

ABSTRACT
To investigate the link between the genomic landscape of cancer cells and immune microenvironment in tumor tissues, we characterized somatic mutations and tumor-infiltrating lymphocytes (TILs) in malignant pleural mesothelioma (MPM), including mutation/neoantigen load, spatial heterogeneity of somatic mutations of cancer cells and TILs (T-cell receptor β (TCRβ) repertoire), and expression profiles of immune-related genes using specimens of three different tumor sites (anterior, posterior, and diaphragm) obtained from six MPM patients. Integrated analysis identified the distinct patterns of somatic mutations and the immune microenvironment signatures both intratumorally and interindividually. MPM cases showed intratumoral heterogeneity in somatic mutations with unique TCRβ clonotypes of TILs that were restricted to each tumor site, suggesting the presence of a neoantigen-related immune response. Correlation analyses showed that higher neoantigen load was significantly correlated with stronger clonal expansion of TILs (p = 0.048) and a higher expression level of an immune-associated cytolytic factor (PRF1 (p = 0.0041) in tumor tissues), suggesting that high neoantigen loads in tumor cells might promote expansion of functional tumor-specific T cells in the tumor bed. Our results collectively indicate that MPM tumors constitute a diverse heterogeneity in both the genomic landscape and immune microenvironment, and that mutation/neoantigen load may affect the immune microenvironment in MPM tissues.
KEYWORDS: Heterogeneity, malignant pleural mesothelioma, neoantigen, T-cell receptor, tumor-infiltrating lymphocytes, whole-exome sequencing
Abbreviations
- CDR3
complementarity determining region 3
- CTLs
cytotoxic T lymphocytes
- HLA
human leucocyte antigen
- MPM
malignant pleural mesothelioma
- PD-1
programmed death-1
- PD-L1
PD-1 ligand-1
- TCR
T-cell receptor
- TILs
tumor-infiltrating lymphocytes.
Introduction
Malignant pleural mesothelioma (MPM) arises from the mesothelial cells in the pleura and approximately 80% of MPM cases are considered to be related to asbestos exposure.1,2 Although the incidence of MPM has been increasing worldwide,3 platinum-based chemotherapy is the only treatment that was scientifically verified to improve overall survival.4
Immune checkpoint blocking antibodies, such as anti-cytotoxic T lymphocyte antigen-4 (CTLA-4) and anti-programmed death-1 (PD-1) antibodies, have shown a durable antitumor immune effect for several types of cancer,5-7 and are under clinical investigation for MPM.8,9 High number of CD8+ tumor-infiltrating lymphocytes (TILs) was suggested as a good prognostic factor in MPM patients,10 indicating that TILs could play a pivotal role in host antitumor response.
Cancer tissues are composed of various types of cells with distinct molecular and phenotypic features.11 Understanding the intratumoral heterogeneity is clinically important since the existence of multiple cancer-cell subclones generated through clonal evolution or environmental adaptation, as well as their surrounding immune microenvironment, could influence the therapeutic effect.11-13 A multiregional sequencing approach, which sequences DNA samples derived from geographically separated regions of a single tumor, has revealed branched evolution and significant intratumoral genetic heterogeneity. The tumor immune microenvironment, including the infiltration of immune cells into the tumor, is also known to be heterogeneous.12 Therefore, it is important to analyze the link between the heterogeneity in a genetic mutational landscape and that in the immune microenvironment across the spatially different tumor regions.
To examine the spatial genetic heterogeneity in MPM cancer tissues and its effect on the immune microenvironment as well as the relationships between somatic mutation/neoantigen load and immune compositions in intratumor sub-regions of tumors, we systemically conducted whole-exome sequencing analysis, prediction of neoantigens, T-cell receptor β chain (TCRβ) sequencing and expression analysis of immune-related genes in three different positions of MPM tumors.
