

Abstract
Background
Collision tumors are uncommon but well described clinical entities composed of distinct tumor histologies occurring within the same anatomic site. Optimal management of patients with collision tumors remains highly variable and depends on clinical characteristics such as the involved tumor types, predominant histology, as well as the extent of disease. Comprehensive genomic profiling is a means of identifying genomic alterations to suggest benefit from targeted therapy.
Case presentation
A 78-year-old woman presented to medical oncology with liver metastases occurring within the background of a 1-year history of uveal melanoma. Biopsy of the liver metastases revealed presence of adenocarcinoma along with nests of malignant melanoma consistent with a collision tumor. The disease was refractory to several lines of conventional cytotoxic chemotherapy, and the patient later developed pulmonary metastases while on chemotherapy. The patient’s tumor tissue was assayed by comprehensive genomic profiling which revealed presence of a TSC1 partial loss. The patient was subsequently initiated on temsirolimus 15 mg intravenously weekly for 4 months. Restaging imaging demonstrated a partial response to therapy by RECIST 1.1 criteria and clinical benefit for 6 months until the patient passed away secondary to unrelated causes.
Conclusions
We report the first case of a collision tumor composed of adenocarcinoma and melanoma with a TSC1 mutation that objectively and durably responded to mTOR inhibition.
Keywords: Collision tumor, Uveal melanoma, TSC1 mutation, mTOR inhibition, Temsirolimus, Carcinoma Undetermined Primary, Melanoma, Next Generation Sequencing, Case Report
Background
Cancer therapy continues to move towards the targeting of molecular signaling pathways; mTOR inhibitors have been well studied since the early 1990s with increasing recognition immunosuppressive and anticancer properties. In particular, the TSC1-TSC2 complex has emerged as an integral signal involved in the inhibition of mTORC1. Inactivating alterations of tumor suppressor genes TSC1 and TSC2 have been implicated in tuberous sclerosis and a wide variety of malignancies in which mTORC1 was found to be highly activated [1]. Promising clinical trials have shown that tumors harboring TSC1 mutations respond to mTOR inhibitors and the clinical significance of such a mutation is highlighted in the present case [1].
We present a patient with a prior diagnosis of uveal melanoma with new metastatic hepatic disease for whom biopsy unexpectedly demonstrated a collision tumor of melanoma and adenocarcinoma of unknown primary. Although collision tumors have been described in literature, primarily between two cutaneous malignancies, a combination of melanoma and adenocarcinoma is extremely rare with only a few reported cases [2–5]. The patient did not respond to several conventional chemotherapy agents but did respond to targeted mTOR inhibition after genomic sequencing revealed the presence of a TSC1 mutation. This case highlights two unique facets in both the phenomenon of collision tumors and the significance of targeted signaling pathway agents where traditional chemotherapy has failed.
Case presentation
A 78-year-old woman presented to our institution for further management of newly diagnosed metastatic liver disease.
The patient initially presented with symptoms of decreased visual acuity of the right eye 1 year prior. Comprehensive physical examination was unrevealing. The patient was referred to ophthalmology and was diagnosed with choroidal melanoma. The tumor initially measured 19.5 x 13.6 mm and involved 50% of the optic nerve head as well as the macula. The patient was treated with radiotherapy along with transpupillary thermotherapy.
As part of routine surveillance for the diagnosis of uveal melanoma, the patient was followed with serial CT scans of the abdomen and pelvis every 3 months. Nine months after the initial diagnosis repeat CT scan revealed multiple liver masses suggestive of metastatic disease involving the lateral segment of the left hepatic lobe. A dominant mass was identified measuring 6.9 x 5.8 cm and the patient was referred for core needle biopsy. Pathology revealed abundant involvement by adenocarcinoma, which stained positive for pankeratin (AE1/AE3), CK7, CK20 (Fig. 1). Additionally, several nests of atypical cells with cytoplasmic pigmentation consistent with malignant melanoma were identified. Immunohistochemical stains showed the malignant pigmented neoplasm to be negative for AE1/AE3 and positive for S100, SOX10, HMB45 (Fig. 1).
Fig. 1.
