

Abstract
BRCA1-associated breast cancer exhibits significantly higher levels of chromosomal abnormalities than sporadic breast cancers. However, the molecular mechanisms regarding the roles of BRCA1 in maintaining genome integrity remain elusive. By using a mouse model deficient for Brca1 full-length isoform (Brca1Δ11/Δ11), we found that Brca1Δ11/Δ11 cells displayed decreased expression of a number of genes that are involved in the spindle checkpoint, including Mad2, which is a key component of spindle checkpoint that inhibits anaphase-promoting complex. We showed that Brca1Δ11/Δ11 cells failed to arrest at metaphase in the presence of nocodazole and underwent apoptosis because of activation of p53. Consistently, reconstitution of Mad2 in Brca1Δ11/Δ11 cells partially restored the spindle checkpoint and attenuated apoptosis. By using UBR60 cells, which carry tetracycline-regulated expression of BRCA1, we demonstrated that BRCA1 binds to transcription factor OCT-1 and up-regulates the transcription of MAD2. Furthermore, we showed that the induction of BRCA1 to endogenous MAD2 or transfected MAD2 luciferase reporter in UBR60 cells was completely inhibited by acute suppression of BRCA1 by RNA interference. These data reveal a role of BRCA1 in maintaining genome integrity by interplaying with p53 and genes that are involved in the spindle checkpoint and apoptosis.
Keywords: Mad2, p53, cell cycle, OCT-1, genetic instability
The spindle checkpoint ensures the astonishing accuracy of chromosome segregation by preventing cells with unaligned chromosomes from exiting mitosis. The molecular components of the spindle checkpoint include at least two evolutionarily conserved protein families, Mad and Bub (1, 2). It was shown that Mad2 binds selectively to unattached kinetochores and is capable of inhibiting anaphase-promoting complex together with BubR1 (3, 4). Consistently, microinjection of Mad2 antibodies yields premature anaphase onset and chromosome missegregation (5). Absence of Mad2 in mouse embryos resulted in accumulation of mitotic errors and apoptosis, leading to early lethality at embryonic day 5 (E5)–E6, whereas haploinsufficiency of Mad2, which produces ≈30% less Mad2 protein, provokes lung tumors after a long latency period (6, 7). Despite the essential role of Mad2 in the spindle checkpoint, it is unclear how the expression of Mad2 is regulated.
Ample experimental evidence indicates that BRCA1 plays essential roles in maintaining genome integrity (8–10). It has been shown that mouse embryos carrying targeted disruptions of Brca1 died at early embryonic developmental stages because of p53-mediated apoptosis triggered by genetic instability (11, 12). Consistently, haploid loss of p53 suppresses the embryonic lethality caused by targeted deletion of Brca1 exon 11 (Brca1Δ11/Δ11) and allows mutant mice survive to adulthood, exhibiting increased tumorigenesis and chromsome abnormalities (13, 14). These observations are consistent with findings that a significantly higher percentage of BRCA1-associated breast cancer than sporadic cancers contained p53 mutations (15). Paradoxically, the Brca1Δ11/Δ11 mouse embryonic fibroblast (MEF) cells exhibited slow growth, fast speed to reach senescence, and the accumulation of aneuploidy (13, 16), suggesting that these cells had undergone abnormal mitosis.
BRCA1 has been shown to associate with many cell-cycle proteins (17). During late G1 and S phases, BRCA1 protein increases and becomes phosphorylated. Upon exiting from M phase, BRCA1 is dephophorylated and its expression decreases (18, 19). This expression pattern prompted intensive studies of BRCA1 in cell-cycle checkpoints, leading to the discoveries of its important roles in centrosome duplication as well as G2/M and S checkpoints (13, 20). However, a role of BRCA1 in the spindle checkpoint is undetermined. Here, we address this issue by using the following three experimental systems: Brca1 mutant MEFs; Cre-loxP, or small interfering RNA-mediated acute Brca1-deletion cell models; and UBR60 cells carrying tet-off regulated BRCA1 expression (21).
Materials and Methods
Cell Culture and Treatment. Primary MEF cells were derived from E14.5 embryos by using a standard procedure. UBR60 cells were cultured in DMEM supplemented with 10% FBS in the presence or absence of 1 μg/ml tetracycline (21). Plasmids bearing GFP-MAD2 or GFP were transfected into Brca1Δ/Δ cells by using Lipofectamine 2000 (Invitrogen). To generate acute Brca1 exon 11-deletion cell lines, linearized pCRE-ERT2 (22) and neo (23) plasmids were cotransfected into Brca1Co/Δ cells. For synchronization, the cells were starved with 0.25% serum, released into culture medium supplemented with 100 ng/ml nocodazole (Sigma), and harvested at 0, 12, 24, 36, 48, and 60 h.
Fluorescence-Activated Cell Sorting (FACS) and Immunofluorescence Analysis. After being fixed with 70% ethanol, the cells were stained with phosphorylated histone H3 Ab (Upstate Biotechnology, Lake Placid, NY) and propidium iodide (Sigma) before being loaded onto the FACSCalibur (Becton Dickinson). To check the lagging chromosome, MEF cells were fixed with methanol. Anti-α-tubulin Ab (Sigma) was applied. Alexa 546-anti-mouse IgG (Molecular Probes) plus 100 ng/ml DAPI (Molecular Probes) was then incubated with cells. Images were captured with a DMRBE epifluorescent microscope (Leica, Deerfield, IL) equipped with a charge-coupled device (CCD) camera (Olympus Megafire, Opelco, Dulles, VA).
Annexin-V-FITC Apoptosis Analysis. Cells were treated with nocodazole for 24 and 36 h. Mitotic shake-off was performed. Mitotic cells were stained with annexin V-EGFP (Clontech). Positive cells were scored under fluorescent microscope. However, the entire cell population was incubated with annexin V-EGFP and analyzed with the FACSCalibur system.
Mitotic Index Analysis. Unsynchronized Brca1Co/ΔCRE-ERT2 clones, parental Brca1Co/Δ cells, and WT-CRE-ERT2 cells were grown on chamber slides in the presence or absence of 1 μM 4-hydroxytamoxifen (4-HT) for 0, 1, 2, 3, 4, 5, 6, 7, and 8 days. At each time point, 100 ng/ml nocodazole was added into the culture for a further 12-h incubation. The cells were stained with DAPI. Mitotic index was determined by scoring ≥1,000 cells under a fluorescent microscope (Olympus 1 × 81, Opelco).
Western Blotting and Immunoprecipitation. Western blot analysis was accomplished by enhanced chemiluminescence detection (Amersham Biosciences). The following antibodies were used: BRCA1 Ab-1 (Oncogene Science), OCT1, cyclin B1, Cdc2 (Santa Cruz Biotechnology), Mad2 (Novus Biologicals, Littleton, CO), Bax (BD Pharmingen), and HRP goat anti-rabbit or mouse IgG (H+L) (Kirkegaard & Perry Laboratories). Nuclear proteins from UBR60 cells were immunoprecipitated with BRCA1 and OCT1 antibodies. The immunoprecipitated complex was analyzed with 8% Novex Tris·glycine gel (Invitrogen).
RT-PCR and TaqMan-PCR Analyses. Total RNA was extracted from embryos or MEF cells. Reverse-transcription reactions were carried out with the First-strand cDNA synthesis kit (Roche). The optimal number of cycles for amplification varies from 22 to 31. The real-time PCR was performed with ABI PRISM 7000 sequence-detection system (Applied Biosystems).
Chromatin Immunoprecipitation (ChIP) Assay. ChIP assays were performed as described (24). Cells were cross-linked with 1% formalin. DNA is extracted from immunoprecipitates of BRCA1 Ab. For PCR, 2 μl from 30 μl of DNA extraction and VENT polymerases (Biolabs, Northbrook, IL) were used.
Human MAD2 Promoter Analysis. The fragment containing 5′-regulatory sequence of human MAD2 promoter (base pairs 1,333–2,421, the translation-initiation codon is at 2,338) was cloned into PGL3-Basic vector (Promega). UBR60 cells were transfected with the plasmids and renilla luciferase pRL-TK vector (Promega). Luciferase analysis was performed with Dual-Luciferase reporterassay system (Promega). Serial deletions were made to search for basal promoter and BRCA1-inducible activity. Point mutations of OCT1 site in fragment of base pairs 2,096–2,297 were done by using the QuickChange site-directed mutagenesis kit (Stratagene).
Biotin–Streptavidin Pull-Down Assay. Three oligonucleotides containing biotin on the 5′ nucleotide of the sense strand were used in the pull-down assays. We incubated 1 μg of each double-stranded oligonucleotide with 300 μg of nuclear protein for 20 min at room temperature. We added 30 μl of poly(dI–dC) preabsorbed streptavidin–agarose beads for 4 h at 4°C. The protein–DNA–streptavidin–agarose complex was analyzed with SDS/PAGE. For more information, see Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Results
Brca1Δ11/Δ11 Cells Exhibited Abnormal Chromosomal Behavior During Mitosis and Were Defective in the Spindle Checkpoint. Analyzing Brca1Δ11/Δ11 MEF cells at passage 2, we found that ≈29% (58/200) of these cells displayed lagging chromosome in metaphase, anaphase, and telophase (Fig. 1A). This abnormality was not observed in >230 examined WT mitotic cells (Fig. 1B). Because all of the MEF cells were derived from E14.5 embryos and had been cultured for at least 3 days before analysis, it is possible that this phenotype was not a direct consequence of the Brca1 mutation. To rule out this possibility, we established a conditional mutant cell model (Brca1Co/Δ) to achieve acute deletion of Brca1 exon 11 by using a 4-HT-inducible Cre-LoxP system (see Fig. 7, which is published as supporting information on the PNAS web site). By analyzing several clones carrying acute deletion of Brca1 exon 11 (Brca1Co/ΔCre-ERT2), we observed similar abnormal chromosome behavior shown in Fig. 1, indicating that these defects were linked directly to the Brca1 deficiency.
Fig. 1.

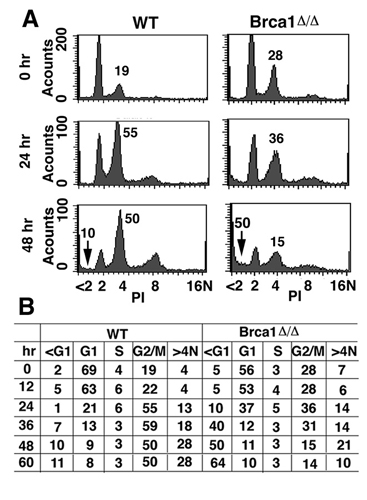
Chromosome lagging and spindle-checkpoint defects associated with Brca1 deficiency during mitosis. (A and B) Images of Brca1Δ/Δ (A) and WT (B) MEF cells at passage 2 stained with DAPI and anti-α-tubulin Ab. Arrows indicate lagging chromosomes. (C) Mitotic index of cells upon nocodazole treatment. Cre-inducible clones (Brca1Co/ΔCRE-ERT2), parental cells (Brca1Co/Δ), and Creinducible WT cells (WT-CRE-ERT2) were grown in the presence of tomaxifen, and nocodazole was added at the indicated time points. At 12 h later, cells were harvested and scored for mitotic index. A steady decline in mitotic index was observed in all three tested inducible clones as the treatment of 4-HT (deletion of Brca1) proceed at 12–96 h. Presence of 4-HT did not alter the response of parental and WT cells to nocodazole-induced mitotic arrest.
It has been shown that a single unattached kinetochore is sufficient to activate the spindle checkpoint (25). Thus, the abnormal chromosome behavior in Brca1Δ11/Δ11 cells suggests that the spindle checkpoint was defective. To verify this phenotype, we treated Brca1Co/Δ11Cre-ERT2 cells in the presence or absence of 4-HT with nocodazole, which is a reagent that depolymerizes microtubules and activates the spindle checkpoint. Parental cells (Brca1Co/Δ) and Cre-expressing WT cells (WT-CRE-ERT2) were also treated as controls. Our data indicated that ≈52% of Brca1Co/Δ11Cre-ERT2 cells were arrested at metaphase 12 h after the nocodazole treatment. The mitotic index of these cells declined dramatically over time after 4-HT treatment (Fig. 1C). In contrast, control cells showed similar mitotic index (≈60%) at all of the time points. These data indicates that, after acute deletion of the conditional allele of Brca1, Brca1Co/Δ11Cre-ERT2 cells did not undergo metaphase arrest and continued to progress through the cell cycle. A failure to undergo metaphase arrest induced by nocodazole or colcemid was also observed in primary and immortalized BrΔ11/Δ11 MEF cells (data not shown).
However, the above experiments did not rule out the possibility that Brca1Δ11/Δ11 cells underwent a profound growth arrest before entering mitosis upon nocodazole treatment. To address this question, we monitored cell growth continuously for their morphology by using a microscope with time-lapse function followed by staining with an Ab for phosphorylated histone H3, which is a marker for mitotic cells. We found that WT cells exhibited a gradually increased population of round and phosphorylated histone-H3-positive cells after nocodazole treatment, indicating that the cells were in the mitosis (Fig. 2A). In Brca1Δ11/Δ11 cells, we found that many round cells, although positive for phosphorylated histone H3, gradually became fragmented (Fig. 2B), suggesting the cells were in mitosis but were dying. To determine whether the cells are going through apoptosis, we shook off the round cells from both WT and Brca1 mutant cells from plates and stained them with annexin V, followed by FACS analysis. After 24 and 36 h of nocodazole treatment, 6% and 12% cells of WT cells were annexin V-positive, respectively. However, 71% and 63% round Brca1Δ11/Δ11 cells were annexin V-positive at the same time points (Fig. 2 D and E). The failure of Brca1Δ11/Δ11 cells to maintain in mitosis and undergo apoptosis were further measured quantitatively by FACS analyses with phosphorylated histone H3 Ab staining (Fig. 2C), and propidum iodide staining (Fig. 8, which is published as supporting information on the PNAS web site), respectively.
Fig. 2.

Morphological and molecular analysis of Brca1Δ11/Δ11 MEF cells. (A and B) Morphology difference between WT (A) and Brca1Δ11/Δ11 (B) primary MEF cells in responding to nocodazole treatment at 24 h. Brca1Δ11/Δ11 MEF cells at mitotic phase exhibited significantly more fragmented (grape-like) cells. Phosphorylated histone H3 Ab staining also indicated that many mutant cells contained fragmented chromosomes. (C) Mitotic index determined by FACS analysis in primary MEF cells by using double staining with propidum iodide and an Ab to phosphorylated histone H3. (D and E) FACS assays of annexin V in WT and Brca1Δ11/Δ11 cells at 24 (D) and 36 (E) h after nocodazole treatment. (F and G) CDK1 kinase assay and cyclin B1 Western blot analysis of WT (F) and Brca1Δ11/Δ11 MEF (G) cells after nocodazole treatment at 0–60 h. CDK1 Western blotting to illustrate the equal amount of kinase was used. (H) Western blot analysis showing cyclin B1 levels in noninducible (29) and inducible (217) clones for Brca1 acute deletion. (I and J) Chromosome spreads showing premature sister-chromatid separation in Brca1 mutant (J) but not in control (I) cells.
Next, we examined the cyclin B1 protein levels and the kinase activity of Cdk1. We first synchronized MEF cells with low serum (0.25%) medium for 96 h, and then the serum starved cells were released into nocodazole-containing medium. The cells progressed into mitosis during the first 24 h, irrespective of their genotypes, as demonstrated by the increase of histone H1 kinase activity and the accumulation of cyclin B1 (Fig. 2 F and G). In WT cells, the CDK1 activity and the cyclin B1 level were maintained in the presence of nocodazole (Fig. 2F), whereas Brca1Δ11/Δ11 cells displayed a sharp drop in both H1 kinase activity and cyclin B1 level (Fig. 2G). This result suggests that Brca1Δ11/Δ11 cells are not capable of inhibiting the anaphase-promoting complex in the presence of spindle damage. Similar decreases in H1 kinase activity and cyclin B1 level were also observed in 4-HT inducible (e.g., clone 217), but not in noninducible (e.g., clone 29), Brca1Co/ΔCre-ERT2 clones (Fig. 2H). Treatment with colcemid resulted in similar phenotypes (data not shown).
A hallmark of cells losing spindle checkpoint is premature sister-chromatid separation (7). To provide further evidence for the defective spindle checkpoint in Brca1Δ11/Δ11 MEFs, we performed chromosome analysis. Our data indicated that Brca1-deficient MEF cells had significantly higher percentages of premature sister-chromatid separation (27/78, 34.6%) than control MEF cells (2/49, 4.1%) (Table 1, which is published as supporting information on the PNAS web site). Furthermore, we analyzed mammary tumor cells derived from Brca1-conditional mutant mice (Brca1Co/Co;MMTV-Cre;p53+/–) (26), and we found that these cells failed to arrest at metaphase upon nocodazole treatment (data not shown) and displayed premature sister-chromatid separation (Fig. 2 I and J, and Table 1). Together, these data provide compelling evidence that Brca1Δ11/Δ11 mutant cells are defective in the spindle checkpoint.
Brca1 Mutant Cells Underwent p53-Mediated Apoptosis. Given the extensive genetic interaction between Brca1 and p53 in multiple biological processes (13, 14, 16, 20, 27), we next test whether the cell death of Brca1Δ11/Δ11 cells after exposure to spindle damaging agents is mediated by activation of p53. Direct comparison between Brca1Δ11/Δ11p53–/– and p53–/– cells at varying time points after nocodazole treatment by FACS analysis revealed similar patterns of DNA contents (Fig. 3 A and B). This observation suggests that the absence of p53 rescues Brca1Δ11/Δ11 cells and allows them to continue cell-cycle progression, leading to the accumulation of cells with ≥8 N ploidy.
Fig. 3.

p53 deficiency suppresses apoptosis in Brca1Δ11/Δ11 cells. (A and B) p53 deficiency suppressed apoptosis of Brca1Δ11/Δ11 cells and allowed accumulation of polyploid cells. (C) Western blot analysis to compare p53 and p21 levels between WT and Brca1Δ11/Δ11 cells.
To provide functional evidence that p53 is activated, we first performed Western blot analysis by using an Ab against p53. Our data indicated that p53 levels steadily increased at 12–36 h after nocodazole treatment and that the high levels of p53 were maintained in Brca1Δ11/Δ11 cells throughout the experiment (60 h) (Fig. 3C). In contrast, p53 expression was very low during the same period in control cells (Fig. 3C). Next, we stained the cells by using an Ab to p21, which is directly activated by p53 (28, 29), and we were able to confirm the hyperactivity of p53 in Brca1Δ11/Δ11 cells but not in control cells (Fig. 3C).
Decreased Mad2 Expression in Brca1Δ11/Δ11 Embryos and MEFs. Our screening of candidate genes also revealed decreased expression of several genes that are known to be involved in the spindle checkpoint, including Bub1, BubR1, Polo-like kinase, ZW-10, and Mad2 (Fig. 4A). We chose to study Mad2 further because of its demonstrated importance in the spindle checkpoint (2, 30). Our analysis revealed that Mad2 was significantly down-regulated in Brca1Δ11/Δ11 mutant embryos compared with WT controls at developmental stages E13.5–E18.5 (Fig. 4 B and C). Furthermore, acute deletion of Brca1 exon11 in inducible clone (clone 217) led to significant reduction of Mad2 48–60 h after addition of 4-HT (Fig. 4D). Decreased expression of Mad2 mRNA also led to decreased Mad2 protein levels, as determined by Western blot analysis (data not shown).
Fig. 4.

