into cytotoxic effectors that curtail the expansion of latently infected-B cells. Here, we describe a novel function for SAP, in collaboration with SLAM family receptors 2B4 and CD48, in regulating the functional responses of naive CD8+ T cells specifically against Ag-presenting-B cells and -B lymphoma cells. Moreover, our findings suggest that the extreme vulnerability of XLP patients to EBV may be a consequence of their naive CD8+ T cells failing to undergo Ag-driven expansion and differentiation upon stimulation by EBV-infected B cells resulting in defective CD8+ T cell immune surveillance.
SLAM family receptors can confer activating or inhibitory signaling that may depend on the relative abundance of SAP and its dual mechanism of action. SAP binding to SLAM receptors can recruit the Src family kinase protein tyrosine kinase FYN to transduce activating signals as well as block the docking of inhibitory phosphatases such as the SH2 domain-containing inositol phosphatase (SHIP)-1 and the SH2 domain containing protein tyrosine phosphatase (SHP)-1. For example, 2B4 ligation has been shown to provide an activating signal to control NK cell effectors, boosting cytotoxicity and cytokine secretion, while the same stimulation inhibits the function of SAP-deficient NK cell effectors from XLP patients.5,7 Thus, the impact of SAP-deficiency on SLAM family receptors may not only disrupt activating signaling but also bolster inhibitory signaling. Studies with human CD8+ T cell clones suggest that SAP functions to block inhibitory SLAM family receptor signaling since antibodies against 2B4 and NTB-A can restore SAP-deficient T cell responses toward Ag-presenting B cells.26 Interestingly, 2B4 has also been proposed to act as an inhibitory receptor to promote CD8+ T cell exhaustion during chronic viral infection.41 By contrast, our findings indicate that 2B4 transmits SAP-dependent activating signals necessary for naive CD8+ T cells to proliferate and differentiate into effectors upon contact with CD48 on B cell APCs (Fig. 3). However, the presence or absence of CD48 on Ag-presenting B cells did not significantly affect the proliferation of SAP-deficient naive CD8+ T cells (data not shown). Consequently, the relative expression of SLAM family receptors to SAP together with the differentiation status of CD8+ T cells may determine whether SLAM receptors convey activating or inhibitory signals.
SLAM family receptors are expressed by virtually all haematopoietic cells, however, their established roles are involved in the regulation of lymphocyte–lymphocyte interactions, controlling the differentiation and effector functions of lymphocytes.5 By contrast, the functionality of lymphocytes interacting with other (non-lymphocyte) immune cells appears to be SAP- and SLAM family receptor-independent. Similarly, we found that 2B4 and SAP expression are required to generate optimal CD8+ T cell responses against B cell or B cell lymphoma APCs but are dispensable when priming is performed by non-B cell APCs, even though these APCs express a variety of SLAM family receptors including CD48. One possibility for this discrepancy is that splenic APCs like DCs are able to provide IL-2 or alternate cytokines that lower the threshold of stimulation required for Ag-driven CD8+ T cell proliferation given that exogenous IL-2 can rescue defective Sh2d1a−/− CD8+ T cell responses toward Ag-presenting B cells (Figs. 2C and D). Alternatively, DCs or other non-B cell APCs may provide sufficient Ag stimulation to Sh2d1a−/− naive CD8 T cells through varied or increased expression of co-stimulatory or adhesion receptors, overriding a dependence on SAP or SLAM family receptors. Moreover, previous work indicates that Ag-mediated CD4+ T cell-DC adhesion depends on integrins LFA-1 and VLA-4 rather than any requirement for SAP and SLAM family receptors, whereas CD4+ T cell–B cell interactions require integrins along with SAP and CD84 for stable cell–cell contact.15,17 Accordingly, DCs may not require SLAM family receptor engagement to activate naive CD8+ T cells due to provision of stimulatory cytokines or differential cytoskeletal or membrane mechanics responsible for adhesion.