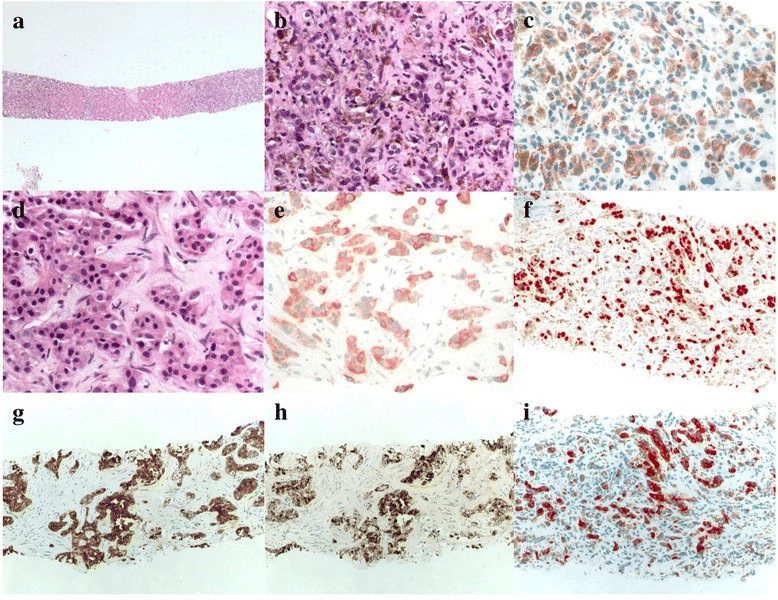
Core-needle liver biopsy. a low-powered field showing melanoma (left) and adenocarcinoma (right). b 40x view of melanoma. c S-100 stain. d 40x of carcinoma. e Pankeratin stain. f SOX10 stain. g CK7 stain. h CK20 stain. i HMB-45 stain
Given the unexpected findings of metastatic adenocarcinoma co-existing with metastatic melanoma, a diagnostic workup was done to identify the site of origin for the adenocarcinoma component. Whole-body PET/CT, and upper and lower endoscopy failed to identify the primary anatomic site. The patient was therefore diagnosed with metastatic collision tumor composed of adenocarcinoma of unknown primary along with malignant melanoma and empiric chemotherapy was initiated. Unfortunately, the disease proved refractory to several lines of conventional cytotoxic chemotherapy. She received two cycles of oral capecitabine (Xeloda, Genentech, South San Francisco, CA; 1000 mg/m2 orally day 1–14 every 21 days). This was discontinued secondary to grade 2–3 gastrointestinal toxicity (nausea, vomiting). The patient subsequently received 8 cycles of gemcitabine (Gemzar, Eli Lilly, Indianapolis, IN; 900 mg/m2 IV day 1, 8 every 21 days;) combined with protein-bound nab-paclitaxel (Abraxane, Celgene, Summit, NJ; 100 mg/m2 IV day 1, 8, 15 every 28 days). Restaging imaging revealed progressive disease in the liver as well as the interval development of metastatic lung nodules.
The patient’s tissue was submitted as a formalin-fixed, paraffin-embedded block to a CLIA-certified, CAP-accredited laboratory (Foundation Medicine, Cambridge, MA) for CGP. DNA was extracted from the tumor specimen. Hybrid-capture-based CGP using next-generation sequencing was performed of the entire coding sequence. This included 236 genes and 47 introns of 19 genes involved in fusions at a depth of X500. Alterations were identified as point mutations, deletions, amplifications, duplications, insertions, rearrangements, and splice variants. These were characterized as known or likely pathogenic changes as reported by the FoundationOne assay.
DNA extracted from the biopsy of the collision tumor submitted for NGS contained both melanoma and adenocarcinoma. Several genomic alterations were identified as follows TSC1 loss (homozygous deletion) of exons 20-23, CDKN2A/B loss, BAP1 (E20fs*52), PBRM1 (R1095fs*39). CGP is a validated approach in detecting base substitutions, short insertions, deletions, copy number alterations and selected fusion products. The technique has been directly compared to established assays including PCR. Test sensitivity approximately 95–99% across various alteration types with high specificity (positive predictive value >99%). Because CGP is a validated technique to identify genetic abnormalities, no additional testing on the tumor sample was clinically indicated [6].
Tuberous sclerosis gene 1 (TSC1) is a tumor suppressor gene found on the long arm of chromosome 9. TSC1 encodes the protein gene product hamartin composed of one transmembrane domain and two coiled-coil domains. The first coiled-coil domain modulates interaction between hamartin and tuberin. A second coiled-coil domain is encoded by exons 17 to 23 which stabilizes the tuberin-hamartin complex [7]. The hamartin-tuberin complex enables the GTPase-activating function of tuberin and is a major regulator of small G-protein Rheb which interacts with mTORC1 [8, 9]. Loss of hamartin, therefore results in increased downstream mTOR activity potentially predicting increased sensitivity to mTOR inhibitors.
Based on the results of CGP the patient began treatment with temsirolimus (Torisel, Pfizer, New York, NY) 15 mg intravenously weekly. The patient was followed with clinical examinations monthly and restaging imaging every 2 months while on therapy. After 4 months of treatment, a restaging CT scan of the chest, abdomen, and pelvis demonstrated a significant partial response to therapy by RECIST 1.1 criteria (Fig. 2). Restaging imaging 2 months later demonstrated stable disease. The patient experienced no dose-limiting toxicities while on therapy. Unfortunately, the patient later developed severe sepsis related to Clostridium Difficile colitis and died from infectious complications 6 months after starting temsirolimus. Informed consent was obtained for the publication of this manuscript and available to the journal editors for review. This manuscript adhered to CARE guidelines for case reports.
Fig. 2.
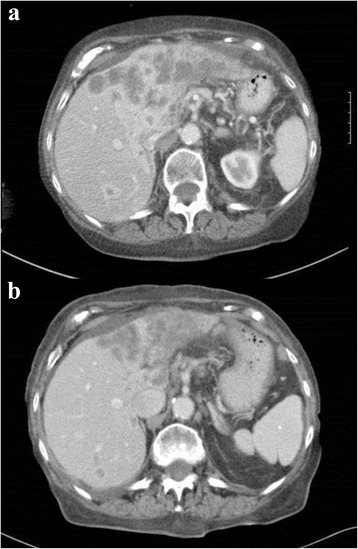
Pre-treatment and post-treatment imaging. a Pre-treatment CT scan of liver. b Post-treatment CT scan of liver
Discussion
A collision tumor is an uncommon occurrence in which two distinct tumor histologies are present in a single anatomic site. Under the World Health Organization (WHO) histological classification guidelines, a collision tumor should comprise at least two different malignant components with no mixed or transitional area between [10, 11]. Several theories have been proposed to explain the pathophysiology of this phenomenon. One mechanism involves the alteration of the tumor microenvironment facilitating colonization or metastases of tumor to that site. An alternative model would be a coincidental meeting of two primary tumors. In contra-distinction, a composite tumor references a tumor that evolves into two distinct histologies from presumably a pluripotent precursor cancer stem cell.
Although cases of synchronous adenocarcinoma and melanoma have been reported, the presentation remains exceedingly rare [2–5]. The clinical behavior and natural history of collision tumors may reflect the biology of the more aggressive tumor involved [11]. In general, both metastatic adenocarcinoma of undetermined primary and metastatic ocular melanoma are associated with poor prognoses. It is unknown whether or not the presence of a collision tumor portends a worse prognosis than either condition alone.
Mutations in TSC1 and TSC2 have been reported in a variety of neoplasms and benign tumors including pulmonary lymphangioleiomyomatosis (LAM), perivascular epitheloid cell tumors (PEComa), urothelial carcinomas, renal cell carcinoma and hepatocellular carcinomas [12]. Mutations in the tuberous sclerosis complex (TSC) has been reported in up to 14.5% of bladder cancer and 28.6% of hepatocellular carcinoma [13, 14]. Alterations resulting in PTEN loss and PIK3CA amplification, which are also targets for mTOR/P13K inhibitors, have been implicated in 7% and 9% respectively, in carcinomas of unknown primary [15]. Of greater clinical relevance, the presence of mutations involving TSC1 or TSC2 may predict a response to downstream inhibition of the mTOR pathway [1, 7].
This patient’s disease had been highly refractory to conventional chemotherapeutic agents, but did demonstrate sensitivity to temsirolimus. In addition, the patient’s CGP demonstrated mutation of BRCA1-associated protein 1 (BAP1) gene, which is frequently inactivated in metastatic uveal melanomas [16]. BAP1 encodes a nuclear ubiquitin carboxyterminal hydrolase, which is a class of deubiquitinating enzymes, and contains binding domains for BRCA1 and BARD1 to form a tumor suppressor complex among other functions [16]. In uveal melanomas, somatic inactivating mutations of BAP1 have been highly associated with onset of metastatic behavior thus suggesting a potential novel target for therapy [16].