BRCA1 positively regulates MAD2 by interacting with its promoter. (A) RT-PCR assay of gene expression in Brca1 mutant and control primary MEF cells. (B and C). Mad2 expression in E13.5–E18.5 embryos revealed by RT-PCR (B) and by TaqMan PCR (C). Expression of Wt embryos were set at 1. (D) RT-PCR analysis of Mad2 expression in Brca1 inducible (clone 217) and noninducible (clone 29) knockout cells in the presence of 4-HT at 0–60 h. (E and F) BRCA1 positively regulate MAD2 in UBR60 cells, as revealed by Western blotting (E) and RT-PCR (F). The relative intensities of bands (normalized with the loading control) were quantified by using quantity one software (Bio-Rad). (G) ChIP assay to show BRCA1 binds to MAD2 promoter in UBR60 cells 48 h in the presence or absence of tet. BRCA1 binds to the following four regions: base pairs 116–619, 599–1,046, 1,025–1,354, and 1,681–2,161. The starting translation codon is at 2,338 (Mad2; GenBank accession no. AB056160). We have also used the same condition to test another promoter (SOX9, last row), and we could not detect any obvious binding.
BRCA1 Up-Regulates Mad2 Expression by Binding to Its Promoter. Next, we tested whether Brca1 might serve as a positive regulator of Mad2 by using UBR60 cells, which carry a tet-off controlled human BRCA1 (21). After withdrawing tetracycline for 24 h, we detected an increase in the BRCA1 protein level, which continued to increase through 48 and 72 h (Fig. 4E). At the same time, MAD2 protein level was elevated by ≈1.7- and 2.2-fold at 48 and 72 h after induction, respectively. RT-PCR analysis revealed that induced expression of BRCA1 increased the mRNA levels of MAD2 (Fig. 4F). These data suggest that BRCA1 is capable of up-regulating the expression levels of MAD2.
To address the relationship between BRCA1 and MAD2 further, ChIP assay with a BRCA1 Ab was performed. PCR assays using the primers that cover 2,421 bp of the 5′ regulatory region of the MAD2 promoter in UBR60 cells under inducible condition (without tetracycline) showed that BRCA1 bound to four regions (i.e., 116–619, 559–1,046, 1,025–1,354, and 1,681–2,161) of the MAD2 promoter (Fig. 4G). Of note, we found that, in the noninducible condition (with tetracycline), the endogenous BRCA1 also interacted with the MAD2 promoter, albeit with reduced intensities as revealed by PCR (Fig. 4G). These data indicate that BRCA1 binds to the promoter of MAD2 in a dosage dependent manner. The endogenous BRCA1 could interact with the promoter of MAD2 was also confirmed in MCF-7 cells (Fig. 9A, which is published as supporting information on the PNAS web site).
To define the minimal MAD2 promoter that contains the essential regulatory element that interacts with BRCA1, we subcloned a DNA fragment including nucleotides 1,333–2,421, which contains one potential BRCA1 interaction site, into a luciferase reporter, pGL3B. Testing serial deletion constructs in UBR60 cells, we found that the element that responds to BRCA1 induction is located between nucleotides 2,096–2,297 (Fig. 5A). We next used matinspector (version 2.2), which is a program that identifies potential DNA-protein interacting sites, to search these 201 nucleotides and identified four nucleotides (2,141–2,144; ACAT), a potential binding motif for the transcription factor OCT1 (Fig. 5B). Next, we tested whether an oligonucleotide containing this site (WT, Fig. 5B) could bind to BRCA1 by a pull-down assay using biotin-labeled oligonucleotides. Our data indicated that the WT oligonucleotide, but not its mutant forms (MT-1 and MT-2, Fig. 5B), bound to BRCA1 (Fig. 5C). Of note, we found that the WT, but not the mutant oligonucleotides, could also pull down OCT1 (Fig. 5C), suggesting a potential interaction between BRCA1 and OCT1. To confirm this result, we performed reciprocal Coimmunoprecipitation and demonstrated that BRCA1 and OCT1 indeed interacted with each other in UBR60 cells (Fig. 5D). We also performed these experiments in MCF-7 cells, and our data indicated that BRCA1 could interact with MAD2 promoter and OCT1 in these cells (Fig. 9 B and C).
Fig. 5.

BRCA1 interacts with MAD2 promoter and positively regulates its function. (A) Luciferase reporter assay of constructs generated by serial deletion. (B) WT and two mutant oligonucleotides from nucleotides 2,131–2,161. The putative OCT1 core like sequence is marked with underlying astral. (C) Biotin-streptavidin pull-down assay. We incubated 1 μg each Biotin labeled oligonucleotide with 300-μg extracts from UBR60 cells 48 h after withdraw of tetracycline. (D) Coimmunoprecipitation assay using antibodies against BRCA1 and OCT1. (C and D) Ten percent of inputs were used in the last lane. (E). Mutation (MT-1) diminished response of MAD2 promoter to BRCA1 induction. (F) Levels of BRCA1 and MAD2 transcripts (Upper) and protein (Lower) in UBR60 cells 48 h after transfection of BRCA1-specific or control RNAi. (G) Depletion of BRCA1 by BRCA1-specific RNAi, but not control RNAi, abolished response of the MAD2 promoter to BRCA1 induction.
Next, we addressed whether the binding between BRCA1 and this site contributes to BRCA1 induction of MAD2 transcription. We introduced the MT-1 mutation (Fig. 5B) into the minimum essential promoter of MAD2 (2,096–2,421, Fig. 5A) and compared its activity with that of the WT promoter. Our data indicated that the mutation significantly decreased induction of MAD2 expression by BRCA1 (Fig. 5E). This assay, however, revealed that BRCA1-mediated induction of MAD2 transcription was moderate, although it was highly reproducible (Fig. 5 A and E). One possibility is that the UBR60 cells already have fair amount of endogenous BRCA1 (Fig. 4 E and F), which could interact with MAD2 promoter at lower levels as revealed by ChIP assay (+tet, Fig. 4H). Thus, endogenous BRCA1 is likely to contribute to MAD2 expression, obscuring the effect of BRCA1 on MAD2 transcription. To verify this issue, we performed RNA interference (RNAi) study to deplete endogenous BRCA1 in UBR60 cells. We showed that transfection with BRCA1-specific small interfering RNA (siRNA), but not control siRNA, resulted in significant decreases in BRCA1 transcripts and protein (Fig. 5F). BRCA1-specific siRNA also reduced MAD2 transcripts and protein levels (Fig. 5F), which is consistent with the observation that Brca1 mutant embryos and MEF contained lower levels of Mad2. Next, we checked activities of the MAD2 reporter by luciferase assay after RNAi treatment. Our data indicated that, in the noninducing condition (+tet), expression of the MAD2 reporter decreased ≈5-fold when UBR60 cells were treated with BRCA1-specific RNAi compared with controls (Fig. 5G). The observation that acute knockdown of BRCA1 significantly decreased expression of MAD2 reporter provides evidence, from another angle, that BRCA1 activates the MAD2 promoter. Next, we performed luciferase assay under inducing conditions (–tet) in the UBR60 cells. Our data revealed that in the presence of BRCA1-specific RNAi, expression of BRCA1 (–tet) failed to induce expression of the MAD2 reporter. In control RNAi-treated cells, the induction remained robust, yielding 7- to 8-fold higher level of expression of the MAD2 reporter compared with that of BRCA1-depleted cells (Fig. 5G).
Overexpression of MAD2 in Brca1Δ11/Δ11 Cells Partially Rescues the Spindle-Checkpoint Defect. If Mad2 down-regulation in Brca1Δ11/Δ11 mutant was a cause for the spindle-checkpoint defect, we would expect that overexpression of Mad2 in Brca1Δ11/Δ11 mutant cells should restore or partially restore the spindle checkpoint. To test this possibility, a plasmid carrying GFP-MAD2 was transfected into immortalized Brca1Δ11/Δ11 cells, which grow much better and have higher transfection efficiency (40%) than primary mutant MEFs. Expression of exogenous MAD2 could be detected by using an Ab to human MAD2 (Fig. 6A). At 48 h after transfection, nocodazole was added into the culture and maintained for 0–72 h. At each time point, the cells were counted for trypan blue staining (Fig. 6B), or analyzed by FACS after stained with phospho-histone H3 Ab to detect mitotic cells (Fig. 6C). As compared with GFP-transfected cells, expression of GFP-MAD2 resulted in a decrease in cell death and an increase in mitotic cells 24–72 h after nocodazole treatment (Fig. 6 B and C). We also examined the mitotic status of GFP-positive cells under the microscope and found that 42% of the GFP-transfected cells had normal chromosome segregation, whereas 58% of them had lagging chromosomes. In contrast, 78% of the GFP-MAD2-transfected cells exhibited normal chromosome segregation, whereas only 22% of them contained lagging chromosomes (Fig. 6D).
Fig. 6.

Overexpression of MAD2 partially rescues spindle-assembly-checkpoint defect in Brca1Δ11/Δ11 MEFs. (A) Western blot analysis showing expression of transfected GFP-MAD2 in Brca1Δ11/Δ11 cells. At 48 h after transfection, these cells were incubated with 100 ng/ml nocodazole for various times, as indicated. (B) Trypan-blue-staining analysis showing the decreased dead-cell population in MAD2-transfected cells. (C) FACS assay showing the increased mitotic population in GFP-MAD2-transfected cells compared with GFP-transfected cells. (D) Mad2-GFP-transfection rescues the lagging chromosome phenotype in normal culture condition.
Discussion
We have provided evidence that BRCA1 plays a critical role in regulating the spindle checkpoint in both mouse and human cells. Numerous studies, from budding yeast to mammalian cells, indicate that several checkpoint proteins (Mad1–3, Bub1, Bub3, BubR1, Mps1, polo-like kinase, and aurora kinase) act coordinately to prevent anaphase entry when the function of the mitotic spindle is compromised (1, 2). Specifically, these proteins detect unattached kinetochores and prevent cell-cycle progression by inhibiting antigen-presenting cell activity, and therefore, play essential roles in maintaining genome integrity. Our study revealed an altered expression of a number of spindle checkpoint components in Brca1Δ11/Δ11 MEFs and WT cells carrying acute deletion of Brca1, including Mad2, polo-like-Kinase, Bub1, BubR1, and ZW-10. Given the critical role of Mad2 in the spindle checkpoint demonstrated in yeast, Xenopus eggs, and mammalian cells (31, 32), we chose to address the possible involvement of Mad2 further. Our data indicated that Brca1 controls the spindle checkpoint, at least in part, by regulating Mad2. By using a tetracyclin-regulated system to express BRCA1 in UBR60 cells (21), we demonstrated that BRCA1 positively regulates MAD2 by interacting, directly or indirectly, with its promoter. Furthermore, overexpression of MAD2 in mutant cells partially overcame the spindle-checkpoint defects. These observations provide strong evidence that MAD2 plays an important role in mediating functions of BRCA1 in the spindle checkpoint.
Notably, the induction of BRCA1 to MAD2 is moderate under our condition. This evidence may suggest the involvement of additional factors in regulating MAD2 expression. Consistent with this observation, we found that BRCA1 and OCT1 are in the same complex that can be pulled down by the WT oligonucleotide, but not mutant oligonucleotides, contained in the promoter of MAD2. This observation suggests that BRCA1 and OCT1 work coordinately in regulating MAD2 expression, although the details remain unclear and warrant further investigation.
BRCA1-associated breast cancer exhibits significantly higher levels of chromosomal abnormalities than sporadic breast cancers (15, 26, 33). The mechanism underlying these alterations can be explained well by the defective spindle checkpoint found in Brca1 mutant cancer cells. Because of the defect of this checkpoint, Brca1Δ11/Δ11 cells would accumulate DNA damage caused by chromosome missegregation and genetic instability. However, based on our finding, the genetic instability would give rise to growth disadvantages for the mutant cells and subject them to apoptosis. However, the genetic instability caused by defective spindle checkpoint could mutate p53 and other tumor suppressor genes, allowing survival of Brca1Δ11/Δ11 cells. We have shown (26, 34) that most (90%) of the tumors derived from Brca1 conditional knockout in mammary gland lost the WT allele of p53. These data are consistent with the finding from human BRCA1-associated breast cancers, which exhibit dramatic chromosome abnormalities and contain more p53 mutations than sporadic breast cancers (15). These observations indicate that the spindle-checkpoint defect associated with Brca1 deficiency in combination with inactivation of p53 plays an essential role in the BRCA1-associated inherited breast cancer.
We have shown that the absence of Brca1 results in the spindle-checkpoint defect that is accompanied by the reduced expression of Mad2 in Brca1 mutant embryos and mutant MEFs. Consistently, induced expression of BRCA1 in UBR60 cells up-regulates MAD2. The defective spindle checkpoint results in chromosome missegregation and premature sister-chromatid separation, leading to p53-mediated apoptosis because p53-deficiency allows mutant cells to survive at expenses of genome integrity. We have shown (13, 16) that p53 deficiency could also rescue embryonic lethality of Brca1Δ11/Δ11 embryos and allow them to develop into adulthood with a high risk of tumorigenesis. These findings not only imply that BRCA1 functions in the spindle checkpoint through modulating MAD2 expression, but they also highlight an important role of p53 in repressing BRCA1-associated tumorigenesis.
Supplementary Material
Acknowledgments
We thank D. A. Haber (Massachusetts General Hospital, Boston) for UBR60 cells, P. P. Chambon (Institut de Genetique et de Biologie Moleculaire et Cellulaire, Institut National de la Santé et de la Recherche Médicale, Paris) for ERT2 plasmids, and O. Cohen-Fix and members of the laboratory of C.-X.D. for critically reading and commenting on the manuscript.
Author contributions: R.-H.W. and C.D. designed research; R.-H.W. performed research; R.-H.W., H.Y., and C.D. analyzed data; and R.-H.W. and C.D. wrote the paper.
Abbreviations: MEF, mouse embryonic fibroblast; 4-HT, 4-hydroxytamoxifen; En, embryonic day n; FACS, fluorescence-activated cell sorting; ChIP, chromatin Coimmunoprecipitation; RNAi, RNA interference.
References
- 1.Cleveland, D. W., Mao, Y. & Sullivan, K. F. (2003) Cell 112, 407–421. [DOI] [PubMed] [Google Scholar]
- 2.Yu, H. (2002) Curr. Opin. Cell Biol. 14, 706–714. [DOI] [PubMed] [Google Scholar]
- 3.Sudakin, V., Chan, G. K. & Yen, T. J. (2001) J. Cell Biol. 154, 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang, Z., Bharadwaj, R., Li, B. & Yu, H. (2001) Dev. Cell 1, 227–237. [DOI] [PubMed] [Google Scholar]
- 5.Gorbsky, G. J., Chen, R. H. & Murray, A. W. (1998) J. Cell Biol. 141, 1193–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dobles, M., Liberal, V., Scott, M. L., Benezra, R. & Sorger, P. K. (2000) Cell 101, 635–645. [DOI] [PubMed] [Google Scholar]
- 7.Michel, L. S., Liberal, V., Chatterjee, A., Kirchwegger, R., Pasche, B., Gerald, W., Dobles, M., Sorger, P. K., Murty, V. V. & Benezra, R. (2001) Nature 409, 355–359. [DOI] [PubMed] [Google Scholar]
- 8.Deng, C. X. (2001) Mutat. Res. 477, 183–189. [DOI] [PubMed] [Google Scholar]
- 9.Venkitaraman, A. R. (2002) Cell 108, 171–182. [DOI] [PubMed] [Google Scholar]
- 10.Zheng, L., Li, S., Boyer, T. G. & Lee, W. H. (2000) Oncogene 19, 6159–6175. [DOI] [PubMed] [Google Scholar]
- 11.Deng, C. X. (2002) Environ. Mol. Mutagen. 39, 171–177. [DOI] [PubMed] [Google Scholar]
- 12.Deng, C. X. & Wang, R. H. (2003) Hum. Mol. Genet. 12, R113–R123. [DOI] [PubMed] [Google Scholar]
- 13.Xu, X., Qiao, W., Linke, S. P., Cao, L., Li, W. M., Furth, P. A., Harris, C. C. & Deng, C. X. (2001) Nat. Genet. 28, 266–271. [DOI] [PubMed] [Google Scholar]
- 14.Bachelier, R., Xu, X., Wang, X., Li, W., Naramura, M., Gu, H. & Deng, C. X. (2003) Oncogene 22, 528–537. [DOI] [PubMed] [Google Scholar]
- 15.Gasco, M., Yulug, I. G. & Crook, T. (2003) Hum. Mutat. 21, 301–306. [DOI] [PubMed] [Google Scholar]
- 16.Cao, L., Li, W., Kim, S., Brodie, B. G. & Deng, C. X. (2003) Genes Dev. 17, 201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng, C. X. & Brodie, S. G. (2000) BioEssays 22, 728–737. [DOI] [PubMed] [Google Scholar]
- 18.Vaughn, J. P., Davis, P. L., Jarboe, M. D., Huper, G., Evans, A. C., Wiseman, R. W., Berchuck, A., Iglehart, J. D., Futreal, P. A. & Marks, J. R. (1996) Cell Growth Differ. 7, 711–715. [PubMed] [Google Scholar]
- 19.Ruffner, H. & Verma, I. M. (1997) Proc. Natl. Acad. Sci. USA 94, 7138–7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu, X., Weaver, Z., Linke, S. P., Li, C., Gotay, J., Wang, X. W., Harris, C. C., Ried, T. & Deng, C. X. (1999) Mol. Cell 3, 389–395. [DOI] [PubMed] [Google Scholar]
- 21.Harkin, D. P., Bean, J. M., Miklos, D., Song, Y. H., Truong, V. B., Englert, C., Christians, F. C., Ellisen, L. W., Maheswaran, S., Oliner, J. D. & Haber, D. A. (1999) Cell 97, 575–586. [DOI] [PubMed] [Google Scholar]
- 22.Metzger, D., Clifford, J., Chiba, H. & Chambon, P. (1995) Proc. Natl. Acad. Sci. USA 92, 6991–6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang, X., Li, C., Xu, X. & Deng, C. (1998) Proc. Natl. Acad. Sci. USA 95, 3667–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shang, Y., Hu, X., DiRenzo, J., Lazar, M. A. & Brown, M. (2000) Cell 103, 843–852. [DOI] [PubMed] [Google Scholar]
- 25.Nicklas, R. B. (1997) Science 275, 632–637. [DOI] [PubMed] [Google Scholar]
- 26.Brodie, S. G., Xu, X., Qiao, W., Li, W. M., Cao, L. & Deng, C. X. (2001) Oncogene 20, 7514–7523. [DOI] [PubMed] [Google Scholar]
- 27.Ouchi, T., Monteiro, A. N., August, A., Aaronson, S. A. & Hanafusa, H. (1998) Proc. Natl. Acad. Sci. USA 95, 2302–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.el-Deiry, W. S., Tokino, T., Velculescu, V. E., Levy, D. B., Parsons, R., Trent, J. M., Lin, D., Mercer, W. E., Kinzler, K. W. & Vogelstein, B. (1993) Cell 75, 817–825. [DOI] [PubMed] [Google Scholar]
- 29.Harper, J. W., Adami, G. R., Wei, N., Keyomarsi, K. & Elledge, S. J. (1993) Cell 75, 805–816. [DOI] [PubMed] [Google Scholar]
- 30.Shah, J. V. & Cleveland, D. W. (2000) Cell 103, 997–1000. [DOI] [PubMed] [Google Scholar]
- 31.Fang, G. (2002) Mol. Biol. Cell 13, 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wassmann, K., Liberal, V. & Benezra, R. (2003) EMBO J. 22, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weaver, Z., Montagna, C., Xu, X., Howard, T., Gadina, M., Brodie, S. G., Deng, C. X. & Ried, T. (2002) Oncogene 21, 5097–5107. [DOI] [PubMed] [Google Scholar]
- 34.Xu, X., Wagner, K. U., Larson, D., Weaver, Z., Li, C., Ried, T., Hennighausen, L., Wynshaw-Boris, A. & Deng, C. X. (1999) Nat. Genet. 22, 37–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}