

ABSTRACT
Kynurenine formation by tryptophan-catabolic indoleamine-2,3-dioxygenase 1 (IDO1) plays a key role in tumor immune evasion and inhibition of IDO1 is efficacious in preclinical models of breast cancer. As the response of breast cancer to immune checkpoint inhibitors may be limited, a better understanding of the expression of additional targetable immunomodulatory pathways is of importance. We therefore investigated the regulation of IDO1 expression in different breast cancer subtypes. We identified estrogen receptor α (ER) as a negative regulator of IDO1 expression. Serum kynurenine levels as well as tumoral IDO1 expression were lower in patients with ER-positive than ER-negative tumors and an inverse relationship between IDO1 and estrogen receptor mRNA was observed across 14 breast cancer data sets. Analysis of whole genome bisulfite sequencing, 450k, MassARRAY and pyrosequencing data revealed that the IDO1 promoter is hypermethylated in ER-positive compared with ER-negative breast cancer. Reduced induction of IDO1 was also observed in human ER-positive breast cancer cell lines. IDO1 induction was enhanced upon DNA demethylation in ER-positive but not in ER-negative cells and methylation of an IDO1 promoter construct reduced IDO1 expression, suggesting that enhanced methylation of the IDO1 promoter suppresses IDO1 in ER-positive breast cancer. The association of ER overexpression with epigenetic downregulation of IDO1 appears to be a particular feature of breast cancer as IDO1 was not suppressed by IDO1 promoter hypermethylation in the presence of high ER expression in cervical or endometrial cancer.
KEYWORDS: DNA methylation; epigenetics; estrogen receptor; immunosuppression; indoleamine-2,3-dioxygenase; tryptophan metabolism
Introduction
To evade immune destruction, tumor cells make use of immunosuppressive mechanisms that originally developed to limit excessive inflammation. Diverse immunosuppressive mechanisms such as secretion of transforming growth factor β (TGFβ), prostaglandin signaling, expression of tolerogenic surface molecules as well as the metabolism of amino acids such as arginine and tryptophan (Trp) are used by tumors to suppress antitumor immune responses. The formation of kynurenine (Kyn) by the Trp-catabolic enzymes indoleamine-2,3-dioxygenase 1 (IDO1) and/or tryptophan-2,3-dioxygenase (TDO2) has emerged as an important metabolic pathway controlling immune escape as well as tumor-intrinsic malignant properties.1-6 Inhibitors of IDO1 and TDO2 are therefore in development for cancer therapy and clinical trials with IDO1 inhibitors in solid tumors in combination with chemo- or immunotherapy are ongoing.3,7
Physiologically, IDO1 plays a role in maintaining maternal-fetal tolerance, and dampening immune responses after immune activation by pro-inflammatory stimuli such as interferon gamma (IFNγ).8 IFNγ represents one of the most potent activators of IDO1 transcription through binding of STAT1 to IFNγ activation sequence elements and interferon regulatory factor 1 (IRF1) to IFN-stimulated response elements (ISRE) in the 5′-flanking region of the IDO1 gene.9 Kyn along with downstream metabolites such as kynurenic acid creates an immunosuppressive microenvironment, due to inhibition of T-cell responses.10 In preclinical cancer models, IDO1 is regulated by tumor suppressor genes such as Bin11 and oncogenes such as KIT,11 while an IDO-AHR-IL-6-STAT3 transcriptional circuit drives constitutive IDO1 expression in ovarian carcinoma and non-small cell lung cancer (NSCLC) cells.12 In contrast, the molecular mechanisms that regulate IDO1 expression in human breast cancer are incompletely understood.
Invasive breast cancer is a heterogeneous disease, classified through gene expression profiling into intrinsic subtypes, which serve as independent predictors of survival.13 The intrinsic subtypes, luminal A and luminal B overexpress ER, while the HER2-enriched and basal-like subtypes are ER-negative.13 Preclinical evidence suggests that the use of pharmacological IDO1 inhibitors to block immunosuppressive Trp metabolism may represent a promising avenue for the treatment of breast cancer.1,4 However, a better understanding of the regulation of IDO1 expression in the different breast cancer subtypes is necessary for selection of patients that may benefit from treatment with IDO1 inhibitors. We therefore investigated the regulation of IDO1 in human breast cancer.
Results
Serum Kyn and tumoral IDO1 expression are lower in ER-positive than in ER-negative breast cancer patients
Analysis of sera obtained at primary breast cancer diagnosis before initial surgery or neoadjuvant chemotherapy revealed reduced Kyn concentrations in ER-positive compared with ER-negative breast cancer patients (Fig. 1A, Table S1). In human breast cancer tissue, prominent IDO1 expression was detected in four out of six ER-negative tumors but not in the nine ER-positive tumors (Fig. 1, B and C, Table S2). To confirm a possible relationship between IDO1 and ER, we analyzed larger data sets of human breast cancer expression data. We found a negative correlation between IDO1 and ESR1 in invasive breast cancer RNASeq data from “The Cancer Genome Atlas” (TCGA, Fig. S1) and across 13 additional publicly available human breast cancer expression data sets (Table S3). Analysis of TCGA data confirmed that IDO1 mRNA expression is significantly lower in ER-positive than in ER-negative breast cancer tissue (Fig. 1D). Correspondingly, in the different breast cancer subtypes, IDO1 is downregulated in the ER-overexpressing luminal A and B subtypes compared with the ER-negative HER2-enriched and basal-like subtypes (Fig. 1E). Using a comprehensive computational model of Trp metabolism based on existing kinetic data for the enzymatic conversions and transporters,14 we investigated whether Kyn concentrations differ in ER-positive and ER-negative tumors as a consequence of the differential IDO1 expression. Integration of breast cancer expression data (TCGA) into our model of Trp metabolism indeed predicted reduced Kyn concentrations in ER-positive compared with ER-negative breast cancer tissue (Fig. 1F).
Figure 1.