Results
Intratumoral genetic heterogeneity in three different sites of MPM tumors
To examine intertumoral and intratumoral genetic heterogeneity in MPM tumors, we performed whole-exome sequencing using genomic DNAs extracted from three different positions (A, anterior; P, posterior; and D, diaphragm) of surgically resected tumors in six MPM patients. We obtained an average sequencing depth of 71.4× per base, and identified a total of 244 non-silent mutations and insertions/deletions (indels) (19–47 non-silent mutations per sample) (Table S1). Among the significantly mutated genes in MPMs reported previously,14-16 including TP53, BAP1, and NF2, TP53 was mutated in three of six MPM cases and BAP1 mutation was detected in one case, but NF2 mutations were not detected in any tumors (Table S1). We selected only non-synonymous mutations for further analysis to investigate the relation between potential neoantigen load and tumor immune microenvironment. Mutational profiles obtained from multiregional sequencing demonstrated high genetic heterogeneity in all six MPM tumors (Fig. 1A). We subsequently predicted potential neoantigen epitopes that harbored the amino acid substitution generated by the somatic mutation and revealed the calculated binding affinity to HLA-A molecules of less than 500 nM. As a result, we found 1–18 potential predicted neoantigens (an average number of 9.2) per tumor portion (Fig. S1).
Figure 1.
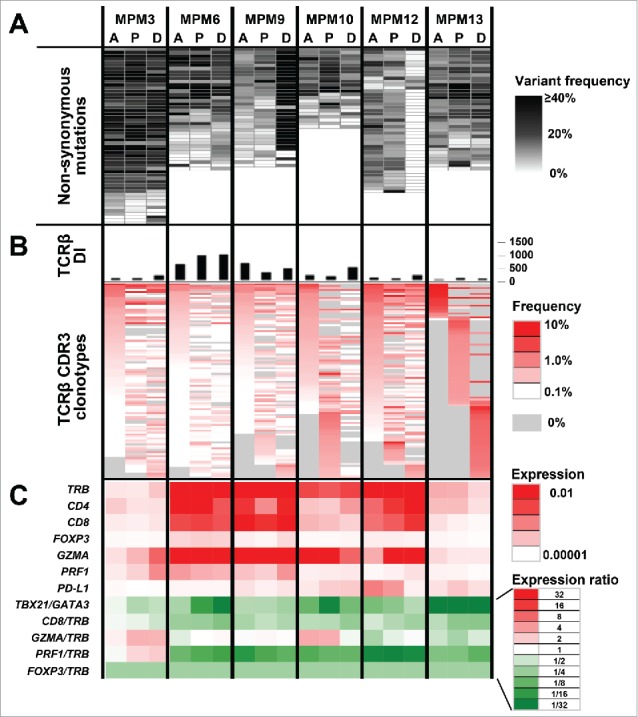
Integrated analysis of MPM tumors for non-synonymous somatic mutations, TCRβ repertoires and expression of immune-related genes Data from three different tumor portions (A: Anterior, P: Posterior, and D: Diaphragm) of six MPM cases are shown. (A) Commonality of non-synonymous mutations identified by whole-exome sequencing. (B) TCRβ diversity index (DI) and heatmaps of CDR3 clonotypes which were sorted according to their frequencies (higher to lower) in the order of tumor portions, A, P, and D. (C) mRNA expression levels of immune-related genes, TCRβ (TRB), CD4, CD8, GATA3, TBX21, FOXP3, GZMA, PRF1, and PD-L1, normalized by GAPDH expression level.
Heterogeneity of TCR repertoire and immune-related gene signature in three different sites of MPM tumors
For TCR repertoire analysis of TILs in the three different positions of MPM tumors, we quantified individual TCRβ clonotypes based on unique V-D-J combinations with complementarity determining region 3 (CDR3) sequences, and calculated the diversity index (DI) of TCRβ to represent the clonality of TILs. We obtained total sequence reads of 586,835 ± 292,275 (average ± one standard deviation), where unique CDR3 clonotypes of 16,700 ± 9,668 were identified in individual tumor samples (Table S2). We found common TCRβ clonotypes in all of three tumor positions, but interestingly we also found unique TCRβ clonotypes in only one tumor position (Fig. 1B). The clonality of TILs was also different among three tumor positions, as represented by differential TCRβ DI values (Fig. 1B). Similarly, expression analysis of immune-related genes showed distinct expression patterns of multiple immune-related genes, such as TCRβ (TRB), CD4, CD8, FOXP3, GATA3, T-bet (TBX21), granzyme A (GZMA), perforin 1 (PRF1), and PD-1 ligand-1 (PD-L1) as well as differential ratios of TBX21/GATA3, CD8/TRB, GZMA/TRB, PRF1/TRB, and FOXP3/TRB, among the three tumor positions (Fig. 1C), suggesting that the immune microenvironment is also spatially heterogeneous in MPM tumors.
Clustering analysis to assess intratumoral heterogeneity between somatic mutations and TCRβ repertoires in MPM tumors
To address the relationship between the intratumoral genetic heterogeneity and differential TCRβ repertoires in the three tumor positions, we conducted an unsupervised clustering analysis by calculating the similarity index (SI) between the data sets of somatic mutations. We detected mutations common in all three positions of the tumors (clonal mutations; Fig. 2), but some mutations present in only one or two tumor positions (subclonal mutations; Fig. 2). Percentage of clonal mutations (observed in one or two portions) was different among patients (average 78.9% ranging from 28.9% to 94.7%). For example, mutational patterns in the MPM12 case were highly heterogeneous with a total of 45 mutations detected in this case, but only 13 mutations were commonly observed in all 3 positions. We also calculated the SI based on TCRβ repertoires of the three tumor portions. Expectedly, clustered patterns based on somatic mutations by dendrogram were quite similar to those based on distribution patterns of TCRβ clonotypes in three cases, MPM3, MPM6, and MPM10 (Fig. 2). These results suggested site-dependent close relationship between somatic mutations and clonality of TILs. Moreover, we observed that certain unique somatic mutations and TCRβ clonotypes were restricted to individual tumor sites, suggesting the presence of unique TIL clones that may recognize the neoantigens derived from site-specific somatic mutations presented on HLA molecules of cancer cells.
Figure 2.
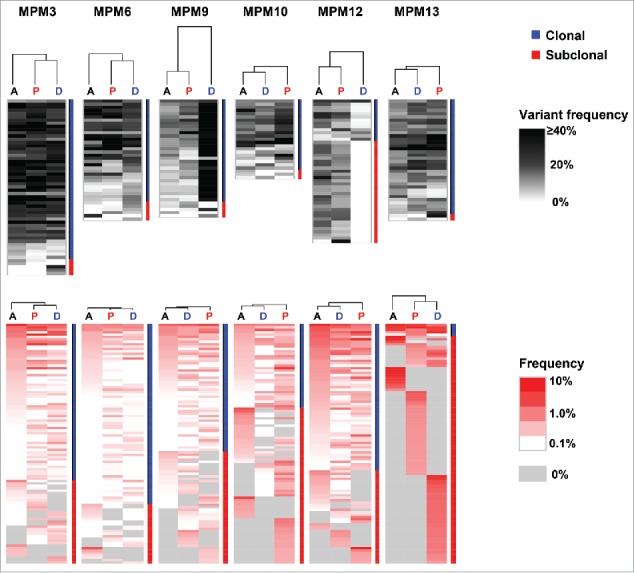
Hierarchial clustering of non-synonymous somatic mutations and TCRβ repertoires Three different MPM tumor portions were hierachically clustered by computing their similarity in the data sets of non-synonymous somatic mutations (upper) or TCRβ repertoires (lower). Vertical length of dendrogam indicates the similarity between two data sets.
Correlation of neoantigen load and immune microenvironment in MPM tumors
To examine a relationship between predicted neoantigens and the immune microenvironment, we performed a correlation analyses for the neoantigen load, TCRβ repertoires and expression levels of multiple immune-related genes. Consequently, we found that a higher neoantigen load was significantly correlated with oligoclonal expansions of TILs, as assessed by TCRβ DI (R = −0.47, p = 0.048; Fig. 3A). The higher neoantigen load showed a weak correlation with a higher ratio of CD8/TRB (high CD8/TRB ratio indicates that CD8+ activity is dominant in αβT cell populations) (R = 0.31, p = 0.20; Fig. 3C), and a strong correlation with a higher ratio of PRF1/TRB (high PRF1/TRB ratio indicates high cytotoxic activity in αβT cell populations) (R = 0.64, p = 0.0041; Fig. 3D). This data represents high levels of cytolytic activity in tumors with higher numbers of somatic mutation/neoantigen. Interestingly, the higher neoantigen load was also correlated with higher ratio of an immune suppressive marker, FOXP3/TRB (R = 0.76, p = 0.00026; Fig. 3E), which indicates the activation of some negative feedback system to protect cancer cells from a host immune attack.
Figure 3.

Correlation analysis between neoantigen load and clonality of TILs or expression levels of immune-related genes Correlation of neoantigen load in the resected MPM tumors (N = 18) to TCRβ diversity index (A), to the expression levels of TRB (B), and to ratios of CD8/TRB (C), PRF1/TRB (D), and FOXP3/TRB (E). Filled rhomboid, square, and triangle indicate anterior, posterior, and diaphragm portions, respectively, in each tumor.
Finally, we examined a relationship between the clonality of TILs and the expression levels of the immune-related genes. As shown in Fig. S2, TCRβ DI tended to be negatively correlated with the expression ratios of CD8/TRB (R = −0.43, p = 0.073), but correlated with that of FOXP3/TRB (R = −0.55, p = 0.018). Considering a strong positive correlation between high neoantigen load and oligoclonality of TILs (Fig. 3A), our findings collectively suggested that such a high neoantigen load in cancer cells might promote clonal expansion of activated T cells and in turn activate the negative feedback system in the tumor bed.
Discussion
The importance of activated cytotoxic T lymphocytes (CTLs), which are critically important in our immune-surveillance system to prevent cancer development and progression, has been proven by recent clinical cancer immunotherapy studies such as adoptive T-cell therapy and immune checkpoint blockades.8,17 However, the role of CTLs or TILs for defining the immune microenvironment in tumors has not been fully elucidated. Moreover, the influence of intratumoral genetic heterogeneity to characterize the immune microenvironment including T cell repertoire in different sites of a single tumor has not been well investigated, although a few studies addressed the relation between tumor heterogeneity and some aspects of the immune microenvironment.12
In this study, we comprehensively examined three different sites of six MPM tumor specimens through whole-exome sequencing, TCR repertoire and expression profile analyses of immune-related genes. First, our results illustrate considerable heterogeneity in patterns of non-synonymous mutations as well as in immune microenvironment signatures (TCRβ repertoires and immune-related gene expression profiles), and it is not only among individual MPM tumors but also among three different tumor sites in a patient (Fig. 1). These findings imply that a single tumor-biopsy specimen may be insufficient to fully characterize the nature of the genetic profile and immune microenvironment of an entire tumor. More interestingly, the clustering analysis showed that there are similarities between mutational patterns and TCRβ clonal patterns in individual tumor positions (Figs. 2 and S3), suggesting some immunogenic non-synonymous mutations might have induced activation and clonal expansion of certain T cells recognizing cancer-specific neoantigens. These results implicate that intratumoral genetic heterogeneity is one of the important determinants to constitute distinct immune microenvironmental conditions.
We also characterized the immune microenvironment of each tumor position through quantifying expression levels of immune-related genes. Correlation analyses revealed that a higher cytolytic activity, represented by the PRF1/TRB ratio in tumor sites, was correlated with higher numbers of somatic mutation/neoantigen and also stronger clonal expansion of TILs (Figs. 3D and S2B and S3D). Interestingly, the FOXP3/TRB ratio, which represents the proportion of immune suppressive regulatory T (Treg) cells among αβ T cell populations, was also higher in tumors (tumor positions) with higher mutation/neoantigen load and in those with lower diversity of TILs (Figs. 3E and S2C and S3E). These findings indicated that an active and suppressive side of the tumor immune microenvironment is well balanced. Once CD8+ T cells are activated and try to eradicate cancer cells, immune suppressive molecules and Treg cells respond and assist cancer cells to escape from the host immune attack.
The immune microenvironment in tumor is defined by various factors including cancer-specific antigens such as neoantigens and cancer-testis antigens, and also various immune-related molecules released from cancer and non-cancerous cells as well as clinical factors including tumor stage, histological subtype and treatment history. Our data indicated significant correlations between the neoantigen load and immune microenvironment factors in tumors from six MPM patients (Fig. 3), but it is certain that a much larger samples are required to stratify tumors by stage, histological subtype and treatment history to further clarify relationships between the neoantigen load and immune profile. We also found an inverse correlation between the mutation load and TRB mRNA level (Fig. S3B), indicating that the tumors with smaller numbers of somatic mutations had more TILs, although they were not clonally expanded (Fig. S3A) and expressed lower levels of PRF1 (Fig. S3C and D). Therefore, this finding implied that quantification of TILs would not be sufficient enough to figure out immune activity of TILs, particularly in the tumors with a smaller number of somatic mutations.