SAP-deficiency compromises immunity toward EBV by diminishing CD8+ T cell function as previous work has shown that CD8+ T cell effectors from XLP patients exhibit decreased cytotoxicity and cytokine secretion when engaging B cells but not other targets.24,27,28 However, our findings suggest that the loss of SAP also undermines the priming of naive CD8+ T cells by B cell APCs and thus, limits the repertoire of T cells specific for infected B cells generated upon EBV infection. In addition, an elegant study showed that SAP is important for the generation or homeostasis of EBV-specific memory CD8+ T cells based on analyses of XLP carrier females (SH2D1A+/−) who possess a mixture of SAP-expressing (SAP+) and SAP-deficient (SAP−) cells due to random X-inactivation.26 Using a SAP antibody and intracellular flow cytometry, most (> 95%) of EBV-specific memory CD8+ T cells found in XLP carriers were shown to be SAP+ while memory CD8+ T cells specific for CMV and influenza were equivalently distributed between SAP+ and SAP− cell subpopulations.26 Collectively, these observations suggest that naive CD8+ T cells require SAP to be primed by EBV-infected B cells and generate long-lived EBV-specific memory CD8+ T cells.
A number of lines of evidence indicate that our murine 2B4-SAP findings may have important implications for CD8+ T cell immunity against EBV and B cell lymphomas. The application of a 2B4 blocking antibody to EBV-infected humanized mice, NOD-Scid IL-2r γ−/− (NSG) mice engrafted with a human immune system, aggravated disease, led to elevated EBV viral loads and increased incidence of B cell lymphomas.42 Furthermore, 2B4 blockade appeared to impede the function of CD8+ T cells since EBV-induced disease was not exacerbated by 2B4 antibody when humanized NSG mice were first depleted of CD8+ T cells.42 In addition, CD48 (aka B-LAST-1) is strongly expressed on the surface of EBV-transformed B cells as well as lymphoid malignancies.43 Interestingly, high levels of soluble CD48 (sCD48) have been detected in patients with acute EBV infection and lymphoproliferative disease.44 Although its significance is unknown, sCD48 may act as a viral immune evasion mechanism by disrupting 2B4–CD48 interactions and inhibiting the priming of naive CD8+ T cells or cytotoxicity of CD8+ T cell effectors. Altogether, our work suggests that modulation of 2B4–CD48 signaling may prove valuable for the clinical management of EBV-associated disease and B cell lymphomas.
Materials and methods
Mice
Sh2d1a−/− mice30 were crossed onto C57BL/6J (WT) background at least 10 generations before mating to OT-I TCR mice.45 WT, OT-I, Thy 1.1+, CD45.1+ and CD48−/− mice46 were purchased from The Jackson Laboratory and housed at The University of British Columbia (UBC) following protocols approved by the UBC Animal Care Committee and the Canadian Council on Animal Care. WT and 2B4−/− OT-I mice47 were kept at Emory University complying with guidelines of their University Animal Care and Use Committee.
Flow cytometry
RBC-depleted splenocytes were suspended in 2% FBS/PBS and incubated with specific antibodies for 15 min on ice. Antibodies against CD8α (53–6.7), CD4 (GK1.5), TCRVα2 (B20.1), CD44 (IM7), Thy1.1 (HIS51), Thy1.2 (30-H12), CD25 (PC61.5), CD45.1 (A20), CD45.2 (104), 2B4 (ebio244F4), CD48 (HM48–1), CD19 (eBio1D3), CD11c (N418), MHC Class I H-2Kb (AF6–88.5.5.3), IL-2 (JES6–1A12), IFNγ (XMG1.2) and CD107a (eBio1D4B) were purchased from eBioscience. For cytokine detection, samples were stimulated with Ag (OVA peptide and APC) or 50 ng/mL PMA and 1 μg/mL ionomycin in the presence of GolgiPlug (BD Biosciences), labeled with surface markers before treatment with fixation/permeabilization buffers (BD Biosciences) and intracellular staining. Sample acquisition was performed using a BD LSR II cytometer and FACSDiva software (BD Biosciences). Sample data were analyzed with Flowjo software version 8.8.6 (Tree Star, Inc.).