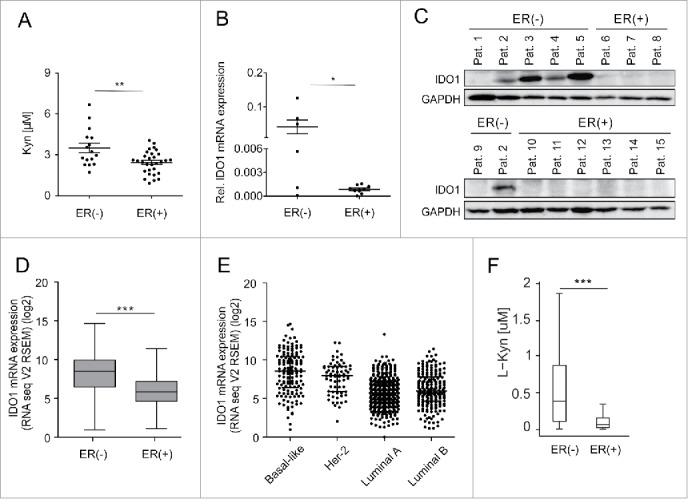
Serum Kyn and tumoral IDO1 expression are lower in ER-positive than ER-negative breast cancer patients. (A) Sera obtained before initial surgery showed significantly lower Kyn concentrations in untreated ER-positive (n = 30) compared with untreated ER-negative breast cancer patients (n = 16, Student's t-test **p < 0.01). (B) In line, IDO1 mRNA normalized to GAPDH (Student's t-test *p < 0.05) and (C) IDO1 protein expression were higher in ER-negative (n = 6) than ER-positive (n = 9) frozen breast cancer samples from distinct patients. (D) TCGA data of breast invasive carcinoma confirm decreased IDO1 expression in ER-positive compared with ER-negative breast cancer tissue (ER(+) n = 343, ER(−) n = 99, Mann–Whitney U test ***p < 0.001). (E) Reduced IDO1 mRNA expression is observed in the ER-positive luminal compared with the mainly ER-negative Her-2 enriched and basal-like intrinsic breast cancer subtypes based on PAM50 classification, (basal-like n = 141, Her2-enriched n = 67, luminal A n = 423, luminal B n = 192). (F) In accordance with a decrease in IDO1 expression a model of Trp metabolism based on TCGA expression data of breast invasive carcinoma predicted lower Kyn concentrations in human ER-positive compared with ER-negative breast cancer (ER(+) n = 311, ER(−) n = 103, ***p < 0.001). Box plots represent the medians and the 75% and 25% percentiles. Whiskers extend to min and max values.
Hypermethylation of the IDO1 promoter downregulates IDO1 expression in ER-positive breast cancer
As DNA methylation is recognized as a potent epigenetic regulator of transcription, we next sought to investigate whether IDO1 promoter methylation contributes to the differential expression of IDO1 in ER-positive and ER-negative breast cancer. Using whole genome bilsulfite sequencing (WGBS) data of ER-positive and ER-negative breast cancer tissue (TCGA),15,16 we identified seven CpGs in the IDO1 promoter overlapping with a region of active chromatin, characterized by corresponding histone modifications and an overlap with a DHS cluster (Fig. 2A, Table S4). Strikingly, methylation of six of these CpGs was enhanced in ER-positive as compared with ER-negative breast cancer (Fig. 2A). As only very few WGBS data of human breast cancer are publically available, we turned to 450k DNA methylation arrays to corroborate our findings in a larger cohort. Although only one of the seven aforementioned CpG sites (cg10262052) in the IDO1 promoter is covered by 450k arrays, analysis of TCGA data revealed significantly higher DNA methylation of cg10262052 in ER-positive as compared with ER-negative human breast cancer tissue (Fig. 2B). Mirroring the IDO1 expression data (Fig. 1E), methylation of this CpG was highest in the ER-positive luminal A and B breast cancer subtypes and lowest in the basal-like subtype (Fig. 2C). Furthermore, methylation of the CpG site in the IDO1 promoter inversely correlated with IDO1 mRNA expression in TCGA 450k and RNASeq data (Fig. 2D), further supporting that DNA hypermethylation in the IDO1 promoter may indeed be involved in the reduction of IDO1 transcription observed in ER-positive breast cancer.
Figure 2.
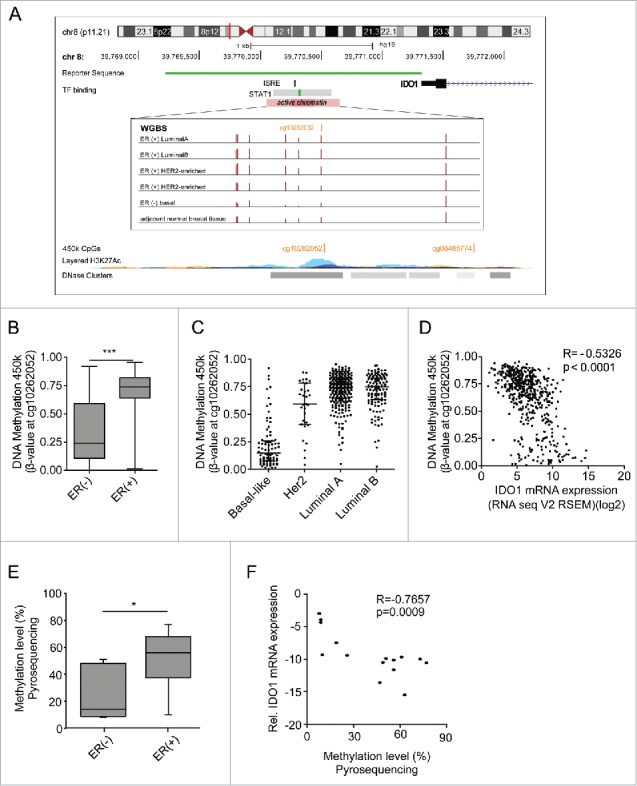
The IDO1 promoter is hypermethylated in ER-positive breast cancer. (A) Schematic representation of regulatory elements in the human IDO1 promoter. In an upstream region of active chromatin (ENCODE ChromHMM E027 and E028, enrichment of H3K27Ac, DNase hypersensitivity cluster) an interferon sensitive response element (ISRE) and a STAT1 binding site overlap with several CpG sites as can be seen in the WGBS data. One of these CpG sites— cg10262052—is covered by 450k arrays (GRCh 37/hg19 assembly). The localization of our IDO1 luciferase reporter gene construct is depicted in green. (B) The DNA methylation level of cg10262052 is significantly lower in ER-negative (n = 99) compared with ER-positive (n = 343) breast cancers (TCGA breast invasive carcinoma, Mann–Whitney U test, ***p < 0.001). Box plots represent the medians and the 75% and 25% percentiles. Whiskers extend to min and max values. (C) DNA methylation of cg10262052 is highest in the ER-positive luminal A, luminal B breast cancer subtypes and lowest in the ER-negative basal-like subtype (basal-like n = 83, Her2-enriched n = 31, luminal A n = 272, luminal B n = 125). (D) The DNA methylation at cg10262052 negatively correlates with IDO1 mRNA expression derived from TCGA breast invasive carcinoma 450k and RNASeq data (n = 511, Spearman's rank correlation). (E) Significant hypermethylation of cg10262052 in ER-positive (n = 9) as compared with ER-negative (n = 6) breast cancer tissue was observed by pyrosequencing (Student's t-test, *p < 0.05). (F) Methylation at this CpG site negatively correlates with IDO1 mRNA expression in these samples (Spearman's rank correlation).
These findings were confirmed by pyrosequencing of DNA extracted from our ER-positive and ER-negative breast cancer tissues. In accordance with the above results, we observed higher DNA methylation of cg10262052 in the ER-positive than in the ER-negative tumors (Fig. 2E). This methylation showed a strong negative correlation with the IDO1 expression of the same samples (Fig. 2F), supporting the notion that methylation of this area is critical for IDO1 expression. Higher DNA methylation of the IDO1 promoter in the ER-positive than in the ER-negative tumors was also corroborated by MassARRAY (Fig. S2A), which correlated nicely with the pyrosequencing data (Fig. S2B).
IDO1 expression and activity is reduced in ER-positive breast cancer cells
To experimentally address the differences in IDO1 expression observed in human breast cancer tissue in vitro, we investigated whether ER also suppresses IDO1 in breast cancer cell lines. We therefore compared IDO1 expression in the ER-positive luminal cell lines, MCF7, BT-474 and ZR-75–1, the ER-negative HER2-enriched cell line HCC1954 and the ER-negative, HER2-negative basal-like cell lines MDA-MB-468 and MDA-MB-231 (Fig. 3). While hardly any IDO1 mRNA was detectable in the ER-positive luminal cells, IDO1 mRNA was expressed at low basal levels in the ER-negative HER2-enriched and basal-like cells (Fig. 3B). At protein level, IDO1 expression was constitutively detectable only in ER-negative HCC1954 cells (Fig. 3D, top). In breast cancer tissue, IDO1 expression appears to be induced by IFNγ released from tumor infiltrating T-cells as interferon-related genes such as IFNG, STAT1 and IRF1 as well as T-cell markers such as CD2, CD3D and LCK were among the genes with the strongest association with IDO1 expression in human invasive breast cancer tissue (Fig. 3A). We therefore also used IFNγ to induce IDO1 in vitro. Induction of IDO1 mRNA expression by IFNγ stimulation was higher in ER-negative than in ER-positive cells (Fig. 3C). IFNγ induced IDO1 protein expression in all the cells examined, albeit to higher levels in ER-negative than in ER-positive cells (Fig. 3D, bottom). In line with their higher IDO1 expression, the ER-negative cells produced more Kyn than the ER-positive cells in response to IFNγ (Fig. 3E). Our results thus indicate that in accordance with the data obtained from human breast cancer tissue, IDO1 expression and Kyn production are higher in ER-negative than ER-positive breast cancer cells. We therefore used these breast cancer cells to experimentally validate the role of the methylation of the IDO1 promoter for IDO1 expression.
Figure 3.
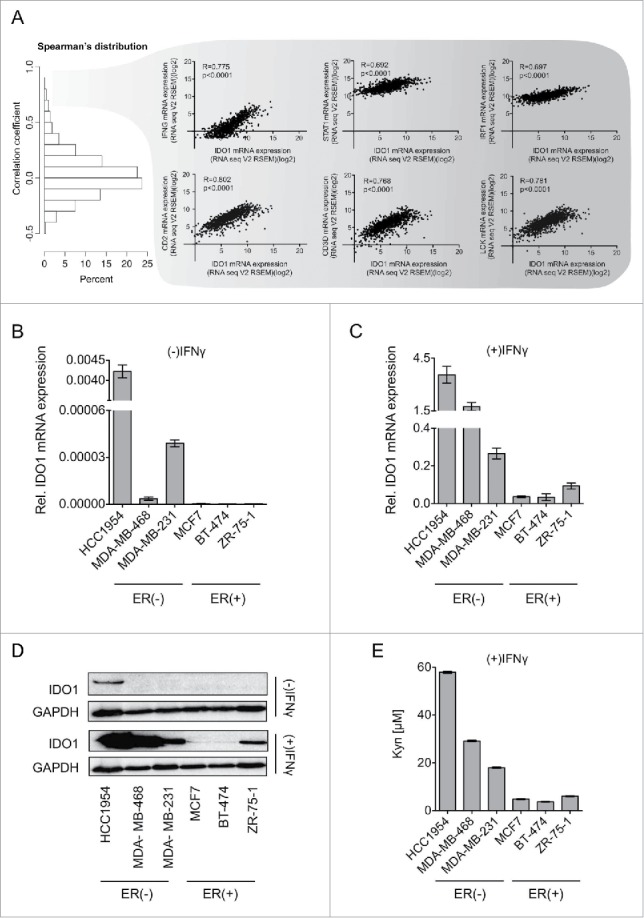
IDO1 expression and Trp metabolism in breast cancer cells. (A) Spearman's rank correlation of gene expression with IDO1 mRNA in TCGA breast invasive carcinoma RNASeq data. IFNG, STAT1 and IRF1, which are components of IFNγ signaling, as well as CD2, CD3D and LCK, which are T-cell markers were among the top IDO1 correlated genes. Their correlation with IDO1 is depicted as scatter plots. Spearman's rank correlation coefficients and corresponding p-values are shown. (B and C) IDO1 mRNA expression normalized to 18S RNA in ER-negative (HCC1954, MDA-MB-468, MDA-MB-231) and ER-positive (MCF7, BT-474 ZR-7–51) breast cancer cell lines, in the absence (B) and presence (C) of IFNγ stimulation (1000 U/mL for 24 h), measured by qRT-PCR. (D) Representative western blots demonstrating that also on the protein level IDO1 is expressed more strongly in ER-negative than ER-positive breast cancer cells. (E) Higher Kyn production was measured by high performance liquid chromatography (HPLC) in the supernatants of ER-negative in comparison to ER-positive cells 48 h after IFNγ stimulation (n = 3). Results are expressed as mean, error bars indicate s.e.m.
In vitro validation of DNA hypermethylation as a regulator of IDO1 expression in breast cancer cells
We first measured the methylation of the IDO1 promoter in our ER-positive and ER-negative cell lines by MassARRAY. While the ER-positive MCF7, ZR-75–1 and BT-474 cells showed high mean methylation of the IDO1 promoter region at CpG1 (cg10262052, chr8:39770526), CpG2 (chr8:39770486) and CpG3 (chr8:39770463), the ER-negative cells HCC1954 and MDA-MB-231 were hypomethylated in comparison (Fig. S3).
To test whether methylation is functionally involved in the inhibition of IDO1 expression, we treated breast cancer cells with the demethylating agent 5-aza-2′-deoxycytidine (5-Aza). 5-Aza increased both IFNγ-induced IDO1 mRNA (Fig. 4A), and IDO1 protein (Fig. 4B) in ER-positive breast cancer cells, which was associated with a concomittant increase in Kyn formation (Fig. 4C). Demethylation of the IDO1 promoter by 5-Aza was confirmed by MassARRAY (Fig. S4). In contrast to the ER-positive cells, no increase in IDO1 expression by 5-Aza treatment was observed in the ER-negative HCC1954 cells (Fig. 4D and E), supporting the hypothesis that methylation of the IDO1 promoter contributes to the lower expression of IDO1 in ER-positive breast cancer cells. To analyze whether methylation of the IDO1 promoter indeed results in decreased IDO1 expression, we cloned a 2.1 kb IDO1 luciferase promoter construct (Fig. 2A) into a CpG-free vector and compared the luciferase activity of the IDO1 promoter methylated by the prokaryotic DNA (cytosine-5)-methyltransferase M.SssI to its unmethylated counterpart (Fig. 4F). The methylated IDO1 promoter suppressed relative luciferase activity in the absence and presence of IFNγ in both ER-positive and ER-negative cells (Fig. 4F). Taken together, our results suggest that the expression of ER in breast cancer tissue is associated with suppression of IDO1 expression through IDO1 promoter hypermethylation.
Figure 4.
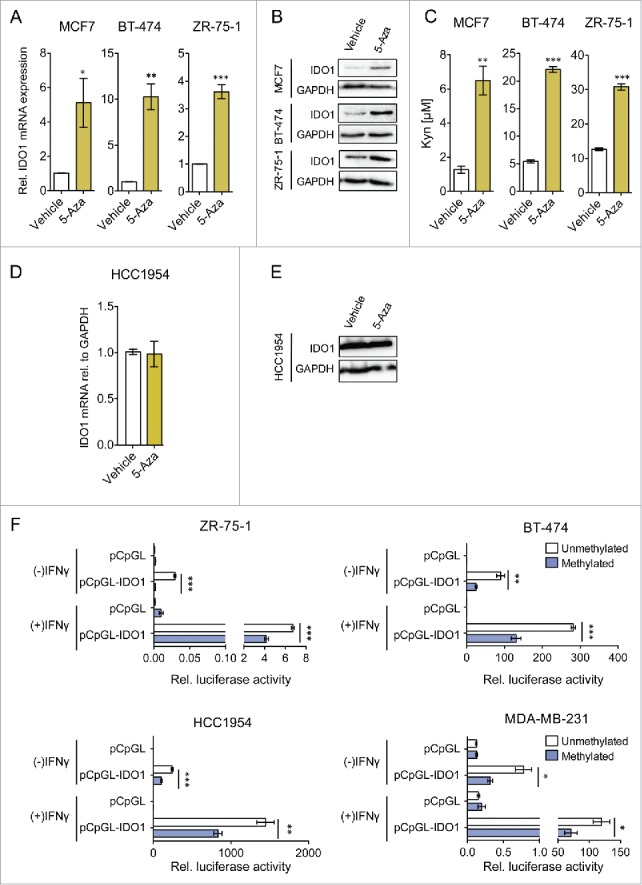
IDO1 expression and activity is suppressed by IDO1 promoter hypermethylation in ER-positive breast cancer cells. Treatment of ER-positive breast cancer cells with the demethylating agent 5-Aza (10 μM, 5 d) followed by stimulation with IFNγ (1000 U/mL, 48 h) significantly increased (A) IDO1 mRNA expression relative to GAPDH and (B) IDO1 protein expression (n = 3, per group). (C) Similarly, Kyn production of cells treated with 5-Aza for 5 d and subsequent IFNγ-stimulation for 48 h (n = 3) measured by HPLC was significantly increased compared with non-5-Aza treated controls (n = 3). 5-Aza treatment of the ER-negative HCC1954 cells neither influenced (D) IDO1 mRNA expression nor (E) IDO1 protein expression. (F) Activity of the unmethylated and the methylated IDO1 promoter was measured by luciferase reporter assay after stimulation with IFNγ (8 h; 20 U/mL for ZR-75–1 and HCC1954, 100 U/mL for BT-474 and MDA-MB-231), indicating that DNA methylation reduces IDO1 expression. Results are expressed as means, error bars indicate s.e.m. Statistical significance was determined by Student's t-tests, *p < 0.05, **p < 0.01, ***p < 0.001.
In cervical and endometrial carcinoma high ESR1 expression is not associated with increased IDO1 promoter methylation and suppression of IDO1 expression
We next analyzed whether high ESR1 expression is associated with low IDO1 expression also in other cancers arising from estrogen-responsive tissues such as cervical and endometrial carcinoma. These cancers differ from breast cancer in that their response rate to endocrine therapy is significantly lower. In contrast to our results in breast cancer, analysis of TCGA data did not reveal an inverse correlation between ESR1 and IDO1 in these tumor entities (Fig. S5A, B). In cervical cancer, IDO1 expression was higher in the cancers expressing ESR1 above the median expression level, than in those with ESR1 expression equal or below the median (Fig. 5A). Methylation of the IDO1 promoter inversely correlated with IDO1 mRNA expression in DNA methylation array and RNASeq data (Fig. 5B), suggesting that DNA-methylation of the IDO1 promoter may reduce IDO1 transcription in these tumors. However, high expression of ESR1 was not associated with enhanced methylation of the IDO1 promoter in cervical cancer (Fig. 5C), possibly explaining why no inverse correlation between ESR1 and IDO1 was observed. In endometrial carcinoma, IDO1 expression did not differ between tumors expressing high or low levels of ESR1 (Fig. 5D). Methylation of the IDO1 promoter did not correlate with IDO1 mRNA expression (Fig. 5E) and high expression of ESR1 was associated with reduced methylation of the IDO1 promoter (Fig. 5F). Collectively, these results suggest that high ESR1 expression is not associated with increased IDO1 promoter methylation and suppression of IDO1 mRNA expression in cervical and endometrial cancer. The suppression of IDO1 expression by IDO1 promoter hypermethylation may thus be characteristic of ER-positive breast cancer.
Figure 5.
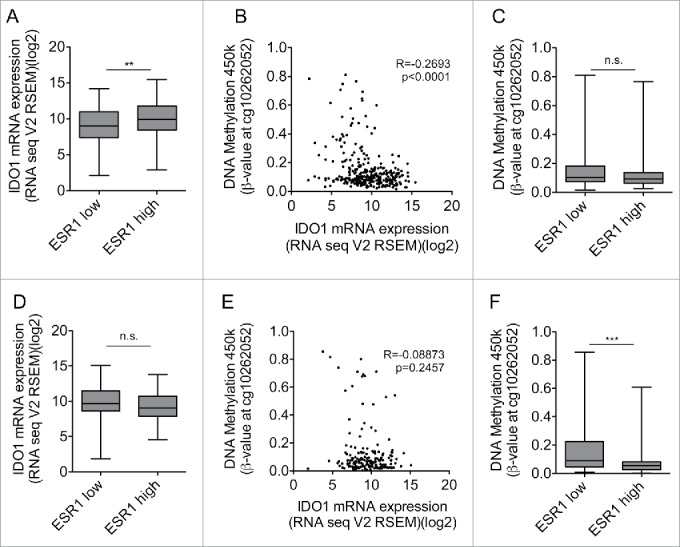
High ESR1 expression is not associated with IDO1 promoter methylation and reduced IDO1 expression in cervical and endometrial carcinoma. (A) IDO1 mRNA expression is higher in ESR1 high (n = 152, higher than the median ESR1 expression) than ESR1 low (n = 153, equal or lower than the median ESR1 expression) cervical cancers (TCGA, cervical squamous cell carcinoma and endocervical adenocarcinoma; Student's t-test, **p < 0.01). (B) The DNA methylation at cg10262052 inversely correlates with IDO1 mRNA expression derived from TCGA (n = 305, Spearman's rank correlation). (C) The DNA methylation level of cg10262052 does not differ between ESR1 low (n = 153) compared with ESR1 high (n = 152) cervical cancers (TCGA, cervical squamous cell carcinoma and endocervical adenocarcinoma, Mann–Whitney U test). (D) IDO1 mRNA expression does not differ between ESR1 low (n = 87, equal and lower than the median ESR1 expression) compared with ESR1 high (n = 86, higher than the median ESR1 expression) human endometrial carcinoma tissues derived from TCGA uterine corpus endometrial carcinoma RNASeq data; Student's t-test. (E) The DNA methylation at cg10262052 does not correlate with IDO1 mRNA expression derived from TCGA uterine corpus endometrial carcinoma (n = 173, Spearman's rank correlation). (F) The DNA methylation level of cg10262052 is lower in ESR1 high (n = 86) compared with ESR1 low (n = 87) endometrial carcinomas (TCGA, uterine corpus endometrial carcinoma, Mann–Whitney U test). Box plots represent the medians and the 75% and 25% percentiles. Whiskers extend to min and max values.
Discussion
The ER plays a pivotal role in the development and progression of breast cancer as it enhances the proliferation, survival and invasion of breast tumor cells.17 Here we show that despite its aforementioned tumor-promoting effects, ER also suppresses a malignant feature of breast cancer cells by inhibiting the induction of IDO1, a key enzyme mediating tumor immune evasion. The role of murine Ido1 in promoting breast cancer has previously been demonstrated in preclinical breast cancer models. Inhibition of Ido1 in combination with the chemotherapeutic agent paclitaxel reduced the growth of spontaneous mammary tumors in MMTV/neu mice,1 while Ido1 knockout protected against lung metastasis after orthotopic engraftment of mice with 4T1 breast carcinoma cells.4 Based on promising preclinical data, clinical trials with IDO1 inhibitors in patients with solid cancer with the aim of eliciting antitumor immunity and enhancing the efficacy of conventional chemotherapy are underway.7 As IDO1 is recognized as a resistance mechanism to immune checkpoint blockade in cancer,18,19 a better understanding of the regulation of IDO1 in human breast cancer may help improve immunotherapeutic strategies for this disease.
Several lines of evidence suggest that there is considerable variation in Trp metabolism in breast cancer patients. In 1967, Rose already described increased excretion of Trp metabolites in the urine of some but not all breast cancer patients analyzed.20 Recently, higher variabilities in Kyn and Kyn/Trp ratios in the plasma of breast cancer patients than in breast cancer-free women were reported21 and breast cancers were found to show differential Trp kinetics by dynamic positron emission tomography (PET).22 These data suggest that different subtypes of breast cancer may differ in Trp metabolism. We found that Kyn, the product of IDO1 enzymatic activity was reduced in the sera of ER-positive compared with ER-negative breast cancer patients (Fig. 1A). In addition, while IDO1 expression was detected in the majority of ER-negative tumors none of the ER-positive tumors expressed relevant IDO1 levels (Fig. 1B and C). Gene expression data showed a negative correlation between IDO1 and ESR1 across 14 breast cancer data sets (Fig. S1 and Table S3) and kinetic modeling of Kyn concentrations,14 revealed lower Kyn concentrations in ER-positive than ER-negative breast cancer (Fig. 1F). While our result of reduced IDO1 expression in ER-positive tumors is in contrast to a study investigating IDO1 expression in breast cancer by immunohistochemistry,23 it is supported by several other studies including a metabolomics study showing increased Kyn levels in ER-negative as compared with ER-positive breast cancer tissue.24-26 In addition, recent evidence suggests that the ER may also be involved in the suppression of Ido1 expression in animal models of breast cancer as treatment of mice with a compound leading to ER expression in otherwise ER-negative mammary tumors resulted in suppression of Ido1 expression.27
In this study, we focused on the regulation of IDO1, but recently also TDO2, which catalyzes the same enzymatic reaction as IDO1 has been found to be expressed in different malignancies including breast cancer.2,3,28 TDO2-mediated AHR activation was shown to protect against anoikis in triple negative breast cancer cells, while the induction of TDO2 by substratum detachment was not significant in ER-positive breast cancer cells.28 In addition, TDO2 expression was found to be higher in ER-negative than ER-positive breast cancer tissue.28 The downregulation of IDO1 by ER signaling identified here therefore does not appear to be counteracted by the activity of TDO2, resulting in a general downregulation of Trp metabolism in the presence of ER.
As the degree of T cell infiltration is highest in ER-negative tumors,29 and IDO1 expression shows a strong correlation with T cell markers and genes involved in IFNγ signaling (Fig. 3A), the difference in IDO1 expression between ER-positive and ER-negative breast cancer may in part be mediated by different levels of inflammatory mediators released by T cells that induce IDO1. However, we show here that also tumor cell intrinsic properties underlie the difference in IDO1 expression between ER-positive and ER-negative breast cancer as IFNγ-induced IDO1 mRNA, IDO1 protein and Kyn production were reduced in ER-positive compared with ER-negative breast cancer cells (Fig. 3, C–E).
The ER is a member of the nuclear receptor family of transcription factors and the mechanisms, through which ER activates gene expression by binding to estrogen responsive elements in the promoters of its target genes are well characterized. In recent years, attention has shifted to repression of gene expression by ER, demonstrating that ER is recruited to ER binding sites of early estrogen-repressed genes, albeit more transiently than for estrogen-stimulated genes.30
Analysis of ENCODE data did not reveal any evidence of direct binding of the ER to the promoter region of IDO1. However, using WGBS, 450k, MassARRAY and pyrosequencing data, we identified CpGs within the STAT1 binding site in the IDO1 promoter (Fig. 2A) that are hypermethylated in ER-positive compared with ER-negative breast cancers (Fig. 2, Fig. S2). Methylation of cg10262052 in the IDO1 promoter is highest in the ER-positive luminal tumors, and lowest in the basal-like subtypes (Fig. 2C), which is mirrored by IDO1 showing the lowest expression in luminal tumors (Fig. 1E). Expression data of human breast cancer tissue as well as our own pyrosequencing data showed an inverse correlation between IDO1 promoter methylation and IDO1 mRNA expression (Fig. 2, D and F). These results suggested that enhanced methylation of the IDO1 promoter in ER-positive breast cancer cells may be involved in suppressing IDO1. In accordance with this notion, treatment with the DNA demethylating agent 5-Aza enhanced IDO1 expression in ER-positive but not in ER-negative cells (Fig. 4A–E, Fig. S4) and the inhibitory effect of IDO1 promoter methylation on IDO1 expression was corroborated by the use of a methylated reporter construct (Fig. 4F).
Identification of distinct DNA methylation between ER-positive and ER-negative breast cancer is in agreement with previous findings showing differences in global DNA methylation patterns between these groups.31 In general, ER-positive tumors have been found to contain more hypermethylated loci than ER-negative tumors32 and ER has been reported to transcriptionally induce DNA (cytosine-5-)-methyltransferase 1 (DNMT1) in breast cancer cells.33 Interestingly, a synergistic induction of IDO1 by IFNγ and the DNA methyltransferase inhibitor zebularine has previously been described in human monocytic cells,34 suggesting that methylation of the IDO1 promoter may inhibit IDO1 expression also in other cell types. We therefore tested whether ER expression also is associated with suppression of IDO1 expression through IDO1 promoter methylation in other cancers that develop from estrogen-dependent tissues, such as cervical and endometrial carcinoma. IDO1 expression has previously been shown to be present in 83% of cervical carcinomas and 94% of endometrial carcinomas.6 Unlike in breast tumors, in endometrial carcinomas, IDO1 was observed to be expressed constitutively within the tumor cells in the absence of T-cell infiltration.35 ER expression has been reported to be required for carcinogenesis in mouse models for HPV-associated cervical cancer and to be frequently expressed in human cervical carcinoma,36 while ER expression in endometrial carcinoma has been described in 32–77% of cases.37 However, in contrast to ER-positive breast cancer, anti-estrogen therapy does not constitute a standard of care for ER-positive cervical or endometrial cancer, suggesting less dependence of these cancers on ER. Interestingly, no inverse correlation was observed between IDO1 and ESR1 mRNA expression in cervical and endometrial carcinoma possibly due to the fact that high ESR1 expression was not associated with hypermethylation of the IDO1 promoter (Fig. 5, C and F). The association of ER overexpression with low IDO1 expression due to IDO1 promoter hypermethylation thus appears to be characteristic for breast cancer (Fig. 6). Higher IDO1 expression in ER-negative breast cancer may contribute to its malignant phenotype as well as bad prognosis, and IDO1 inhibitors may thus be a promising avenue for the treatment of ER-negative breast cancer.
Figure 6.
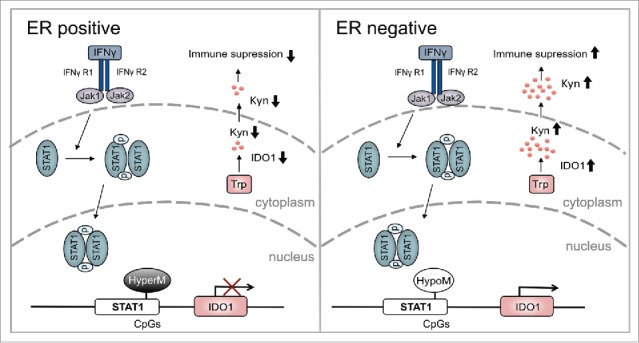
Overview of the modulation of IDO1 by ER in breast cancer. In ER-positive breast cancer, hypermethylation (HyperM) of CpGs in the IDO1 promoter reduces IDO1 expression. Reduced IDO1 expression results in less production of immunosuppressive kynurenine. In contrast, in ER-negative breast cancer the CpGs in the IDO1 promoter are hypomethylated (HypoM) leading to stronger induction of IDO1, increased production of Kyn and finally enhanced suppression of antitumor immune responses.
Methods
Measurement of Kyn concentrations in human sera by ELISA
Serum samples from a total of 46 patients with histologically confirmed invasive breast cancer were obtained from the Department of Gynecology and Obstetrics at the University Hospital of Dresden, Germany. Clinical characteristics are documented in Table S1. This sample set consisted of (i) sera from 30 untreated ER-positive patients and (ii) 16 sera from untreated ER-negative patients. Informed written consent was obtained from all patients. The study was approved by the Local Ethics Committee (EK 74032013) and was performed according to the declaration of Helsinki.
Kyn was measured using the IDK® IDO activity ELISA K7726 (Immundiagnostik AG) according to the manufacturer's instructions.
Human breast cancer tissue samples
Six ER-negative and nine ER-positive breast cancer tissue cryosamples were provided by the tissue bank of the National Center for Tumor Diseases (NCT, Heidelberg, Germany) in accordance with the regulations of the tissue bank and the approval (206/2005 and 207/2005) of the ethics committee of Heidelberg University. Clinical characteristics are documented in Table S2. RNA was isolated using the Qiagen RNeasy Mini Kit according to the manufacturer's instruction for RNA isolation from tissue. Protein was isolated by lysing the frozen tissues in RIPA buffer supplemented with complete protease inhibitor (Roche) and phosphatase inhibitor (Sigma-Aldrich). qRT-PCR measurements and Western blot were performed as described below.
Modeling of Trp metabolism
TCGA breast invasive carcinoma RNASeq data were applied to a comprehensive kinetic model of Trp metabolism as described previously14 For each patient, a separate model was generated resulting in patient-specific concentrations. The Trp concentration used as input for the model was 5 µM, corresponding to the median concentration of free Trp measured in the blood of healthy donors.
Analysis of expression data
TCGA breast invasive carcinoma (TCGA, provisional), TCGA Uterine Corpus Endometrial Carcinoma (TCGA, provisional) and TCGA Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma (TCGA, provisional) 450k array data were downloaded from TCGA and RNASeq data were downloaded from the cBio Cancer Genomics Portal (www.cbioportal.org).38,39 RNA seq V2 RSEM values were log2 transformed. Patients' ER status, based on immunohistochemistry, and PAM50 classification were derived from the UCSC cancer genomics browser.40,41 For correlation analyses of IDO1 with ESR1 microarray data was downloaded from the R2:Genomic analysis and Visualization Platform (http://hgserver1.amc.nl) and Gene Expression Omnibus (GEO).
Cell culture
All breast cancer cell lines were obtained from ATCC and cultivated in phenol red-free DMEM (Gibco) supplemented with 10% FCS (Gibco), 100 U/mL penicillin 100 µg/mL streptomycin (Gibco), 2 mM glutamine (Gibco) and 1 mM sodium pyruvate (Gibco). HCC1954 were cultivated in RPMI (Gibco) supplemented with 10% FCS (Gibco). IDO1 expression was equal whether cells were cultured in DMEM or RPMI. Cell lines were confirmed to be mycoplasma-free and were authenticated by Multiplex Cell Authentication (Multiplexion).
Drug and cytokine treatment
5-Aza-2′ deoxycytidine (5-Aza; A3656, Sigma-Aldrich, 200 mM stock) was dissolved in DMSO. Cells were incubated with 1 µM or 10 µM 5-Aza for 5 d and the medium was replaced with fresh medium containing 5-Aza every 24 h, subsequently 1000 U/mL IFNγ (14–8319, eBiosciences) were added for 48 h.
RNA isolation and quantitative (q)RT-PCR
Total RNA was isolated using the Qiagen RNeasy Mini Kit (Qiagen). One microgram RNA was reverse-transcribed using the High Capacity cDNA reverse transcriptase kit (Applied Biosystems). qRT-PCR was performed in quadruplicates with the SYBR Select Master Mix (Thermo Scientific) using a StepOnePlus real-time PCR system (Applied Biosystems). Relative quantification was done using the 2ΔΔCt method. GAPDH or 18S RNA were used for normalization. qRT-PCR primers were designed using Primer Blast (NCBI) and were separated by at least one intron on the genomic DNA.
Primers sequences:
IDO1 forward: 5′-GATGTCCGTAAGGTCTTGCC -3′
reverse: 5′-TCCAGTCTCCATCACGAAAT-3′
GAPDH forward: 5′-CTCTCTGCTCCTCCTGTTCGAC-3′
reverse: 5′-TGAGCGATGTGGCTCGGCT-3′
18S RNA forward: 5′-GATGGGCGGCGGAAAATAG-3′
reverse: 5′-GCGTGGATTCTGCATAATGGT-3′
Protein isolation and western blot
Cells were harvested and lysed in ice-cold lysis buffer, consisting of TRIS-HCl (50 mM, pH 8,0) containing 50 mM NaCl, 1% NP-40, 2.5 mM EDTA, 10% glycerin, 200 mM dithiothreitol, 100 µM PMSF, complete protease inhibitor (Roche) and phosphatase inhibitor (Sigma). Protein concentrations were measured by Bradford Assay (BioRad, Hercules, CA, USA) at 595 nm. 30 to 60 µg of protein were diluted in Laemmli buffer, denatured for 5 min at 95°C, loaded and separated by 12% SDS-PAGE. Proteins were transferred onto 0.2 µm nitrocellulose membranes, blocked with either 5% milk or 5% BSA for 1 h, and stained with the primary antibodies diluted in blocking solution overnight. Primary antibodies: rabbit anti-human IDO1 (#AG-25A-0029-C100, Adipogene, 1:1000) and mouse anti-human GAPDH (#39–8600, Thermo Scientific, 1:1000). Membranes were stained with appropriate HRP-linked secondary antibodies (GE Healthcare, #GENA9340–1M, #GENXA931, 1:5000) for 2 h at RT. Signals were visualized using ECL (GE Healthcare). Images were captured using the BioRad ChemiDoc MP system with Image Lab 5.1 software.
Kyn measurement by High Performance Liquid Chromatography (HPLC)
For 1 mL of cell culture supernatant, 162.8 µL of 72% trichloroacetic acid were added, and mixed briefly. Samples were centrifuged at maximum speed for 12 min, and the supernatants were analyzed using a Dionex Ultimate® 3000 UHPLC (Thermo Scientific). Chromatographic separation was achieved on a reversed phase Accucore™ aQ column (Thermo Scientific) with 2.6 µm particle size with a gradient mobile phase consisting of 0.1% trifluoroacetic acid (TFA) in water (A) and 0.1% TFA in acetonitrile (B). Kyn was detected based on comparison with the standard, its retention time and UV emission spectrum at 365 nm. Results were analyzed using the Chromeleon™ 7.2 Chromatography Data System (Thermo Scientific).
Pyrosequencing
Genomic DNA (gDNA) was isolated using the QIAamp DNA Mini Kit (Qiagen). 1 µg gDNA was bisulfite converted using the EZ DNA-methylation Kit (ZymoResearch) and DNA methylation of cg10262052 was assessed on a PyroMark Q48 Autoprep station (Qiagen) following the manufacturers' instructions. The pyrosequencing assay was designed on the reverse complement strand using the PyroMark Assay Design version 2.0.1.15 software (Qiagen) with the reverse primer biotinylated at the 5′end (forward primer 5′-GTAAGTTTGTGGTTTATTTTAGAGGTATTG-3′, reverse primer [biotin]-5′-ACTATTTCTCTTTTCTCCTTTTAATCA-3′, sequencing primer 5′-GGAAGTTAAAGAAGAAATTAAG-3′). PCR was performed using the PyroMark PCR Kit (Qiagen) following the manufacturer's recommendations with an annealing temperature of 52°C.
DNA methylation analysis by MassARRAY
Quantitative DNA methylation analyses were performed on the mass spectrometry-based Agena Bioscience MassARRAY platform. A PCR primer pair for the IDO1 promoter region (chr8:39771416–39771555, covering cg10262052 and two neighboring CpGs) was designed with a T7 promoter tag (y = cagtaatacgactcactatagggagaaggct) on the reverse primer for in vitro transcription and a 10-mer tag (x = aggaagagag) on the forward primer (forward: x-AGGAAGTTAAAGAAGAAATTAAG; reverse: y- CACAATTTAATTTATTTCAAATAC). Bisulfite-treated DNA was PCR amplified using HotStarTaq DNA Polymerase (Qiagen) with the following cycling program: 95°C for 15 min, followed by 45 cycles of 94°C for 30 sec, 56°C for 30 sec, 72°C for 1 min and a final elongation step at 72°C for 5 min on a T1 Thermocycler (Biometra). The PCR product was in vitro transcribed and cleaved by RNase A using the EpiTyper T Complete Reagent Set (Sequenom) and subjected to MALDI-TOF mass spectrometry analysis to determine methylation patterns as described previously.42 DNA methylation standards (0%, 20%, 40%, 60%, 80% and 100% methylated genomic DNA) were used to control for potential PCR bias. Validity of the derived DNA methylation values was confirmed by correlation of expected and measured DNA methylation values (IDO1: R2 = 0.98, Fig. S6).
Cloning of the IDO1 reporter plasmid
To generate an IDO1 reporter plasmid lacking CpGs in the backbone, the 2.1 kb promoter region of IDO1 was PCR-amplified using Phusion polymerase with high fidelity buffer (Thermo Scientific) with the following primers:
forward: 5′-ACACTAGTTAGAGCAAAACGCTGAGTTCTG -3′
reverse: 5′-ACCCATGGCCATTCTTGTAGTCTGCTCCTCT-3′
The PCR product was cloned into SpeI and NcoI sites of the pCpGL basic vector (kind gift from M. Rehli). The resulting plasmid (pCpGL-IDO1) was grown in PIR1-competent E.coli under Zeocin (Invitrogen) selection. Insertion of the IDO1 promoter was confirmed using colony PCR and sequencing.
In vitro methylation assay
Methylated IDO1 reporter plasmid was prepared by incubating 10 µg of plasmid with 20 µL of the prokaryotic DNA(cytosine-5)-methyltransferase M.SssI (Thermo Scientific) and 100 µM S-adenosylmethionine (Thermo Scientific) in the presence of M.SssI buffer at 37°C for 1 h. The enzyme was inactivated by incubation at 65°C for 20 min. The methylation status was confirmed by digesting the plasmid using the methylation-sensitive HpaII restriction enzyme.
Luciferase reporter gene assay
5 × 104 cells were transfected with 150 ng of pCpGL-IDO1 or pCpGL empty vector with pRL-TK renilla using Fugene (Promega). 48 h after transfection, cells were treated with 20 or 100 U/mL IFNγ for 8 h and directly lysed in passive lysis buffer (PromoCell). Luciferase activity was assayed using the PromoKine Luciferase Reporter Assay Kit III (PromoCell) and measured using PheraStar FS (BMG Lab Tech) with the MARS program 3.01.R2 software. Relative luciferase activity was defined as the mean of firefly luciferase/renilla luciferase ratios.
Statistical analyses
Statistical analyses were performed using GraphPad Prism version 5.00 (GraphPad Software), SigmaPlot version 12 (Systat Software) and Statistica for Windows, Version 10 (Statsoft). Correlation analyses were based on Spearman's rank analysis. Two-tailed Student's t-tests were used for single comparisons. Where applicable, rank sum analysis by Mann–Whitney U was conducted. All data are expressed as mean ± s.e.m. Statistical significance is assumed at p < 0.05.
Supplementary Material
Disclosure of potential conflicts of interest
Karl-Heinz Kellner is the CEO of Neuroimmun GmbH, which produces and sells kits for the measurement of kynurenine. The other authors have declared that no conflict of interest exists.
Acknowledgments
We thank Sascha Schäuble for support with graphics and Christoph Weigel and Martina Ott for technical advice. We thank Reka Toth for help with the WGBS data and Anna S. Berghoff for critical reading of the manuscript. The breast cancer tissue samples were provided by the tissue bank of the National Center for Tumor Diseases (NCT, Heidelberg, Germany). The results are in part based upon data generated by The Cancer Genome Atlas managed by the NCI and NHGRI. In addition, NIH Roadmap Epigenomics Project data were used to evaluate chromatin states (http://nihroadmap.nih.gov/epigenomics/).
Funding
This work was supported by grants from the BMBF e:Med initiative (GlioPATH, 01ZX1402) to SL, ST and CAO and an Otto Bayer Fellowship to DLD. ST was supported by the Helmholtz Initiative on Personalized Medicine (iMed). LFSP is supported by scholarships from the University of Costa Rica (UCR) and Costa Rica's Ministry of Science, Technology and Telecommunications (MICITT). SRM and KH are supported by a Helmholtz International Graduate School for Cancer Research Fellowship.
Author contributions
DLD and CAO designed the study. DLD, SRM, IA, KH, ST and CAO developed methodology, DLD, SRM, SBC, IA, LFSP, BB, MK, LT, SL, KH and OM acquired data, DLD, DW, IH, CG, ST and CAO analyzed and interpreted data. PW, JDK, KK, SS, CP and MP provided clinical and material support. DLD, ST and CAO wrote the manuscript. All the authors read, reviewed and revised the manuscript.
References
- 1.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med 2005; 11:312-9; PMID:15711557; http://dx.doi.org/ 10.1038/nm1196 [DOI] [PubMed] [Google Scholar]
- 2.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M et al.. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011; 478:197-203; PMID:21976023; http://dx.doi.org/ 10.1038/nature10491 [DOI] [PubMed] [Google Scholar]
- 3.Pilotte L, Larrieu P, Stroobant V, Colau D, Dolusic E, Frederick R, De Plaen E, Uyttenhove C, Wouters J, Masereel B et al.. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci U S A 2012; 109:2497-502; PMID:22308364; http://dx.doi.org/ 10.1073/pnas.1113873109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith C, Chang MY, Parker KH, Beury DW, DuHadaway JB, Flick HE, Boulden J, Sutanto-Ward E, Soler AP, Laury-Kleintop LD et al.. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov 2012; 2:722-35; PMID:22822050; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 2003; 9:1269-74; PMID:14502282; http://dx.doi.org/ 10.1038/nm934 [DOI] [PubMed] [Google Scholar]
- 6.Theate I, van Baren N, Pilotte L, Moulin P, Larrieu P, Renauld JC, Hervé C, Gutierrez-Roelens I, Marbaix E, Sempoux C et al.. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol Res 2015; 3:161-72; PMID:25271151; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0137 [DOI] [PubMed] [Google Scholar]
- 7.Vacchelli E, Aranda F, Eggermont A, Sautes-Fridman C, Tartour E, Kennedy EP, Platten M, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 2014; 3:e957994; PMID:25941578; http://dx.doi.org/ 10.4161/21624011.2014.957994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol 2016; 37:193-207; PMID:26839260; http://dx.doi.org/ 10.1016/j.it.2016.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chon SY, Hassanain HH, Gupta SL. Cooperative role of interferon regulatory factor 1 and p91 (STAT1) response elements in interferon-gamma-inducible expression of human indoleamine 2,3-dioxygenase gene. J Biol Chem 1996; 271:17247-52; PMID:8663541; http://dx.doi.org/ 10.1074/jbc.271.29.17247 [DOI] [PubMed] [Google Scholar]
- 10.Opitz CA, