

ABSTRACT
The Wilms' tumor oncogene protein (WT1) is a highly validated tumor antigen for immunotherapy. WT1-targeted immunotherapy has been extensively explored in multiple human trials in various cancers. However, clinical investigations using WT1 epitopes have generally focused on two peptides, HLA-restricted to HLA-A*02:01 or HLA-A*24:02. The goal of this study was to identify new epitopes derived from WT1, to expand the potential use of WT1 as a target of immunotherapy. Using computer-based MHC-binding algorithms and in vitro validation of the T cell responses specific for the identified peptides, we found that a recently identified HLA-A*24:02-binding epitope (239–247), NQMNLGATL (NQM), was also a strong CD8+ T cell epitope for HLA-A*02:01 molecule. A peptide second position Q240L substitution (NLM) or Q240Y substitution (NYM), further enhanced the T cell responses in both HLA-A*02:01 positive and HLA-A*24:02 positive healthy donors. Importantly, T cells stimulated with the new analog peptides displayed heteroclitic cross-reactivity with the native NQM sequence and were able to kill HLA-matched WT1-positive tumor cell lines and primary leukemia blasts. In addition, longer native and heteroclitic HLA-DR.B1-binding peptides, comprising the nine amino acid NQM or NLM sequences, could induce T cell response that recognized the CD8+ epitope NQM, suggesting the processing and the presentation by HLA-A*02:01 molecules of the CD8+ T cell epitope embedded within it. Our studies suggest that the analog peptides NLM and NYM could be potential candidates for future immunotherapy targeting WT1 positive cancers in the context of HLA-A*02:01 and A*24:02 positive populations.
KEYWORDS: Analog peptides, cancer vaccines, CTL response, immunotherapy, WT1
Introduction
WT1 is a zinc finger transcription factor that is normally expressed in mesodermal tissues during embryogenesis. In normal adult tissue, WT1 expression is limited to low levels in CD34+ haematopoietic stem cells, and renal podocytes; WT1 is overexpressed in leukemias of multiple lineages and a wide range of solid tumors.1-3 More recently, WT1 expression has been reported to be a marker of minimal residual disease in leukemia. Increasing transcript levels in patients with acute myeloid leukemia (AML) in morphologic remission have been predictive of overt clinical relapse.4,5 The lack of haematopoietic progenitor cell suppression by WT1 anti-sense treatment or WT1-specific CTLs suggests that WT1 is a highly selective cancer target for immunotherapy.6 Furthermore, antibodies to WT1 were detected in patients with haematopoietic malignancies and solid tumors, indicating that WT1 is a non-tolerizing antigen.7
Immunotherapy targeting WT1 has been extensively explored in pre-clinical studies and in numerous clinical trials in patients with hematological malignancies and solid tumors. Since WT1 is an intracellular protein that cannot be targeted by conventional monoclonal antibody (mAb) therapy, generating WT1-specific cytotoxic CD8+ T cell (CTL) responses that recognize peptides presented on cell surface by MHC class I molecule has been a major goal for WT1-targeted therapy. Active immunization with WT1-derived peptides, DNA and dendritic cell (DC) vaccines and adoptive transfer of WT1 epitope-specific T cells have been widely used as experimental approaches in a variety of human cancers. In some cases, clinical benefits have been shown to be associated with T cell responses.8-11 Most recently, the use of TCR mimic monoclonal antibodies to WT1 epitopes has been explored as another approach.12-15
To date, two HLA class I peptides have been the focus of much study worldwide. WT1 126–134 (RMFPNAPYL) is presented by human leukocyte antigen (HLA) HLA-A*02:01, a common HLA type in Caucasians and WT1 235–243 (CMTW), presented by HLA-A*24:02, a frequent HLA type in Japanese, other Asian, and Latino populations. Both peptides have been shown to be processed and presented by their restricting HLA-alleles to induce cytotoxic CD8+ T cells, capable of killing WT1-positive tumor cells.1,8,9 To enhance the immunogenicity of the native RMFPNAPYL (RMF) peptide, we generated an analog peptide by substituting the first amino acid with tyrosine, R126Y or YMFPNAPYL (YMF) peptide, which increased the stability of peptide binding to HLA-A*02:01 molecule. This peptide has been shown to induce more potent CD8+ T cell response, which recognizes the native peptide and kill the WT1-expressing leukemia cells.16 We have demonstrated that the WT1 YMF peptide vaccine induces immune responses in a high proportion of patients with both AML and thoracic malignancies in our phase I and II trials.9,17 Long survival was also observed in some AML patients who received the vaccinations.8,9,18 Similarly, an analog peptide for WT1–235 made by substitution of the second amino acid with tyrosine (M236Y or CYT) has been used in clinical trials in leukemias and various solid tumors and has demonstrated both immunological and clinical responses.1 These results are encouraging and have provided strong evidence and a rational for therapeutic targeting of the WT1-derived T cell epitopes for leukemias and other human cancers.
To help generate and sustain a long-lasting memory CD8+ T cell response, a strategy to include CD4+ T cell epitopes in vaccine design has received much attention and promising results. Detection of Th1-biased IgG1 Ab specific for WT1 protein in the sera of patients with AML implies that WT1-specific CD4+ T helper responses are present in these patients. WT1 class II peptides specific for multiple HLA-DRB1* haplotypes have been identified with helper function for CD8+ T cell responses, and in some cases, with direct CD4+ T cell cytotoxicity against the cancer.19-21
Human cancers commonly express multiple antigens (Ags). Targeting multiple Ags would reduce the chances of tumor escape in the setting of antigen loss variants. In the search for new T cell epitopes for the WT1 oncoprotein, a new WT1-derived peptide, NQMNLGATL, has recently been identified,22 as an immunogenic epitope for CD8+ T cells in the context of HLA-A*24:02 molecule. Here, we found that this peptide can also be presented by HLA-A*02:01, thereby also inducing strong T cell responses within this restriction. We generated an analog heteroclitic peptide by substitution of the glutamine at the second peptide position with leucine, a canonical anchor for HLA-A*02:01. This Q240L modification of the peptide further enhanced T cell responses in healthy donors with HLA-A*02:01 molecule. Importantly, T cells stimulated with the new analog peptide cross-reacted with its native peptide sequence and were able to kill HLA-matched leukemia cell lines and primary leukemia blasts. This new peptide could be a potential candidate for T cell-based therapy in HLA-A*02:01 or HLA-A*24:02 positive populations.
Results
Predicted binding of the native and its analog peptides to HLA-A*02:01 and HLA-A*24:02
Using a pool of 15 mer overlapping peptides spanning human WT1 protein to sensitize human T cells in vitro, the sequence 239–247 (NQMNLGTAL) has recently been identified as an immunogenic CD8+ T cell epitope in the context of HLA-A*24:02.22 In order to generate analog peptides with stronger immunogenicity, we first screened the prediction scores of the native peptide and possible analogs with various amino acid substitutions in the position 2 and 9 (class I anchor residues), using four online available databases (BIMAS, RANKPEP, SYFPEITHI and Net MHC) (Table 1). The predicted binding scores from all four databases showed better predicted binding of the native NQM peptide to HLA-A*02:01 than to HLA-A*24:02 molecule. When the glutamine at position 2 was substituted by leucine, the binding score to HLA-A*24:02 remained at a similar level by all prediction programs. However, a significantly stronger binding score was predicted for HLA-A*02:01 and 15-fold higher affinity. On the other hand, when the glutamine at the position 2 was substituted by tyrosine, binding score to HLA-A*24:02 was dramatically improved in three of four algorithms, showing about 90-fold increased binding by BIMAS prediction and 100-fold higher affinity. However, binding to HLA-A*02:01 generally decreased. All three peptides were predicted to be cleaved at the C-terminus by RANKPEP algorithm, suggesting the potential processing of the peptide fragment. We also checked the binding score by substitution with various amino acids at position 9 but none of them showed a significant improvement in binding compare with the substitution at the position 2. Therefore, the two analog peptides NLMNLGTAL and NYMNLGTAL (Q240L and Q240Y) were selected for further studies (Table 1).
Table 1.
Peptide binding to HLA-A*02:01 or HLA-A*24:02 was predicted by four different algorithms as indicated. Peptide binding to HLA-DR.B1 was predicted by SYFPETHI algorithm. All software can be found in the Materials and methods.
| HLA-class-II-binding scores by SYFPETHI prediction algorithm. | ||||||||
|---|---|---|---|---|---|---|---|---|
| BIMAS |
SYFPETHI |
RANK-PEP |
Net MHC (kd, nM) |
|||||
| Sequences (p239–247) | HLA-A*02:01 | HLA-A*24:02 | HLA-A*02:01 | HLA-A*24:02 | HLA-A*02:01 | HLA-A*24:02 | HLA-A*02:01 | HLA-A*24:02 |
| NQMNLGATL | 8.014 | 7.200 | 16 | 10 | 34 | 10.482 | 917 | 4343 |
| NLMNLGATL | 79.041 | 7.2 | 26 | 10 | 78 | 8.948 | 59 | 6771 |
| NYMNLGATL | 0.011 | 360.000 | 16 | 20 | 41 | 23.573 | 13011 | 56 |
HLA-class-II-binding scores by SYFPETHI prediction algorithm.
| Sequences (p238–252) | DR.B1-0101 | DR.B1-0301 | DR.B1-0401 | DR.B1-0701 | DR.B1-1101 | DR.B1-1501 |
|---|---|---|---|---|---|---|
| wNQMNLGATLkgvaa | 17 | 13 | 14 | 16 | 13 | 24 |
| wNLMNLGATLkgvaa | 17 | 13 | 14 | 16 | 13 | 24 |
Binding of the peptides to HLA-A*02:01 and HLA-A*24:02 molecules
The immunogenicity of MHC class I-restricted peptides requires the capacity to bind and stabilize MHC class I molecules on the live cell surface. Moreover, the computer predictions are typically no more than 70–80% accurate; therefore we sought direct measurement of the strength of the interaction between the peptides and the HLA-A*02:01 molecules using a conventional binding and stabilization assay that uses the TAP deficient HLA-A*02:01 human T2 cells. T2 cells lack TAP function and consequently are defective in properly loading class I molecules with antigenic peptides generated in the cytosol. The association of exogenously added peptides with thermolabile, empty HLA-A*02:01 molecules stabilizes them and results in an increase in the level of surface HLA-A*02:01 recognizable by specific anti-HLA-A*02:01 mAb such as BB7.2.
The T2-binding assay showed that native NQM peptide did not stabilize the HLA-A2 expression on T2 cells (Fig. 1A). However, the NLM analog peptide stabilized the HLA-A2 molecule in a dose-dependent manner (Fig. 1B). Similar to the native peptide NQM, the NYM peptide, which showed no consistent predicted increase in binding HLA-A2 (Table 1), did not increase HLA-A2 expression (Fig. 1C). These data confirmed that NLM peptide is a stronger binder to HLA-A*0201, as predicted by the computer-based algorithm.
Figure 1.
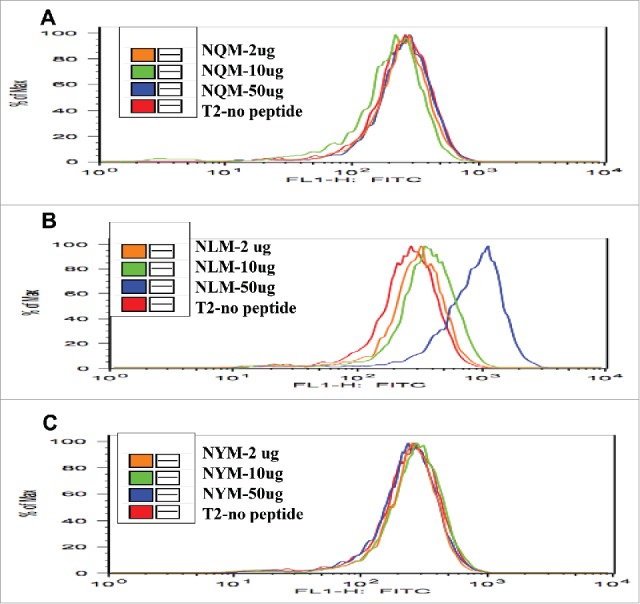
T2 stabilization assay with NQM peptides and analogs. The binding of NLM peptide to HLA-A2 molecule was stronger than NQM and NYM peptides. Native NQM, heteroclitic NLM or NYM peptides were pulsed onto T2 cells at the indicated concentrations as described in the Materials and methods and shown in the inset. The stabilization of the HLA-A2 molecule by the peptides was measured by the expression of HLA-A2 molecule using an anti-HLA-A2 mAb, clone BB7, conjugated to FITC, and recorded as fluorescence on the x-axis.
Induction of peptide-specific cytotoxic T lymphocyte (CTL) response in the context of HLA-A*02:01 and A*24:02 molecules
Although affinity for MHC molecules is necessary for peptide presentation, it is not always sufficient for T cell recognition and response. The T cells may be tolerant or anergic to the epitope, or the reactivity may be absent from the TCR repertoire. Therefore, using a well-established stimulation protocol in vitro,16 we investigated whether the new synthetic WT1 analogs could stimulate peptide-specific T cell response in both HLA-A*02:01 and A*24:02 donors.
To induce peptide-specific T cell response in healthy human donors, three to five in vitro stimulations were performed and the specificity of T cell response was measured by IFN-gamma production, when challenged with individual peptide (Fig. 2). We first compared the NQM peptide with the analog peptide YMF of the WT1 epitope RMF peptide, in eliciting T cell response in HLA-A*02:01+ donors. Both NQM and YMF peptides induced peptide-specific T cell response after three stimulations (Fig. 2A); responses were significantly enhanced after five stimulations (Fig. 2B). (Please note scale differences in panels.) The NQM-stimulated T cells recognized the native NQM peptide and also its analog peptides NLM and NYM, but not unrelated WT1 sequences RMF or YMF. Similarly, the YMF peptide induced strong T cell response against YMF and RMF, but not any of the WT1 239 peptides or the WT1 235 peptide CMTW. Importantly, the data demonstrated that the NQM peptide is a strong T cell stimulator in the context of HLA-A*02:01 molecule, comparable to the well-studied YMF peptide. Peptide-specific T cell responses induced by NQM and NLM peptide were further tested and confirmed in multiple HLA-A*02:01+ donors. The strength of the T cell response induced by NQM and NLM was highly variable among the donors used (compare panels A–D). In some donors, responses were far lower for NQM than NLM (Fig. 2C), and in others the reverse was true (Fig. 2D). This was seen as well in the cytotoxicity and Elispot assays described in other figures below. The differences did not appear to be related to heterozygosity of the A02 allele. However, both peptide-induced T cell responses were specific for the stimulating peptides. In addition, there was a strong cross-reactivity between native and analog peptides. The NYM peptide, could also induce peptide-specific IFN-gamma secretion after repeated stimulation of T cells from a donor of HLA-A*02:01. However, the response was generally weaker than NQM and NLM peptides in multiple HLA-A*02:01+ donors.
Figure 2.
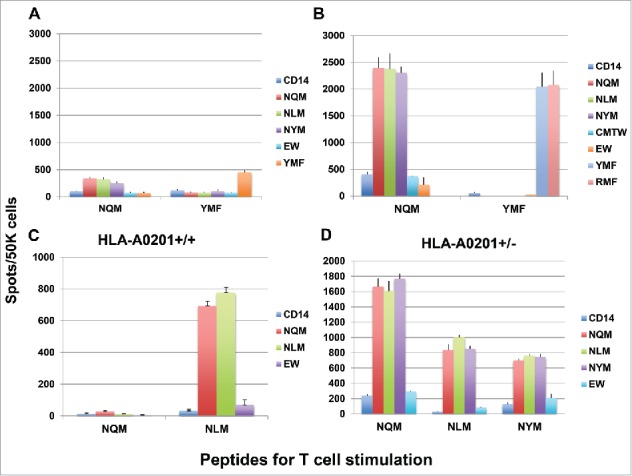
Peptide-specific T cell responses measured by gamma interferon ELISPOT assays. (A and B) The NQM induces peptide-specific T cell response, comparable to the peptide YMF, an analog peptide for RMF epitope. CD3 T cells from a healthy HLA-A*02:01+ donor were stimulated with either NQM or YMF peptide for three (A) or five (B) rounds. The peptide-specific response was measured by the IFN-gamma Elispot assay by pulsing autologous CD14+ cells (antigen-presenting cells, APC) with individual peptide. Each data point represents a mean +/− SD from triplicate cultures. (C and D) The NLM peptide induced strong peptide-specific T cell responses which cross-reacted to the native sequence NQM in a HLA-A*02:01 homozygous donor. CD3 T cells from a healthy HLA-A*02:01 homozygous donor were stimulated with either NQM or NLM peptide three times (C). Similarly, T cells from a HLA-A*02:01 heterozygous donor were stimulated with NQM, NLM, or NYM three times (D) and the peptide-specific response was measured by the IFN-gamma secretion upon challenging with individual peptide. Each data point represents the mean +/− SD from triplicate cultures. Data are representative of results from ten experiments from six donors.
We next tested if NQM or NLM peptide could generate CTLs that could recognize and specifically kill NQM-expressing target cells. NLM-stimulated CTLs showed cytotoxicity against T2 cells pulsed with NQM peptide, as compare with T2 cells alone or pulsed with control HLA-A2-binding EW peptide. NQM peptide also showed cytotoxicity against T2 pulsed with NQM peptide, but the net killing was weaker than NLM-induced CTLs (Fig. 3A vs. 3B). To verify recognition of the naturally processed NQM epitope, CTLs were generated from HLA-A*02:01+ donors by stimulating with NLM peptide. NLM-induced CTLs recognized and lysed WT1+HLA-A*02:01+ leukemia cell line 697, but not WT1+HLA-A*02:01− AML cell line HL-60, nor HLA-A*02:01+ WT1-negative leukemia cells SKLY-16 (Fig. 3C). Importantly, the CTL were also able to kill leukemia blasts from a HLA-A*02:01+ patient with AML, but not the blasts from a patient who is non HLA-A*02:01 nor A*24:02 (Fig. 3D). When T cells from a donor who is HLA- A*02:01 homozygous were stimulated with NQM, NLM or NYM peptide, the best killing against WT1+ HLA-A*02:01+ leukemia cell lines 697 and BV173 was seen by T cells stimulated with NLM peptide, followed by NQM, but not NYM peptide (Figs. 4A, B and C). We further confirmed that NLM-stimulated T cells were able to lyse AML blasts in a HLA-A0201-restricted manner and that IFN-gamma pre-treatment enhanced the cytotoxicity (red line), suggesting that the NQM epitope could be better processed and presented by HLA-class I molecule (Fig. 4D), after IFN-gamma treatment.
Figure 3.
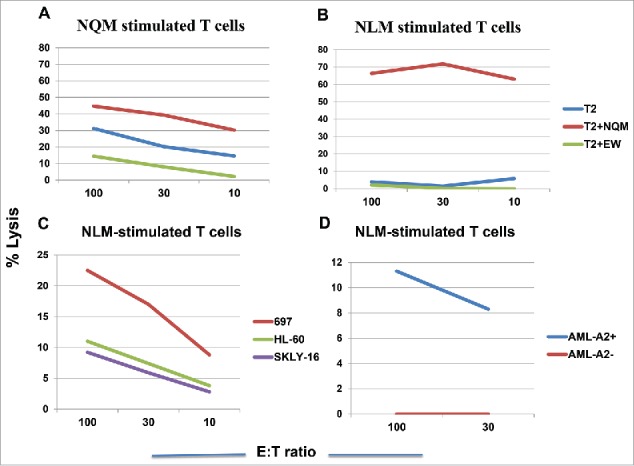
NLM peptide induced peptide-specific cytotoxicity by T cells that recognizes NQM native sequence. T cells from a HLA-A*02:01+ donor were stimulated with either NQM (A) or NLM peptide (B ) for five times and the cytotoxicity was measured by standard 51Cr-release assay against T2 cells pulsed with NQM peptide. Similarly, CD3T cells from two different donors who are HLA-A*02:01+, were stimulated with analog NLM peptide for five rounds and the cytotoxicity was measured against WT1+ HLA-A*02:01+ leukemia cells 697, WT1- HLA-A*02:01+ leukemia cells SKLY-16, WT1+ HLA-A*02:01− leukemia cell line HL-60 (C) and leukemia blasts derived from a HLA-A*02:01+ or negative patient with AML (D). Each data point represents the mean from triplicate micro-well cultures. Data represent results from seven experiments from three donors in panels A, B and C, and three experiments from three pairs of samples from AML patients.
Figure 4.