Additionally, noted on NGS were alterations in CDKN2A/2B as well as PBRM1. CDKN2A/2B encode tumor suppressor gene products cyclin-dependent kinase 4 inhibitor A/B. This gene encodes a cycle-dependent kinase inhibitor that prevents activation of the CDK kinases thereby inhibiting cell cycle G1 progression [17, 18]. Interestingly, germ line mutations in CDKN2A have been associated with familial melanoma syndromes and may have reflected genetic alterations from this patient’s uveal melanoma. PBRM1 encodes Polybromo-1 (BAF180) which is a subunit of ATP-dependent chromatin-remodeling complex. It has an additional role as a cofactor in transactivation of nuclear hormone receptors [19]. This abnormality is most closely associated with clear cell renal cell carcinoma occurring in up to 30% of cases. In addition, it has been reported in up to 2–4% of colorectal adenocarcinoma [20, 21]. Unfortunately, no FDA approved therapies targeting CDKN2A/B or PBRM1 are currently available. If additional targeted agents had been available, combination therapy may have been an important treatment consideration in management of this patient’s disease.
In our patient, disease progression on multiple lines of cytotoxic chemotherapy suggests that targeted inhibition of mTOR pathway was critical in inducing tumor response. Because tissue composed of both adenocarcinoma and melanoma was submitted for NGS, it is unknown whether the genetic alterations observed were unique to either tumor histology. Given the radiographic response to therapy with mTOR inhibitor, no repeat biopsy was clinically indicated. Nonetheless, the potential of detecting significant therapeutic targets implies that patients with refractory collision tumors would benefit from genomic profiling.
Conclusion
To our knowledge, this is the first case of a collision tumor composed of adenocarcinoma and melanoma with a mutation of the TSC1 gene locus. The objective clinical response achieved with targeted inhibition of the mTOR pathway further highlights the clinical significance of the genetic alteration identified by NGS. As a result, NGS may have a potential utility in identifying actionable targets in collision tumors failing to response to traditional cytotoxic chemotherapy. Further investigation into the identification of driver genetic mutations in collision tumors and potential therapeutic targets is warranted.
Acknowledgements
We thank Dr. Eric Chao, radiology and Dr. John Hughes, pathology for their contribution in selecting images for the case.
Funding
None.
Availability of data and material
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Authors’ contributions
The manuscript was written by MB, JZ, and CC. SA, RM and AS reviewed the manuscript and contributed to planning of the review. All the authors have read and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from the patient for publication of this Case Report. A copy of the written consent is available for review by the Editor of this journal.
Ethics approval and consent to participate
Not applicable to this submission
Abbreviations
- BAP1
BRCA1 associated protein 1
- BARD1
BRCA1 Associated RING Domain 1
- BRCA1
Breast cancer type 1 susceptibility protein
- CDKN2A/B
Cyclin-dependent kinase inhibitor 2A/B
- CGP
Comprehensive genomic profiling
- CK
Cytokeratin
- CLIA
Clinical Laboratory Improvement Act
- CT
Computerized tomography
- GNAQ
G protein subunit alpha q
- GNAS
G protein subunit alpha S
- GTPase
Guanosine-5’-triphosphate hydrolase
- HMB45
Human Melanoma Black common marker to confirm melanoma
- mTOR
Mammalian target of rapamycin
- P13K
Phosphatidylinositide 3-kinases
- PET/CT
Positron emission tomography computerized tomography
- PIK3CA
Phosphatidylinositol-4, 5-bisphosphate 3-kinase catalytic subunit alpha
- PRM1
Protein polybromo-1
- PTEN
Phosphatase and tensin homolog
- RECIST
Response Evaluation Criteria in Solid Tumors
- S100 protein
Common marker for neural tissues and melanoma
- SOX
Sry-related HMG box
- TSC
Tuberous sclerosis
Contributor Information
Munveer S. Bhangoo, Email: bhangoo.munveer@scrippshealth.org
Jenny Y. Zhou, Email: jennyzhou822@gmail.com
Siraj M. Ali, Email: sali@foundationmedicine.com
Russell Madison, Email: rmadison@foundationmedicine.com.
Alexa B. Schrock, Email: aschrock@foundationmedicine.com
Carrie Costantini, Email: Costantini.Carrie@scrippshealth.org.