Recent studies have demonstrated a possibility of personalized immunotherapies using neoantigen-based cancer vaccines and adoptive T-cell therapies using TCR-engineered autologous T cells.18,19 Previous reports also indicated that tumors with a higher number of somatic mutations and/or predicted neoantigens revealed better clinical responses to immune checkpoint blockades.12,20,21 This further supports that cytotoxic T cells are likely to be induced by neoantigens. In this aspect, a strong correlation between higher neoantigen load and clonally enriched TILs (Fig. 3A) implies that TCRs in these expanded T cells might provide the useful information for generation of cytotoxic T cells that would be able to recognize neoantigens.
In conclusion, our integrated analysis of three different MPM tumor positions demonstrated a very high complexity in the context of intratumoral heterogeneity, as well as significant associations among somatic mutation/neoantigen load, clonality of TILs, and expression levels of genes related to immune responses. Our data also imply that identification of common clonally-expanded TILs, which may recognize common somatic mutations across the different tumor sites including metastatic locations, might serve to improve therapeutic strategies, although obtaining biopsy samples from multiple (metastatic) sites remains challenging.
Materials and methods
Patient samples
Six MPM patients received EPD (extended pleurectomy and decortication) operation in the University of Chicago Medical Center. From each patient, we obtained blood as well as tumor tissues from three different sites (anterior, posterior, and diaphragmatic positions). Detailed clinical information of the MPM patients is summarized in Table 1. All samples were obtained according to the study protocol (IRB 15-0128) and informed consent procedures, which were approved by the University of Chicago Institutional Review Board.
Table 1.
Characteristics of malignant pleural mesothelioma patients.
| Patient ID | Sex | Age | Histology | TNM stage | Occupational asbestos exposure | Previous chemotherapy |
|---|---|---|---|---|---|---|
| MPM3 | M | 76 | Epithelioid | T3N2 | No | No |
| MPM6 | F | 77 | Epithelioid | T3N0 | No | No |
| MPM9 | M | 69 | Epithelioid | T3N2 | No | Yes |
| MPM10 | M | 72 | Biphasic | T2N1 | No | No |
| MPM12 | M | 77 | Biphasic | T3N0 | Yes | No |
| MPM13 | M | 72 | Biphasic | T4N0 | No | No |
Whole-exome sequencing
From frozen tumor tissues, genomic DNA and total RNA were extracted using AllPrep DNA/RNA mini kit (Qiagen, Valencia, CA, USA). Genomic DNA was also extracted from peripheral blood mononuclear cells (PBMCs) as germline control DNA. Whole-exome libraries were prepared from 1,000 ng of genomic DNA using SureSelectXT Human All Exon V5 kit (Agilent Technologies, Santa Clara, CA, USA). The prepared whole-exome libraries were sequenced by 100-bp paired-end reads on a HiSeq2500 Sequencer (Illumina, San Diego, CA, USA).
Read mapping and variant calling
After base quality filtering by excluding low-quality reads (base quality of < 20 for more than 80% of bases) using FASTX toolkit (http://hannonlab.cshl.edu/fastx_toolkit/), sequence reads were mapped to the human reference genome GRCh37/hg19 using Burrows-Wheeler Aligner (BWA) (v0.7.10).22 Possible PCR duplicated reads were removed using Picard v1.91 (http://broadinstitute.github.io/picard/). Read pairs with a mapping quality of < 30 and with mismatches more than 5% of read length were also excluded. Somatic variants (single nucleotide variations (SNVs) and indels) were called using Fisher's exact test-based method with the following parameters, (i) base quality ≥ 15, (ii) sequence depth ≥ 10, (iii) variant depth ≥ 4, (iv) variant frequency in tumor ≥ 10%, (v) variant frequency in normal < 2%, and (vi) Fisher p value < 0.05.23 SNVs and indels were annotated based on RefGene using ANNOVAR.24
To analyze intratumoral heterogeneity, we merged the variant data of all three portions into a single file, made variant position lists, and then called variants at these positions using the following criteria: (i) base quality ≥ 15, (ii) sequence depth ≥ 10 in all samples, (iii) variant depth ≥ 2, and (iv) variant frequency in normal 2% as reported previously.25
HLA genotyping
PCR amplicon-based high-resolution HLA-A genotyping on MiSeq (Illumina) was performed in Scisco Genetics, Inc. (Seattle, WA, USA).26
Neoantigen prediction
The whole-exome sequencing data from a total of 18 tumor samples were used for the neoantigen prediction, for which we examined all 8- to 11-mer peptides harboring each substituted amino acid by applying the filtering with the predicted binding affinity to HLA-A of < 500 nM, using NetMHCv3.4 and NetMHCpanv2.8 software.27-30
TCR sequencing
The libraries for TCRβ sequencing were prepared using the methods described previously.27,31 Briefly, during the synthesis of cDNA from total RNA, we added 5′ rapid amplification of cDNA end adapter using SMART cDNA library construction kit (Clontech, Mountain View, CA, USA). The TCRβ was amplified by PCR using a reverse primer specific to the constant region and a forward primer for the SMART adapter. After adding Illumina sequence adapter with barcode sequences using the Nextera XT Index kit (Illumina), the prepared libraries were sequenced by 300-bp paired-end reads on the Illumina MiSeq platform, using MiSeq Reagent v3 600-cycels kit (Illumina).
TCR repertoire analysis
Sequencing analysis was performed using Tcrip software.31 Obtained sequencing reads were mapped to the TCR reference sequences obtained from IMGT/GENE-DB (http://www.imgt.org)32,33 using Bowtie2 aligner (version 2.1.0),34 and the CDR3s were decomposed. The inverse Simpson's DI was used to evaluate the TCRβ clonality according to , where N is the total number of sequences, ni is the number of sequences belonging to the ith clonotype, and K is the total number of clonotypes.35 To present TCRβ repertoire of each sample, we used the Excel program (Microsoft, Redmond, WA, USA) to generate bar graphs and heatmaps.
Gene expression analysis
The expression levels of nine immune-related genes, TRB, CD4, CD8, FOXP3, GATA3, TBX21, GZMA, PRF1, and PD-L1, were measured by quantitative real-time RT-PCR using TaqMan gene expression assays (Thermo Fisher Scientific, Carlsbad, CA, USA). All mRNA expression levels were normalized to GAPDH expression level (Hs02758991_g1).
Clustering analysis
To examine similarity (or distance) of data sets from the three different tumor portions, we conducted unsupervised hierarchical clustering analysis using Cluster 3.0 and TreeView software.36 Briefly, the similarity metric was computed by the Pearson correlation coefficient of each somatic mutation or TCRβ clonotype, which then generated SI between two data sets based on the mean of all pairwise distances between two items (average linkage method). According to the SI and clustered nodes, dendrogram figures were generated by TreeView software.
Statistical analysis
Pearson correlation (R) was used to analyze the association between all parameters examined. Statistical analysis was performed using the R statistical environment version 3.3.0. p value of < 0.05 was considered to be statistically significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Drs. Rui Yamaguchi, Seiya Imoto, and Satoru Miyano at The University of Tokyo for developing the algorithm of TCR repertoire analysis and helpful support in data management. The super-computing resource (http://sc.hgc.jp/shirokane.html) was provided by Human Genome Center, Institute of Medical Science, The University of Tokyo.
Author contributions
Y.N. designed and supervised the project; H.I. performed TCR sequencing and quantitative RT-PCR experiments, and wrote the manuscript with K.K.; K.K. and J.P. analyzed data and interpreted data; A.H. performed pathological review of tumor specimens; S.O. and M.Z. assisted experiments; Y.N. and J.P. edited the manuscript; W.V. provided the samples and clinical information, and supervised the project.
References
- 1.Spirtas R, Heineman EF, Bernstein L, Beebe GW, Keehn RJ, Stark A, Harlow BL, Benichou J. Malignant mesothelioma: attributable risk of asbestos exposure. Occup Environ Med 1994; 51:804-11; PMID:7849863; http://dx.doi.org/ 10.1136/oem.51.12.804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, Cox NJ, Dogan AU, Pass HI, Trusa S et al.. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet 2011; 43:1022-5; PMID:21874000; http://dx.doi.org/ 10.1038/ng.912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frank AL, Joshi TK. The global spread of asbestos. Ann Glob Health 2014; 80:257-62; PMID:25459326; http://dx.doi.org/ 10.1016/j.aogh.2014.09.016 [DOI] [PubMed] [Google Scholar]
- 4.Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, Gatzemeier U, Boyer M, Emri S, Manegold C et al.. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol 2003; 21:2636-44; PMID:12860938; http://dx.doi.org/ 10.1200/JCO.2003.11.136 [DOI] [PubMed] [Google Scholar]
- 5.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K et al.. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366:2455-65; PMID:22658128; http://dx.doi.org/ 10.1056/NEJMoa1200694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol 2016; 34:539-73; PMID:26927206; http://dx.doi.org/22658127 10.1146/annurev-immunol-032414-112049 [DOI] [PubMed] [Google Scholar]
- 7.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al.. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443-54; PMID:22658127; http://dx.doi.org/ 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marcq E, Pauwels P, van Meerbeeck JP, Smits EL. Targeting immune checkpoints: new opportunity for mesothelioma treatment? Cancer Treat Rev 2015; 41:914-24; PMID:26433514; http://dx.doi.org/ 10.1016/j.ctrv.2015.09.006 [DOI] [PubMed] [Google Scholar]
- 9.Calabro L, Morra A, Fonsatti E, Cutaia O, Amato G, Giannarelli D, Di Giacomo AM, Danielli R, Altomonte M, Mutti L et al.. Tremelimumab for patients with chemotherapy-resistant advanced malignant mesothelioma: an open-label, single-arm, phase 2 trial. Lancet Oncol 2013; 14:1104-11; PMID:24035405; http://dx.doi.org/ 10.1016/S1470-2045(13)70381-4 [DOI] [PubMed] [Google Scholar]
- 10.Yamada N, Oizumi S, Kikuchi E, Shinagawa N, Konishi-Sakakibara J, Ishimine A, Aoe K, Gemba K, Kishimoto T, Torigoe T et al.. CD8+ tumor-infiltrating lymphocytes predict favorable prognosis in malignant pleural mesothelioma after resection. Cancer Immunol Immunother 2010; 59:1543-9; PMID:20567822; http://dx.doi.org/ 10.1007/s00262-010-0881-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015; 27:15-26; PMID:25584892; http://dx.doi.org/ 10.1016/j.ccell.2014.12.001 [DOI] [PubMed] [Google Scholar]
- 12.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT et al.. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016; 351:1463-9; PMID:26940869; http://dx.doi.org/ 10.1126/science.aaf1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andor N, Graham TA, Jansen M, Xia LC, Aktipis CA, Petritsch C, Ji HP, Maley CC. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med 2016; 22:105-13; PMID:26618723; http://dx.doi.org/ 10.1038/nm.3984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo G, Chmielecki J, Goparaju C, Heguy A, Dolgalev I, Carbone M, Seepo S, Meyerson M, Pass HI. Whole-exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer Res 2015; 75:264-9; PMID:25488749; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-1008 [DOI] [PubMed] [Google Scholar]
- 15.De Rienzo A, Archer MA, Yeap BY, Dao N, Sciaranghella D, Sideris AC, Zheng Y, Holman AG, Wang YE, Dal Cin PS et al.. Gender-specific molecular and clinical features underlie malignant pleural mesothelioma. Cancer Res 2016; 76:319-28; PMID:26554828; http://dx.doi.org/ 10.1158/0008-5472.CAN-15-0751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, Gnad F, Nguyen TT, Jaiswal BS, Chirieac LR et al.. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet 2016; 48:407-16; PMID:26928227; http://dx.doi.org/ 10.1038/ng.3520 [DOI] [PubMed] [Google Scholar]