Proliferation assays
Ten thousand sorted WT, Sh2d1a−/− and 2B4−/− OT-I CD8+ T cells (∼99% CD8α+ CD11c−) were stimulated with indicated OVA concentrations and 2 × 105 sorted B cell (∼99% CD19+ CD11c−)- or B cell-depleted splenocyte-APCs (∼98% CD19−) with or without exogenous IL-2 (100 U/mL). B lymphoma cells expressing GFP (B-GFP) or OVA (B-OVA) were derived from terminally ill Eμ-myc transgenic mice transfected with expression cassettes containing GFP alone or a membrane-bound ovalbumin, passaged through mice and cryogenically preserved.34,35 For in vitro assays, B lymphoma cells were cultured from lymph nodes of transplanted mice and expanded as cell lines in DMEM containing 10% FCS, non-essential amino acids, 100 U/mL of penicillin/streptomycin and 55 μM β-mercaptoethanol (all Thermo Fisher Scientific). Antitumor proliferation assays were performed by stimulating 104 CFSE-labeled WT, Sh2d1a−/− and 2B4−/− OT-I CD8+ T cells with 105 B lymphoma cells, 104 B16-OVA or 104 NOP12 cells. Thymidine incorporation assays and CFSE labeling were performed as described previously.48,49 For blocking experiments, B cell, non-B cell and B lymphoma APCs were pre-coated with 10 μg/mL of SLAM family receptor CD48 (HM48–1), Ly9 and Ly108 antibodies or isotype-matched control antibodies for 1 h at 37° C.
CD8+ T cell adoptive transfers, B lymphoma challenge and bacterial infections
Age- and sex-matched CD45.1+ mice were infused with 5 × 105 CFSE-labeled wild type or Sh2d1a−/− OT-I CD8+ T cells (Thy1.1+CD45.2+) one day before intravenous injection of 5 × 105 OVA-expressing B cell lymphoma cells. For bacterial infections, recipient mice received 10,000 WT and Sh2d1a−/− CD8+ T cells one day before infection with 2 × 103 CFU of LM-OVA as described.50,51
Statistical analysis
Statistical significance was determined by unpaired, two-tailed Student's t-tests using Prism 6 software.
Supplementary Material
Disclosure of potential conflict of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Xiaoxia Wang, Huilian Qin and Maylene Wagener for technical assistance, Lisa Xu (BCCH Flow Cytometry Core Facility) for cell sorting, Drs. Ninghai Wang and Cox Terhorst (Harvard Medical School) for Ly108−/− spleens and Sh2d1a−/− breeder mice and Drs. Sandro Prato and Jose Villadangos (U. of Melbourne) for OVA-expressing B cell lymphoma cells. We are thankful to Drs. Rusung Tan (Sidra Medical and Research Center) and Troy E. Baldwin (U. of Alberta) for feedback.
Funding
This work was supported by funding from the Canadian Institutes of Health Research (MOP-10266) and Sidra Medical and Research Center (F14–01832).
Author contributions
YHH and JJP designed the research; YHH, KT, SYT and SK performed experiments; YHH, KWH and JJP analyzed data; KWH and MLF contributed vital reagents; YHH and JJP wrote the manuscript; YHH, KWH and JJP edited the manuscript.