References
- 1.Wander S, Hennessy B, Slinglerland J. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121(4):1231–1241. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim A, Grant B, Avramovic J, Ho YH, Wallace C. Synchronous primary anorectal melanoma and sigmoid adenocarcinoma: a case report. Int Surg. 2015;100(5):814–7. doi: 10.9738/INTSURG-D-14-00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dias AR, Pinto RA, Mory E, Silva IC, Siqueira SA, Nahas SC, Cecconello I, Wexner SD. Synchronous collision malignant melanoma and adenocarcinoma of the rectum. Tech Coloproctol. 2010;14(2):181–4. doi: 10.1007/s10151-010-0571-6. [DOI] [PubMed] [Google Scholar]
- 4.Ueyama T, Tsuru T, Tsuneyoshi M, Sueishi K, Sibuya T, Fukuda T. Primary collision neoplasm of malignant melanoma and adenocarcinoma in the lung. A case report. Pathol Res Pract. 1993;189(2):178–83. doi: 10.1016/S0344-0338(11)80089-0. [DOI] [PubMed] [Google Scholar]
- 5.Inoshita T, Laurain AR, Youngberg GA, Musil G. Metastasis of bronchogenic carcinoma to the skin involved by melanoma. Arch Pathol Lab Med. 1984;108(7):595–8. [PubMed] [Google Scholar]
- 6.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–31. [DOI] [PMC free article] [PubMed]
- 7.Sun W, Zhu YJ, Wang Z, Zhong Q, Gao F, Lou J, Gong W, Xu W. Crystal structure of the yeast TSC1 core domain and implications for tuberous sclerosis pathological mutations. Nat Commun. 2013;4:2135. doi: 10.1038/ncomms3135. [DOI] [PubMed] [Google Scholar]
- 8.Han JM, Sahin M. TSC1/TSC2 signaling in the CNS. FEBS Lett. 2011;585:973–80. doi: 10.1016/j.febslet.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–34. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirono S, Tani M, Terasawa H, Kawai M, Ina S, Uchiyama K, et al. A collision tumor composed of cancers of the bile duct and ampulla of Vater - immunohistochemical analysis of a rare entity of double cancer. Hepatogastroenterology. 2008;55:861–4. [PubMed] [Google Scholar]
- 11.Stukavec J, Jirasek T, Mandys V, Denemark L, Havluj L, Sosna B, Kosmahl M, Zadorova Z. Poorly differentiated endocrine carcinoma and intraductal papillary-mucinous neoplasm of the pancreas: description of an unusual case. Pathol Res Pract. 2007;203:879–84. doi: 10.1016/j.prp.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 12.Northrup H, Koenig MK, Pearson DA, Au KS. Tuberous sclerosis complex. In GeneReviews. Edited by Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K. Seattle (WA): University of Washington, Seattle; 1993-2016. [http://www.ncbi.nlm.nih.gov/books/NBK1220/?report=printable]. Accessed on 22 Apr 2016. [PubMed]
- 13.Knowles MA, Habuchi T, Kennedy W, Cuthbert-Heavens D. Mutation spectrum of the 9q34 tuberous sclerosis gene TSC1 in transitional cell carcinoma of the bladder. Cancer Res. 2003;63:7652–7656. [PubMed] [Google Scholar]
- 14.Huynh H, Hao HX, Chan SL, Chen D, Ong R, Soo KC, Pochanard P, Yang D, Ruddy D, Liu M, Derti A, Balak MN, Palmer MR, Wang Y, Lee BH, Sellami D, Zhu AX, Schlegel R, Huang A. Loss of tuberous sclerosis complex 2 (TSC2) is frequent in hepatocellular carcinoma and predicts response to mTORC1 inhibitor everolimus. Mol Cancer Ther. 2015;14(5):1224–35. doi: 10.1158/1535-7163.MCT-14-0768. [DOI] [PubMed] [Google Scholar]
- 15.Ross JS, Wang K, Gay L, Otto GA, White E, Iwanik K, Palmer G, Yelensky R, Lipson DM, Chmielecki J, Erlich RL, Rankin AN, Ali SM, Elvin JA, Morosini D, Miller VA, Stephens PJ. Comprehensive genomic profiling of carcinoma of unknown primary site: new routes to targeted therapies. JAMA Oncol. 2015;1(1):40–49. doi: 10.1001/jamaoncol.2014.216. [DOI] [PubMed] [Google Scholar]
- 16.Harbour JW, Onken MO, Roberson EDO, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, Bowcock AM. Freqeunt mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330(6009):1410–1413. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gazzeri S, Gouyer V, Vourch C, Brambilla C, Brambilla E. Mechanisms of p16INK4A inactivation in non-small-cell lung cancers. Oncogene. 1998;16(4):497–504. doi: 10.1038/sj.onc.1201559. [DOI] [PubMed] [Google Scholar]
- 18.Roussel MF. The INK4 family of cell cycle inhibitors in cancer. Oncogene. 1999;18(38):5311–7. doi: 10.1038/sj.onc.1202998. [DOI] [PubMed] [Google Scholar]
- 19.Lemon B, Inouye C, King DS, Tijan R. Selectivity of chromatin-remodeling cofactors for ligand-activated transcription. Nature. 2001;414(6866):924–8. doi: 10.1038/414924a. [DOI] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research Network Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas Research Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]