- 17.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol 2016; 13:273-90; PMID:26977780; http://dx.doi.org/ 10.1038/nrclinonc.2016.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E et al.. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 2013; 19:747-52; PMID:23644516; http://dx.doi.org/ 10.1038/nm.3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348:69-74; PMID:25838375; http://dx.doi.org/ 10.1126/science.aaa4971 [DOI] [PubMed] [Google Scholar]
- 20.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS et al.. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014; 371:2189-99; PMID:25409260; http://dx.doi.org/ 10.1056/NEJMoa1406498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS et al.. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348:124-8; PMID:25765070; http://dx.doi.org/ 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25:1754-60; PMID:19451168; http://dx.doi.org/ 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M et al.. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011; 478:64-9; PMID:21909114; http://dx.doi.org/ 10.1038/nature10496 [DOI] [PubMed] [Google Scholar]
- 24.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38:e164; PMID:20601685; http://dx.doi.org/ 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leisegang M, Engels B, Schreiber K, Yew PY, Kiyotani K, Idel C, Arina A, Duraiswamy J, Weichselbaum RR, Uckert W et al.. Eradication of large solid tumors by gene therapy with a T-cell receptor targeting a single cancer-specific point mutation. Clin Cancer Res 2016; 22:2734-43; PMID:26667491; http://dx.doi.org/ 10.1158/1078-0432.CCR-15-2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson WC, Pyo CW, Vogan D, Wang R, Pyon YS, Hennessey C, Smith A, Pereira S, Ishitani A, Geraghty DE. An integrated genotyping approach for HLA and other complex genetic systems. Hum Immunol 2015; 76:928-38; PMID:26027777; http://dx.doi.org/ 10.1016/j.humimm.2015.05.001 [DOI] [PubMed] [Google Scholar]
- 27.Choudhury N, Kiyotani K, Yap KL, Campanile A, Antic T, Yew PY et al.. Low T-cell receptor diversity, high somatic mutation burden, and high neoantigen load as predictors of clinical outcome in muscle-invasive bladder cancer. Eur Urol Focus 2016; 2:445-52; http://dx.doi.org/ 10.1016/j.euf.2015.09.007 [DOI] [PubMed] [Google Scholar]
- 28.Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics 2008; 24:1397-8; PMID:18413329; http://dx.doi.org/ 10.1093/bioinformatics/btn128 [DOI] [PubMed] [Google Scholar]
- 29.Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 2009; 61:1-13; PMID:19002680; http://dx.doi.org/ 10.1007/s00251-008-0341-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Røder G, Peters B, Sette A, Lund O et al.. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One 2007; 2:e796; PMID:17726526; http://dx.doi.org/ 10.1371/journal.pone.0000796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang H, Yamaguchi R, Liu X, Daigo Y, Yew PY, Tanikawa C, Matsuda K, Imoto S, Miyano S, Nakamura Y. Quantitative T cell repertoire analysis by deep cDNA sequencing of T cell receptor alpha and beta chains using next-generation sequencing (NGS). Oncoimmunology 2014; 3:e968467; PMID:25964866; http://dx.doi.org/ 10.4161/21624011.2014.968467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giudicelli V, Chaume D, Lefranc MP. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res 2005; 33:D256-61; PMID:15608191; http://dx.doi.org/ 10.1093/nar/gki010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lefranc MP, Giudicelli V, Kaas Q, Duprat E, Jabado-Michaloud J, Scaviner D, Ginestoux C, Clément O, Chaume D, Lefranc G. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res 2005; 33:D593-7; PMID:15608269; http://dx.doi.org/ 10.1093/nar/gki065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012; 9:357-9; PMID:22388286; http://dx.doi.org/ 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venturi V, Kedzierska K, Turner SJ, Doherty PC, Davenport MP. Methods for comparing the diversity of samples of the T cell receptor repertoire. J Immunol Methods 2007; 321:182-95; PMID:17337271; http://dx.doi.org/ 10.1016/j.jim.2007.01.019 [DOI] [PubMed] [Google Scholar]
- 36.Juan HF, Huang HC. Bioinformatics: microarray data clustering and functional classification. Methods Mol Biol 2007; 382:405-16; PMID:18220245; http://dx.doi.org/ 10.1007/978-1-59745-304-2_25 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.