References
- 1.Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD. The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol 2015; 33:787-821; PMID:25706097; http://dx.doi.org/ 10.1146/annurev-immunol-032414-112326 [DOI] [PubMed] [Google Scholar]
- 2.Coffey AJ, Brooksbank RA, Brandau O, Oohashi T, Howell GR, Bye JM, Cahn AP, Durham J, Heath P, Wray P et al.. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet 1998; 20:129-35; PMID:9771704; http://dx.doi.org/ 10.1038/2424 [DOI] [PubMed] [Google Scholar]
- 3.Nichols KE, Harkin DP, Levitz S, Krainer M, Kolquist KA, Genovese C, Bernard A, Ferguson M, Zuo L, Snyder E et al.. Inactivating mutations in an SH2 domain-encoding gene in X-linked lymphoproliferative syndrome. Proc Natl Acad Sci U S A 1998; 95:13765-70; PMID:9811875; http://dx.doi.org/ 10.1073/pnas.95.23.13765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sayos J, Wu C, Morra M, Wang N, Zhang X, Allen D, van Schaik S, Notarangelo L, Geha R, Roncarolo MG et al.. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature 1998; 395:462-9; PMID:9774102; http://dx.doi.org/ 10.1038/26683 [DOI] [PubMed] [Google Scholar]
- 5.Cannons JL, Tangye SG, Schwartzberg PL. SLAM family receptors and SAP adaptors in immunity. Annu Rev Immunol 2011; 29:665-705; PMID:21219180; http://dx.doi.org/ 10.1146/annurev-immunol-030409-101302 [DOI] [PubMed] [Google Scholar]
- 6.Dong Z, Veillette A. How do SAP family deficiencies compromise immunity? Trends Immunol 2010; 31:295-302; PMID:20650688; http://dx.doi.org/ 10.1016/j.it.2010.05.008 [DOI] [PubMed] [Google Scholar]
- 7.Tangye SG. XLP: clinical features and molecular etiology due to mutations in SH2D1A encoding SAP. J Clin Immunol 2014; 34:772-9; PMID:25085526; http://dx.doi.org/ 10.1007/s10875-014-0083-7 [DOI] [PubMed] [Google Scholar]
- 8.Fischer A. Human primary immunodeficiency diseases. Immunity 2007; 27:835-45; PMID:18093537; http://dx.doi.org/ 10.1016/j.immuni.2007.11.012 [DOI] [PubMed] [Google Scholar]
- 9.Turvey SE, Bonilla FA, Junker AK. Primary immunodeficiency diseases: a practical guide for clinicians. Postgrad Med J 2009; 85:660-6; PMID:20075404; http://dx.doi.org/ 10.1136/pgmj.2009.080630 [DOI] [PubMed] [Google Scholar]
- 10.Filipovich AH, Zhang K, Snow AL, Marsh RA. X-linked lymphoproliferative syndromes: brothers or distant cousins? Blood 2010; 116:3398-408; PMID:20660790; http://dx.doi.org/ 10.1182/blood-2010-03-275909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nichols KE, Ma CS, Cannons JL, Schwartzberg PL, Tangye SG. Molecular and cellular pathogenesis of X-linked lymphoproliferative disease. Immunol Rev 2005; 203:180-99; PMID:15661030; http://dx.doi.org/ 10.1111/j.0105-2896.2005.00230.x [DOI] [PubMed] [Google Scholar]
- 12.Milone MC, Tsai DE, Hodinka RL, Silverman LB, Malbran A, Wasik MA et al.. Treatment of primary Epstein-Barr virus infection in patients with X-linked lymphoproliferative disease using B-cell-directed therapy. Blood 2005; 105:994-6; PMID:15494422; http://dx.doi.org/ 10.1182/blood-2004-07-2965 [DOI] [PubMed] [Google Scholar]
- 13.Bond J, Shahdadpuri R, Mc Mahon C, O'Marcaigh A, Cotter M, Smith O. Successful treatment of acute Epstein-Barr virus infection associated with X-linked lymphoproliferative disorder with rituximab. Pediatr Blood Cancer 2007; 49:761-2; PMID:17066466; http://dx.doi.org/ 10.1002/pbc.21081 [DOI] [PubMed] [Google Scholar]
- 14.Crotty S, Kersh EN, Cannons J, Schwartzberg PL, Ahmed R. SAP is required for generating long-term humoral immunity. Nature 2003; 421:282-7; PMID:12529646; http://dx.doi.org/ 10.1038/nature01318 [DOI] [PubMed] [Google Scholar]
- 15.Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature 2008; 455:764-9; PMID:18843362; http://dx.doi.org/ 10.1038/nature07345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Veillette A, Zhang S, Shi X, Dong Z, Davidson D, Zhong MC. SAP expression in T cells, not in B cells, is required for humoral immunity. Proc Natl Acad Sci U S A 2008; 105:1273-8; PMID:18212118; http://dx.doi.org/ 10.1073/pnas.0710698105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cannons JL, Qi H, Lu KT, Dutta M, Gomez-Rodriguez J, Cheng J, Wakeland EK, Germain RN, Schwartzberg PL. Optimal germinal center responses require a multistage T cell:B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity 2010; 32:253-65; PMID:20153220; http://dx.doi.org/ 10.1016/j.immuni.2010.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chung B, Aoukaty A, Dutz J, Terhorst C, Tan R. Signaling lymphocytic activation molecule-associated protein controls NKT cell functions. J Immunol 2005; 174:3153-7; PMID:15749842; http://dx.doi.org/ 10.4049/jimmunol.174.6.3153 [DOI] [PubMed] [Google Scholar]
- 19.Pasquier B, Yin L, Fondaneche MC, Relouzat F, Bloch-Queyrat C, Lambert N, Fischer A, de Saint-Basile G, Latour S. Defective NKT cell development in mice and humans lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product. J Exp Med 2005; 201:695-701; PMID:15738056; http://dx.doi.org/ 10.1084/jem.20042432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nichols KE, Hom J, Gong SY, Ganguly A, Ma CS, Cannons JL, Tangye SG, Schwartzberg PL, Koretzky GA, Stein PL. Regulation of NKT cell development by SAP, the protein defective in XLP. Nat Med 2005; 11:340-5; PMID:15711562; http://dx.doi.org/ 10.1038/nm1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong Z, Cruz-Munoz ME, Zhong MC, Chen R, Latour S, Veillette A. Essential function for SAP family adaptors in the surveillance of hematopoietic cells by natural killer cells. Nat Immunol 2009; 10:973-80; PMID:19648922; http://dx.doi.org/ 10.1038/ni.1763 [DOI] [PubMed] [Google Scholar]
- 22.Bottino C, Falco M, Parolini S, Marcenaro E, Augugliaro R, Sivori S, Landi E, Biassoni R, Notarangelo LD, Moretta L et al.. NTB-A [correction of GNTB-A], a novel SH2D1A-associated surface molecule contributing to the inability of natural killer cells to kill Epstein-Barr virus-infected B cells in X-linked lymphoproliferative disease. J Exp Med 2001; 194:235-46; PMID:11489943; http://dx.doi.org/ 10.1084/jem.194.3.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parolini S, Bottino C, Falco M, Augugliaro R, Giliani S, Franceschini R, Ochs HD, Wolf H, Bonnefoy JY, Biassoni R et al.. X-linked lymphoproliferative disease. 2B4 molecules displaying inhibitory rather than activating function are responsible for the inability of natural killer cells to kill Epstein-Barr virus-infected cells. J Exp Med 2000; 192:337-46; PMID:10934222; http://dx.doi.org/ 10.1084/jem.192.3.337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hislop AD, Palendira U, Leese AM, Arkwright PD, Rohrlich PS, Tangye SG, Gaspar HB, Lankester AC, Moretta A, Rickinson AB. Impaired Epstein-Barr virus-specific CD8+ T-cell function in X-linked lymphoproliferative disease is restricted to SLAM family-positive B-cell targets. Blood 2010; 116:3249-57; PMID:20644117; http://dx.doi.org/ 10.1182/blood-2009-09-238832 [DOI] [PubMed] [Google Scholar]
- 25.Zhao F, Cannons JL, Dutta M, Griffiths GM, Schwartzberg PL. Positive and negative signaling through SLAM receptors regulate synapse organization and thresholds of cytolysis. Immunity 2012; 36:1003-16; PMID:22683123; http://dx.doi.org/ 10.1016/j.immuni.2012.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palendira U, Low C, Chan A, Hislop AD, Ho E, Phan TG, Deenick E, Cook MC, Riminton DS, Choo S et al.. Molecular pathogenesis of EBV susceptibility in XLP as revealed by analysis of female carriers with heterozygous expression of SAP. PLoS Biol 2011; 9:e1001187; PMID:22069374; http://dx.doi.org/ 10.1371/journal.pbio.1001187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dupre L, Andolfi G, Tangye SG, Clementi R, Locatelli F, Arico M, Aiuti A, Roncarolo MG. SAP controls the cytolytic activity of CD8+ T cells against EBV-infected cells. Blood 2005; 105:4383-9; PMID:15677558; http://dx.doi.org/ 10.1182/blood-2004-08-3269 [DOI] [PubMed] [Google Scholar]
- 28.Sharifi R, Sinclair JC, Gilmour KC, Arkwright PD, Kinnon C, Thrasher AJ, Gaspar HB. SAP mediates specific cytotoxic T-cell functions in X-linked lymphoproliferative disease. Blood 2004; 103:3821-7; PMID:14726378; http://dx.doi.org/ 10.1182/blood-2003-09-3359 [DOI] [PubMed] [Google Scholar]
- 29.Czar MJ, Kersh EN, Mijares LA, Lanier G, Lewis J, Yap G, Chen A, Sher A, Duckett CS, Ahmed R et al.. Altered lymphocyte responses and cytokine production in mice deficient in the X-linked lymphoproliferative disease gene SH2D1A/DSHP/SAP. Proc Natl Acad Sci U S A 2001; 98:7449-54; PMID:11404475; http://dx.doi.org/ 10.1073/pnas.131193098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu C, Nguyen KB, Pien GC, Wang N, Gullo C, Howie D, Sosa MR, Edwards MJ, Borrow P, Satoskar AR et al.. SAP controls T cell responses to virus and terminal differentiation of TH2 cells. Nat Immunol 2001; 2:410-4; PMID:11323694; http://dx.doi.org/ 10.1038/ni0901-823 [DOI] [PubMed] [Google Scholar]
- 31.Chen G, Tai AK, Lin M, Chang F, Terhorst C, Huber BT. Signaling lymphocyte activation molecule-associated protein is a negative regulator of the CD8 T cell response in mice. J Immunol 2005; 175:2212-8; PMID:16081788; http://dx.doi.org/ 10.4049/jimmunol.175.4.2212 [DOI] [PubMed] [Google Scholar]
- 32.Crotty S, McCausland MM, Aubert RD, Wherry EJ, Ahmed R. Hypogammaglobulinemia and exacerbated CD8 T-cell-mediated immunopathology in SAP-deficient mice with chronic LCMV infection mimics human XLP disease. Blood 2006; 108:3085-93; PMID:16788096; http://dx.doi.org/ 10.1182/blood-2006-04-018929 [DOI] [PubMed] [Google Scholar]
- 33.McArdel SL, Terhorst C, Sharpe AH. Roles of CD48 in regulating immunity and tolerance. Clin Immunol 2016; 164:10-20; PMID:26794910; http://dx.doi.org/ 10.1016/j.clim.2016.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985; 318:533-8; PMID:3906410; http://dx.doi.org/ 10.1038/318533a0 [DOI] [PubMed] [Google Scholar]
- 35.Prato S, Mintern JD, Lahoud MH, Huang DC, Villadangos JA. Induction of antigen-specific effector-phase tolerance following vaccination against a previously ignored B-cell lymphoma. Immunol Cell Biol 2011; 89:595-603; PMID:21079642; http://dx.doi.org/ 10.1038/icb.2010.131 [DOI] [PubMed] [Google Scholar]
- 36.Brown DM, Fisher TL, Wei C, Frelinger JG, Lord EM. Tumours can act as adjuvants for humoral immunity. Immunology 2001; 102:486-97; PMID:11328383; http://dx.doi.org/ 10.1046/j.1365-2567.2001.01213.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sio A, Chehal MK, Tsai K, Fan X, Roberts ME, Nelson BH, Grembecka J, Cierpicki T, Krebs DL, Harder KW. Dysregulated hematopoiesis caused by mammary cancer is associated with epigenetic changes and hox gene expression in hematopoietic cells. Cancer Res 2013; 73:5892-904; PMID:23913828; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-0842 [DOI] [PubMed] [Google Scholar]
- 38.Zenewicz LA, Shen H. Innate and adaptive immune responses to Listeria monocytogenes: a short overview. Microbes Infect 2007; 9:1208-15; PMID:17719259; http://dx.doi.org/ 10.1016/j.micinf.2007.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edelson BT. Dendritic cells in Listeria monocytogenes infection. Adv Immunol 2012; 113:33-49; PMID:22244578; http://dx.doi.org/ 10.1016/B978-0-12-394590-7.00006-3 [DOI] [PubMed] [Google Scholar]
- 40.Prato S, Zhan Y, Mintern JD, Villadangos JA. Rapid deletion and inactivation of CTLs upon recognition of a number of target cells over a critical threshold. J Immunol 2013; 191:3534-44; PMID:24018271; http://dx.doi.org/ 10.4049/jimmunol.1300803 [DOI] [PubMed] [Google Scholar]
- 41.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 2009; 10:29-37; PMID:19043418; http://dx.doi.org/ 10.1038/ni.1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chijioke O, Marcenaro E, Moretta A, Capaul R, Munz C. Role of the 2B4 Receptor in CD8+ T-Cell-Dependent Immune Control of Epstein-Barr Virus Infection in Mice With Reconstituted Human Immune System Components. J Infect Dis 2015; 212:803-7; PMID:25722295; http://dx.doi.org/ 10.1093/infdis/jiv114 [DOI] [PubMed] [Google Scholar]
- 43.Thorley-Lawson DA, Schooley RT, Bhan AK, Nadler LM. Epstein-Barr virus superinduces a new human B cell differentiation antigen (B-LAST 1) expressed on transformed lymphoblasts. Cell 1982; 30:415-25; PMID:6291768; http://dx.doi.org/ 10.1016/0092-8674(82)90239-2 [DOI] [PubMed] [Google Scholar]
- 44.Smith GM, Biggs J, Norris B, Anderson-Stewart P, Ward R. Detection of a soluble form of the leukocyte surface antigen CD48 in plasma and its elevation in patients with lymphoid leukemias and arthritis. J Clin Immunol 1997; 17:502-9; PMID:9418191; http://dx.doi.org/ 10.1023/A:1027327912204 [DOI] [PubMed] [Google Scholar]
- 45.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell 1994; 76:17-27; PMID:8287475; http://dx.doi.org/ 10.1016/0092-8674(94)90169-4 [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez-Cabrero J, Wise CJ, Latchman Y, Freeman GJ, Sharpe AH, Reiser H. CD48-deficient mice have a pronounced defect in CD4(+) T cell activation. Proc Natl Acad Sci U S A 1999; 96:1019-23; PMID:9927686; http://dx.doi.org/ 10.1073/pnas.96.3.1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu D, Krummey SM, Badell IR, Wagener M, Schneeweis LA, Stetsko DK et al.. 2B4 (CD244) induced by selective CD28 blockade functionally regulates allograft-specific CD8+ T cell responses. J Exp Med 2014; 211:297-311; PMID:24493803; http://dx.doi.org/ 10.1084/jem.20130902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Priatel JJ, Chen X, Huang YH, Chow MT, Zenewicz LA, Coughlin JJ, Shen H, Stone JC, Tan R, Teh HS. RasGRP1 regulates antigen-induced developmental programming by naive CD8 T cells. J Immunol 2010; 184:666-76; PMID:20007535; http://dx.doi.org/ 10.4049/jimmunol.0803521 [DOI] [PubMed] [Google Scholar]
- 49.Priatel JJ, Teh SJ, Dower NA, Stone JC, Teh HS. RasGRP1 transduces low-grade TCR signals which are critical for T cell development, homeostasis, and differentiation. Immunity 2002; 17:617-27; PMID:12433368; http://dx.doi.org/ 10.1016/S1074-7613(02)00451-X [DOI] [PubMed] [Google Scholar]
- 50.Foulds KE, Zenewicz LA, Shedlock DJ, Jiang J, Troy AE, Shen H. Cutting edge: CD4 and CD8 T cells are intrinsically different in their proliferative responses. J Immunol 2002; 168:1528-32; PMID:11823476; http://dx.doi.org/ 10.4049/jimmunol.168.4.1528 [DOI] [PubMed] [Google Scholar]
- 51.Priatel JJ, Chen X, Zenewicz LA, Shen H, Harder KW, Horwitz MS, Horwitz MS, Teh HS. Chronic immunodeficiency in mice lacking RasGRP1 results in CD4 T cell immune activation and exhaustion. J Immunol 2007; 179:2143-52; PMID:17675473; http://dx.doi.org/ 10.4049/jimmunol.179.4.2143 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.