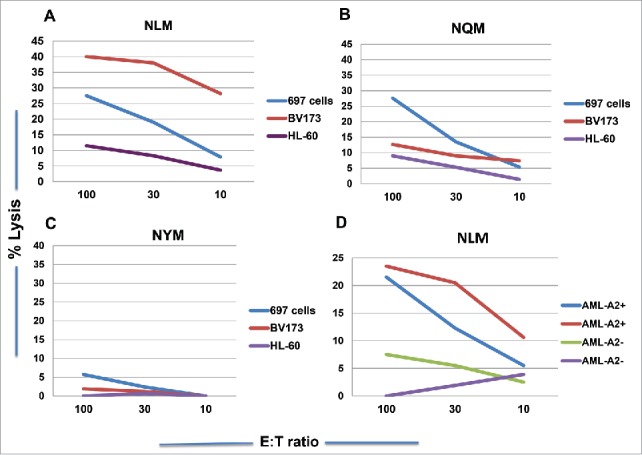
NLM peptide induced stronger T cell cytotoxicity against WT1+ leukemia cells in the context of HLA-A*02:01 molecule. CD3T cells from a donor who is HLA-A*02:01 homozygous were stimulated with analog NLM peptide (A), native NQM peptide (B) and NYM peptide (C) for five rounds and the cytotoxicity was measured against WT1+ HLA-A*02:01+ leukemia cells 697 and BV173 and WT1+ HLA-A*02:01− leukemia cell line HL-60. The T cells also lysed AML blasts from a patient who is WT1+ HLA-A*02:01+ (blue line) and IFN-gamma treatment further enhanced the cytotoxicity (red line). No killing was observed against the AML blasts from a patient who is WT1+, but HLA-A*02:01 negative (green line: untreated and purple line: with IFN-gamma) (D). Each data point represents the mean from triplicate micro-well cultures. Data represent results from seven experiments from three donors in panels A, B and C, and three experiments from three pairs of samples from AML patients.
We next assessed if the analog peptide for HLA-A*24:02, NYM, could induce CTLs in the context of HLA-A*24:02 molecule. Indeed, NYM peptide induced a strong peptide-specific T cell response showing by IFN-gamma secretion, which cross-reacted with native sequence NQM, after three stimulations. In contrast, the NQM and NLM peptides induced little response, or a far weaker response, respectively; both of these peptides have favorable affinity for HLA-A*02:01 molecule rather than A*24:02 (Fig. 5A). Importantly, both NQM and NYM peptides induced CTLs that killed a HLA-A*24:02+ WT1+ mesothelioma cell line, and a HLA-A*24:02+ B cell leukemia BA25, but not the A*24:02 negative HL-60 cell line (Figs. 5B, C and D). These data demonstrated that NYM analog peptide is a strong epitope for CD8+ T cells in the context of HLA-A*24:021 molecule.
Figure 5.
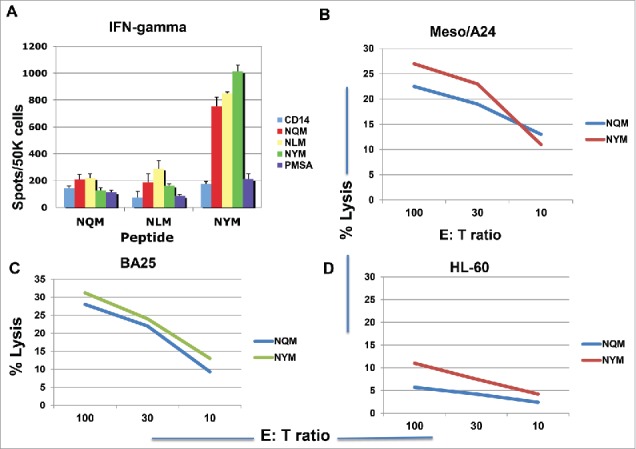
Peptide-specific T cell responses and cytotoxicity in HLA- A*24:02+ donors. CD3 T cells from a healthy HLA-A*24:02 homozygous donor were stimulated with NQM, NLM or NYM peptides for three rounds. The peptide-specific T cell response was measured by the IFN-gamma secretion upon challenging with individual peptide (A). Each data point represents the mean +/− SD from triplicate cultures. CD3 T cells from two different donors were stimulated with either NQM or NYM peptide for four to five rounds and the cytotoxicity was measured by 51Cr-release assay against WT1+ HLA- A*24:02+ mesothelioma cell line (B) leukemia cell line BA25 (C) and the A*24:02 negative line HL60 (D). Each data point represents the mean from triplicate micro-well cultures. Data are representative of 12 experiments from five HLA-A*24:02+ donors.
Induction of T cell responses by HLA-DR. B1 peptides that recognize NQM CD8+ T cell epitopes
It has been shown19,20 that a peptide combining both CD4+ and CD8+ epitopes can be more effective than the single class I epitope in eliciting effective immune response for vaccine design, because CD4+ T cells can help CD8+ CTL by fully activating DCs through the CD40/CD40L signaling as well as by producing IL-2 and IFN-gamma. In addition, if T cells stimulated with longer peptides recognize the shorter CD8+ epitopes, embedded within long peptides, it would confirm the processing and the presentation of the CD8+ T cell epitopes from the larger protein. Therefore, we designed two HLA-DR.B1-binding peptides, that span the NQM and NLM epitopes, respectively, and bind to multiple HLA-DR.B1 alleles at various degrees, predicted by the SYFPEITHI algorithms (Table 1). The HLA-DR.B1 peptides spanning NQM and NLM short peptides were designated as DR238-native and DR238-heteroclitic, respectively.
We investigated whether the inclusion of either NQM or NLM in DR.B1–238 native or heteroclitic peptide would induce both CD4+ and CD8+ T cell responses by stimulating CD3 T cells with both DR peptides, from a donor who is HLA-A*02:01+/A*24:02+ and DR.B1 0801/1001. The DR238-native peptide induced peptide-specific IFN-gamma secretion directed primarily to itself, and the DR238-heteroclitic analog peptide induced strong T cell responses against the CD8+ epitopes NQM, NLM, NYM and also both native and heteroclitic DR238 peptides (Fig. 6 A). This suggests that the long peptide could simultaneously induce both CD4+ and CD8+ T cell responses and more importantly, the NQM CD8+ epitopes are processed and presented by HLA-A*02:01 and A*24:02 molecules, as the donor is HLA-A*02:01 and A*24:02 positive. The ability of inducing both CD4+ and CD8+ T cell responses by both DR238 peptides was confirmed in multiple donors with various HLA-DR.B1 haplotypes. When T cells from a donor, who was homozygous HLA-A*02:01 and DR.B1 0701/1101+, were stimulated with DR238 native peptide for five rounds, a strong peptide-specific HLA-DR-restricted response was induced. The addition of anti-HLA-DR antibody blocked the peptide-specific response (Fig. 6B). The T cells were also able to kill WT1+HLA-A*02:01+ AML cell line SET-2 but not HL-60 (Fig. 6C). The DR238-heteroclitic peptide also induced a HLA-class-I-restricted T cell response from the same donor, because the anti-HLA-class I mAb abrogated the IFN-gamma secretion against CD8+ epitopes NQM and NLM (Fig. 6D). Consistently, the T cells killed HLA-A*02:01+ leukemia cells 697 cells, but not HL-60 cells (Fig. 6E).
Figure 6.
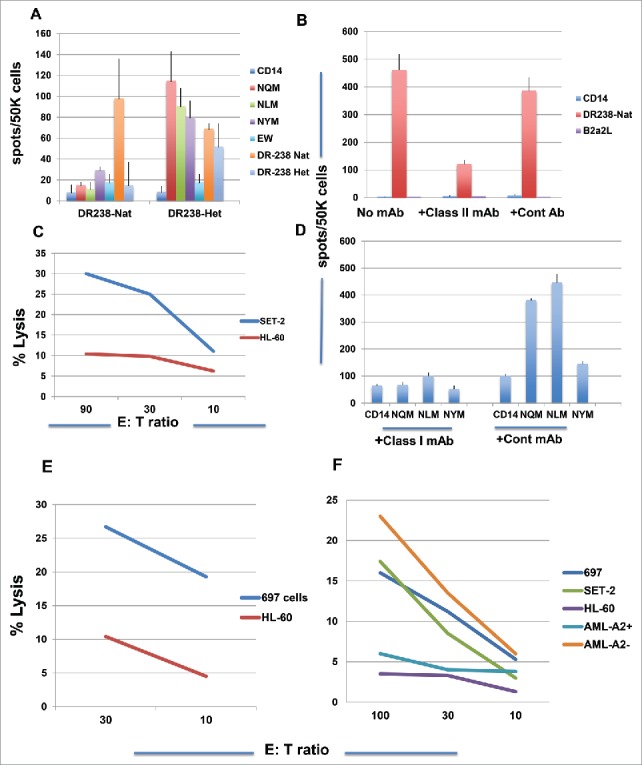
WT1 derived HLA-DR.B1 peptide induced T cell responses that recognized both class I and class II peptides. (A) CD3 T cells from a donor (HLA-A*02:01+/A*24:02+) were stimulated with the DR-238 native or analog peptide five times and the peptide-specific response was measured by IFN-gamma Elispot assay against individual peptides . Each data point represents the mean +/− SD from triplicate cultures. Similarly, CD3 T cells from a donor (HLA-A*02:01+/+) were stimulated with DR-238 native peptide (B) or DR-238 heteroclitic peptide (D) for three rounds, and the IFN-gamma secretion was measured with or without antibodies specific for the HLA-DR or its isotype control mAb (Cont mAb) (B) or HLA-ABC (D). HLA-A*02:01-restricted T cells cytotoxicity was measured against WT1+ leukemia cells SET-2 (C) and 697 (E) after five rounds of stimulation of the T cells. Fig. 6F Cytotoxicity of T cells stimulated five rounds from a donor who is HLA-A0201+ and DR.B1–15xx was measured against WT1+ HLA-A*02:01+ leukemia cells 697, SET-2, WT1+ HLA-A*02:01− leukemia cells HL-60 and AML blasts from patients who are HLA-A*02:01+ or negative. Each data point represents the mean from triplicate cultures. Data represent results from seven experiments from four donors.
These results demonstrated that short WT1–239 fragment is processed and presented to be recognized by HLA-A*02:01 molecule. Finally, when T cells from a donor, who is HLA-A*02:01+ and DR.B1 07xx/15xx, were stimulated with DR238 analog peptide for four rounds, a CTL response was generated against WT1+, HLA-A*02:01+ tumor cells 697, SET-2, but not HLA-A*02:01 negative HL-60 cells. The T cells also killed AML blasts from a HLA-A*02:01 negative patient, who shares the DR.B1 allele with the donor T cells (DR.B1 15xx). As shown in Table 1, this peptide is predicted to have binding affinity to HLA-DR.B1–15:01. The data suggest possible HLA-DR.B1-restricted T cell killing. We do not have explanation for the non-cytotoxicity against the AML blasts from a patient who was WT1+HLA-A*02:01+ in this experiment, but the primary samples are often variable (Fig. 6F).
Discussion
Clinical success of immune checkpoint inhibitors and adoptive transfer of T cells specific for tumor-associated antigens has firmly demonstrated that the immune system can effectively control subsets of a wide range of cancers.23 Recent technological advances in therapeutic platforms using chimeric antigen receptors in T cells (CAR T cells) and bi-specific antibodies significantly improved the efficacy of immunotherapy.24,25 Using T cell receptor mimic monoclonal antibodies (TCRm mAbs), it is now possible to target intracellular tumor antigens after presentation of their peptides on the cell surface with more powerful and versatile approaches.12-15,26 There is now renewed interest in developing cancer vaccines. In pre-clinical mouse models, therapeutic vaccination against neoantigens had the same efficacy as checkpoint blockade therapy.27-29 Thus, it may be possible to achieve effective vaccine therapy in conjunction with immunomodulation. Neoantigens as targets are typically limited to individual patients, making this strategy difficult to expand widely to large populations. Therefore, it is crucial to identify tumor-specific, shared targets in a broader range of cancers. WT1 is a promising target because of its abundant and wide range expression in many human cancers, cancer stem cells, and its limited expression in normal tissues. In a National Cancer Institute pilot project aimed at prioritizing cancer antigens, WT1 was identified as the highest priority antigen.30 Clinical trials of immunotherapy directed to WT1 in leukemias and other malignancies have shown that WT1-specific T cell response could be induced and objective clinical responses have been observed in some patients.1,8,18,31 Currently, there are numerous immunotherapy trials focusing on WT1 as a target, by variety of approaches ranging from peptide vaccination to adoptive T cell transfer (Clinicaltrials.gov). Multiple HLA class I and class II-restricted T cell epitopes of WT1 have been studied, but most work has been focused on two HLA class I peptides: WT1–126 in the context of HLA-A*02:01 and WT1–235, in the context of HLA-A*24:02. In addition, the WT1–332 epitope has been widely accepted as a CD4+ T cell epitope that can be presented by multiple HLA-DR.B1 alleles.21,32 A vaccine consisting of four peptides with multiple class 1 and class 2 epitopes is advancing into late phase trials in AML and mesothelioma.9,17 The identification of additional epitopes that are restricted by other HLA alleles would be important to expand WT1-targeted immunotherapy to more patients.
In a search of new T cell epitopes derived from WT1, 41 new epitopes that are presented by multiple HLA class I or class II alleles were identified using a pool of overlapping 15-mer peptides spanning the entire amino acid sequences of WT1 protein. The peptide, WT1–239–247, (NQM) has been shown to induce T cell response in the context of HLA-A*24:02, that were cytotolytic against peptide loaded autologous antigen-presenting cells (APCs) and more importantly, leukemia blasts.22 Using computational algorithms to predict the binding and C-terminal cleavage of peptides to HLA-class I, we found that this epitope preferentially binds to HLA-A*02:01 molecule, and is cleaved in C-terminus, suggesting a high probability of the presentation of the epitope. Substitution of the position 2 with leucine (NLM) or tyrosine (NYM), predicted enhanced binding to HLA-A*02:01 or A*24:02 molecules, respectively. We directly confirmed stronger binding of the peptide NLM to HLA-A2 molecule in vitro and demonstrated stimulation and cytolysis of T cells with the synthetic peptides. Both native NQM and NLM peptides elicited CD8+ T cell responses in HLA-A*02:01+ healthy donors that were able to specifically lyse tumor cells lines that were both WT1 and HLA-A*02:01 positive, demonstrating the heteroclitic properties of the analog peptides. The NQM peptide also induced T cell responses in HLA-A*24:02+ donors, but to a lesser degree than the response seen in HLA- A*02:01+ donors. On the other hand, the NYM peptide, as predicted by algorithm, induced a robust CD8+ T cell responses in the context of HLA-A*24:02 molecule and was able to kill tumor cells expressing both HLA-A*24:02 and WT1. These results demonstrated that the synthetic NQM and NLM peptides could be new epitopes for WT1-targeted immunotherapy primarily in HLA-A*02:01 population, while NYM peptide could be used primarily in HLA-A*24:02+ donors.
We also designed HLA-DR-binding peptides, in which either the native NQM sequence or analog CD8+ epitope NLM sequence are embedded. Inducing long-lasting and robust CD8+ T cells requires CD4+ T cell help, which provides cytokines and licensing DCs to present antigen more efficiently. In addition to providing this indirect help for CD8+ T cells, direct antitumor activity of CD4+ T cells has also been demonstrated repeatedly recently, including CD4+ T killing against leukemia cells.20 Recent clinical studies in adoptive transfer of neoantigen-specific T cell have also highlighted the importance of CD4+ T cells in eliminating and controlling tumor growth and metastasis. Most importantly, CD8+ or CD4+ T cell epitopes must be processed and presented on the surface of tumor cells. By stimulating CD3 T cells with longer peptides, it is possible to test CD8+ T cell response against the nested shorter sequences that are presented by HLA class I molecules. Indeed, the HLA-DR.B1–238 native and heteroclitic peptides induced specific T cell responses against both HLA-DR.B1- and HLA-A*02:01-binding epitopes, suggesting a simultaneous induction of CD4+ and CD8+ T cell responses, and the required processing and presentation of the shorter sequences NQM and NLM. Interestingly, we also observed that HLA-DR-238 analog peptides induced a HLA-DR.B1–15-restricted cytotoxic T cell responses against AML blasts from a patient who is not HLA-A*02:01. These data suggest that in this instance the CD4+ T cells are the cytolytic effectors that were recognizing the CD4+ epitopes presented by HLA-DR.B1–15xx molecule.
In summary, the WT1 NQM epitope, its longer version, and two analog peptides may be used to broaden the applicability of WT1-based treatment strategies. These epitopes can be used as targets of cancer vaccines, adoptive T cell therapy, TCRm antibodies, or CAR-T cells derived from TCRm mAb.
Materials and methods
Peptides. All peptides were purchased and synthesized by Genemed Synthesis, Inc. (San Antonio, TX). Peptides were sterile with purity of more than 70% to 90%. The peptides were dissolved in DMSO and diluted in saline at 5 mg/mL and stored at −80°C. Amino acid sequences and predicted binding of putative CD4+epitopes to HLA-DRB1 molecules, HLA-A*02:01 and HLA-A*24:02 were identified using the predictive algorithm of the SYFPEITHI, RANKPEP and BIMAS, and were confirmed with the Net MHC algorithm. HLA-DR.B1 peptides were selected using the predictive algorithm of the SYFPEITHI. Control peptides used are as follows: for HLA-DR.B1: BCR.ABL-derived peptide B2A2L (IVHSATGFKQSSKALQRPVASDFEP); for HLA-A*02:01: Ewing sarcoma-derived peptide EW (QLQNPSYDK) and for HLA-A*24:02: prostate-specific membrane antigen (PMSA)-derived peptide 624–632 (TYSVSFDSL).
Samples, cell lines, cytokines and antibodies. After informed consent on Memorial Sloan-Kettering cancer Center (MSKCC) Institutional Review Board-reviewed protocols, PBMCs from HLA-typed healthy donors and patients were obtained by Ficoll density centrifugation. Human leukemia cell lines SET-2, BV173, 697, SKLY-16, BA25, HL-60, and a mesothelioma cell line Meso/A24 were used as a targets for measuring cytotoxicity of T cells. WT1 expression and HLA phenotyping were described previously.20 Human granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin (IL)-1β, IL-4, IL-6, IL-15, tumor necrosis factor (TNF)-α and prostglandin E2 (PGE2) were purchased from R&D Systems (Minneapolis, MN). β 2-microglobulin (b2-m) and human recombinant interferon (IFN)-gamma were purchased from Sigma (St. Louis, MO). In some cases, AML blasts were pre-treated with human recombinant IFN-gamma (100 ng/mL) for 2 d to upregulate HLA expression, when used as targets for 51Cr-release assays. The antibodies used for immunofluorescence assays including mAbs to human CD3, CD4+, CD8+, HLA-A2 (clone BB7.2) and isotype controls were obtained from BD Biosciences (San Diego, CA). The antibodies specific for HLA class II DR, DQ and DP L243 and HLA-class I ABC W6/32 were obtained from the mAb core facility at MSKCC. Cell isolation kits for CD14 and CD3 were purchased from Miltenyi Biotec. (Bergisch Gladbach, Germany).
T2 assay for peptide binding. T2 cells (transporter-associated protein TAP deficient, HLA-A*02:01+) were incubated overnight at 37°C at 1×106 cells/mL in FCS-free RPMI medium supplemented with 10 ug/mL human β2m (Sigma, St Louis, MO, USA) in the absence (negative control) or presence peptides at various final concentrations (50, 10 and 2 ug/mL). Brefeldin A (Sigma) at 5 ug/mL was added to the cultures for the final 2 h of incubation. Then, T2 cells were washed and stained with anti-HLA-A2.1 (BB7.2) mAb conjugated to FITC for 30 min at 4°C and followed by washing with staining buffer (PBS plus 1% FBS and 0.02% azaid). The expression of the HLA-A2 on the cell surface was measured by flow cytometry on a FACScalibur (Becton Dickinson) and analyzed with FlowJo 9.6.3 software.
In vitro stimulation and human T-cell cultures. After informed consent on Memorial Sloan-Kettering Cancer Center Institutional Review Board approved protocols, peripheral blood mononuclear cells (PBMCs) from HLA-typed healthy donors were obtained by Ficoll density centrifugation. CD14+ monocytes were isolated by positive selection using mAb to human CD14 coupled with magnetic beads (Miltenyi Biotec) and were used for the first stimulation of T cells. The CD14− fraction of PBMC were used for isolation of CD3, by negative immunomagnetic cell separation using a pan T cell isolation kit (Miltenyi Biotec). The purity of the cells was always more than 98%. T cells were stimulated for 7 d in the presence of RPMI 1640 supplemented with 5% autologous plasma (AP), 20 ug/mL synthetic peptides, 1 ug/mL B2-m, and 10 ng/mL IL-15. Monocyte-derived DCs were generated from CD14+ cells, by culturing the cells in RPMI 1640 medium supplemented with 1% AP, 500 units/mL recombinant IL-4, and 1,000 units/mL GM-CSF. On days 2 and 4 of incubation, fresh medium with IL-4 and GM-CSF was either added or replaced half of the culture medium. On day 5, 20 ug/mL class II peptide was added to the immature DCs, for the processing. On day 6, maturation cytokine cocktail was added. On day 7 or 8, T cells were re-stimulated with mature DCs, with IL-15. In most cases, T cells were stimulated three times in the same manner, using either DCs or CD14+ cells as APCs. A week after final stimulation, the peptide-specific T cell response was examined by IFN-gamma enzyme-linked immunospot (ELISPOT) assay and the cytotoxicity was tested, by 51chromium (Cr)-release assay.
IFN-gamma ELISPOT. HA-Multiscreen plates (Millipore) were coated with 100 uL of mouse anti-human IFN-gamma antibody (10 ug/mL; clone 1-D1K; Mabtech) in PBS, incubated overnight at 4°C, washed with PBS to remove unbound antibody, and blocked with RPMI 1640/10% autologous plasma (AP) for 2 h at 37°C. Purified CD3+ T cells (> 98% pure) were plated with either autologous CD14+ (10:1 E: APC ratio) or autologous DCs (30:1 E: APC ratio). Various test peptides were added to the wells at 20 ug/mL. Negative control wells contained APCs and T cells without peptides or with irrelevant peptides. Positive control wells contained T cells plus APCs plus 20 ug/mL phytohemagglutinin (PHA, Sigma). All conditions were done in triplicates. Microtiter plates were incubated for 20 h at 37°C and then extensively washed with PBS/0.05% Tween and 100 uL/well biotinylated detection antibody against human IFN-g (2 ug/mL; clone 7-B6–1; Mabtech) was added. Plates were incubated for an additional 2 h at 37°C and spot development was done as described.16 Spot numbers were read and determined by Zellnet Consulting Inc.
51Chromium release assay. The presence of specific CTLs was measured in a standard chromium release assay as described.16 Briefly, target cells alone, or pulsed with 50 ug/mL of synthetic peptides for 2 h (in some cases for over-night) at 37°C, are labeled with 50 uCi/million cells of Na2 51CrO4 (NEN Life Science Products, Inc.). After extensive washing, target cells are incubated with T cells at various E:T ratios. All conditions were done in triplicate. Plates were incubated for 4–5 h at 37°C in 5% CO2. Supernatant fluids were harvested and radioactivity was measured in a gamma counter. Percentage specific lysis was determined from the following formula: [(experimental release − spontaneous release)/(maximum release – spontaneous release)] × 100%. Maximum release was determined by lysis of radiolabeled targets in 1% SDS.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr Annamalai Selvakumar and Alice Yeh for their expert HLA typing and Dr Sylvia Zellenet for Elispot evaluation.
Funding
This study was supported by: NIH RO1 CA55349, PO1 CA23766, P30 CA008748, MSK Experimental Therapeutics Center, Sellas Life Sciences, The lymphoma Foundation, the Tudor Fund.
References
- 1.Oka Y, Tsuboi A, Elisseeva OA, Nakajima H, Fujiki F, Kawakami M, Shirakata T, Nisjida S, Hosen N, Oji Y, et al.. WT1 peptide cancer vaccine for patients with hematopoietic malignancies and solid cancers. Scientific World J 2007; 7:649-665; PMID:17619750; http://dx.doi.org/ 10.1100/tsw.2007.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mundlos S, Pelletier J, Darveau A, Bachmann M, Winterpacht A, Zabel B. Nuclear localization of the protein encoded by the Wilms' tumor gene WT1 in embryonic and adult tissues. Development 1993; 119:1329-41; PMID:8306891 [DOI] [PubMed] [Google Scholar]
- 3.Keilholz U, Menssen HD, Craiger A, Menke A, Oji Y, Oka Y, Scheinbenbogen C, Stauss H, Thiel E, Sugiyama H. Wilms' tumor gene 1 (WT1) in human neoplasia. Leukemia 2005; 19:1318-13231; PMID:15920488; http://dx.doi.org/ 10.1038/sj.leu.2403817 [DOI] [PubMed] [Google Scholar]
- 4.Inoue K, Sugiyama H, Ogawa H, Nakagawa M, Yamagami T, Miwa H, Kita K, Hiraoka A, Masaoka T, Nasu K, et al.. WT1 as a new prognostic factor and a new marker for the detection of minimal residual disease in acute leukemia. Blood 1994; 84(9):3071-3079; PMID:7949179 [PubMed] [Google Scholar]
- 5.Ogawa H, Tamaki H, Ikegami K, Soma T, Kawakami M, Tsuboi A, Kim EH, Hosen N, Murakami M, Fujioka T, et al.. The usefulness of monitoring WT1 gene transcripts for the prediction and management of relapse following allogeneic stem cell transplantation in acute type leukemia. Blood 2003; 101 (5):1698-1704; PMID:12406915; http://dx.doi.org/ 10.1182/blood-2002-06-1831 [DOI] [PubMed] [Google Scholar]
- 6.Yarnagarni T, Sugiyarna H, Inoue K, Ogawa H, Tatekawa T, Hirata M, Kudoh T, Akiyarna T, Murakami A, Maekawa T, et al.. Growth Inhibition of Human Leukemic Cells by WTl (Wilms Tumor Gene) Antisense Oligodeoxynucleotides: Implications for the Involvement of WT1 in Leukemogenesis. Blood 1996; 87:2878-2884; PMID:8639907 [PubMed] [Google Scholar]
- 7.Gaiger A, Carter L, Greinix H, Carter D, McNeill PD, Houghton RL, Cornellison CD, Vedvick TS, Skeiky YA, Cheever MA. WT1- specific serum antibodies in patients with leukemia. Clin Cancer Res 2001; 7 (suppl 3):761-765; PMID:11309320 [PubMed] [Google Scholar]
- 8.Keiholz U, Letsch A, Busse A, Asemissen AM, Bauer S, Blau IW, Hofmann WK, Uharel L, Thiel E, Scheinbenbogen C. A clinical and immunologic phase 2 trial of Wilms tumor gene product (WT1) peptide vaccination in patients with AML and MDS. Blood 2009; 113:6541-6548; PMID:19389880; http://dx.doi.org/ 10.1182/blood-2009-02-202598 [DOI] [PubMed] [Google Scholar]
- 9.Maslak P, Dao T, Krug LM, Channel S, Korontsvit T, Zakhaleva V, Zhang R, Wolchok J, Yuan JD, Pinella-Ibarz J, et al.. Vaccination with synthetic analog peptides derived from WT1 oncoprotein induces T cell responses in patients with complete remission from acute myeloid leukemia (AML). Blood 2010; 116 (2):171-179; PMID:20400682; http://dx.doi.org/ 10.1182/blood-2009-10-250993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Tendeloo VF, Van de Velde A, Van Driessche A, Cools N, Anguille S, Ladell K, Gostick E, Vermeulen K, Pieters K, NiJs G, et al.. Induction of complete and molecular remission in acute myeloid leukemia by Wilm's tumor 1 antigen-targeted dendritic cell vaccination. PNAS 2010; 107 (31):13824-13829; PMID:20631300; http://dx.doi.org/ 10.1073/pnas.1008051107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaise C, Buchan SL, Rice J, Marguet J, Rouard H, Kuentz M, Vittes GE, Molinier-Frenkel V, Farcet JP, Stauss HJ, et al.. DNA vaccination induces WT1-specific T-cell responses with potential clinical relevance. Blood 2008; 112 (7):2956-2964; PMID:18502835; http://dx.doi.org/ 10.1182/blood-2008-02-137695 [DOI] [PubMed] [Google Scholar]
- 12.Dao T, Yan S, Veomett N, Pankov D, Zhou L, Krontsvit T, Scott AC, Witten JA, Maslak P, Casey E, et al.. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl Med. 2013; 5 (176):176ra33 PMC3963696; http://dx.doi.org/ 10.1126/scitranslmed.3005661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veomett N, Dao T, Dubrovsky L, Liu H, Xiang JY, Whitten JA, Park SM, Korontsvit T, Zakhaleva V, Curcio M, et al.. Improved Therapeutic Efficacy of an Fc-Enhanced TCR-Like Antibody to the intracellular WT1 Onco-Peptide. Clin Cancer Res 2014; 20 (15):40360-46; PMID:24850840; http://dx.doi.org/24723681 10.1158/1078-0432.CCR-13-2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubrovsky L, Pankov D, Brea EJ, Dao T, Scott AC, Liu C, Scheinberg DA. A TCR-mimic antibody to WT1 bypasses tyrosine kinase inhibitor resistance in human BCR-ABL+ leukemias. Blood 2014; 123(21):3296-304; PMID:24723681; http://dx.doi.org/ 10.1182/blood-2014-01-549022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dao T, Pankov D, Scott AC, Korontsvit T, Zakhaleva V, Xu YY, Xiang JY, Yan S, Guerreiro M, Veomett N, et al.. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Bio Tech 2015; 33 (10):1079-1086; PMID:2638957; http://dx.doi.org/16990779; 10.1038/nbt.3349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinilla-Ibarz J, May RJ, Korontsvit T, Gomez M, Kappel B, Zakhaleva V, Zhang RH, Scheinberg DA. Improved human T-cell responses against synthetic HLA-A*02:01 analog peptides derived from the WT1 oncoprotein. Leukemia 2006; 20 (11):2025-2033; PMID:16990779; http://dx.doi.org/ 10.1038/sj.leu.2404380 [DOI] [PubMed] [Google Scholar]
- 17.Krug LM, Tao Dao, Brown A, Maslak P, Travis W, Bekele S, Korontsvit T, Zakhaleva V, Wolchok J, Yuan JD, et al.. WT1 peptide vaccinations induce CD4 and CD8 T cell immune responses in patients with mesothelioma and non-small cell lung cancer. Cancer Immunol Immunother 2010; 59 (10):1467-1479; PMID:20532500; http://dx.doi.org/ 10.1007/s00262-010-0871-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsuboi A, Oka Y, Kyo T, Katayama Y, Elisseva OA, Kawakami M, Nishida S, Murano A, Nakajima H, Hosen N, et al.. Long-term WT1 peptide vaccination for patients with acute myeloid leukemia with minimal residual disease. Leukemia 2012; 26:1410-1413; PMID:22157809; http://dx.doi.org/ 10.1038/leu.2011.343 [DOI] [PubMed] [Google Scholar]
- 19.Melief CJM and van der Burg SH. Immunotherapy of established (pre) malignant disease by synthetic long peptide vaccines. Nature Rev 2008; 8:351-360; PMID:18418403; http://dx.doi.org/15845894; 10.1038/nrc.2373 [DOI] [PubMed] [Google Scholar]
- 20.Guo Y, Niiya H, Azuma T, Uchida N, Yakushijin Y, sakai I, Takahashi M, Senju S, Nishimura Y, Yasukawa M. Direct recognition and lysis of leukemia cells by WT1-specific CD4 T lymphocytes in an HLA-class II-restricted manner. Blood 2005; 106 (4):1415-8; PMID:15845894; http://dx.doi.org/ 10.1182/blood-2005-01-0413 [DOI] [PubMed] [Google Scholar]
- 21.May RJ, Dao T, Pinilla-Ibarz J, Korontsvit T, Zakhaleva V, Zhang RH, Maslak P, Scheinberg DA. Peptide epitopes from the Wilms tumor 1 oncoprotein stimulate CD4+ and CD8+ T cells that recognize and kill human malignant mesothelioma tumor cells. Clin Cancer Res 2007; 13:4547-4555; PMID:17671141; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-0708 [DOI] [PubMed] [Google Scholar]
- 22.Doubrovina E, Carpenter T, Pankov D, Selvakumar A, Hasan A, O'Reilly RJ. Mapping of novel peptides of WT1 and presenting HLA alleles that induce epitope-specific HLA-restricted T cells with cytotoxic activity against WT1 (+) leukemias. Blood 2012; 123 (8):1633-46; http://dx.doi.org/ 10.1182/blood-2011-11-394619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015; 328(6230):56-61; PMID:25838373; http://dx.doi.org/23550147; 10.1126/science.aaa8172 [DOI] [PubMed] [Google Scholar]
- 24.Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov 2013; 3 (4):388-398; PMID:23550147; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frankel SR, Baeuerle PA. Targeting T cells to tumor cells using bispecific antibodies. Curr Opin Chem Bio 2013; 17:385-392; PMID:23623807; http://dx.doi.org/21296998; 10.1016/j.cbpa.2013.03.029 [DOI] [PubMed] [Google Scholar]
- 26.Sergeeva A, Alatrash G, he H, Ruissard K, Lu S, Wygant J, McIntyre BW, Ma Q, Li D, St John L, Clise-Wwyer K, Molldrem JJ. An anti-PR1/HLA-A2 T-cell receptor-like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood 2011; 117 (16):4262-4272; PMID:21296998; http://dx.doi.org/ 10.1182/blood-2010-07-299248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova YJ, Hundal J, Arthur CD, Krebber WJ, et al.. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014; 515:577-581; PMID:25428507; http://dx.doi.org/ 10.1038/nature13988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, et al.. A dendritic cell vaccine increases the breadth and diversity of melanoma noeantigen-specific T cells. Science 2015; 348 (6236):803-806; PMID:25837513; http://dx.doi.org/ 10.1126/science.aaa3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al.. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014; 515:572-576; PMID:25428506; http://dx.doi.org/ 10.1038/nature14001 [DOI] [PubMed] [Google Scholar]
- 30.Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, Mellman I, Prindiville SA, Viner JL, Weiner LM, et al.. The prioritization of cancer antigens: A national Cancer Institute pilot project for the acceleration of translational research. Clin Cancer Res 2009; 15:5323-37; PMID:19723653; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-0737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, Duerkopp N, Roberts IM, Pogosov GL, Ho WY, et al.. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Tansl Med. 2013; 5 (174):174ra27; http://dx.doi.org/ 10.1126/scitranslmed.3004916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anguille S, Fujiki F, Smith EL, Oji Y, Lion E, Berneman ZN, Sugiyama H. Identification of a Wilms' tumor 1-derived immunogenic CD4 (+) T-cell epitope that is recognized in the context of common Caucasian HLA-DR haplotypes. Leukemia 2013; 27(3):748-750; PMID:22929521; http://dx.doi.org/ 10.1038/leu.2012.248 [DOI] [PubMed] [Google Scholar]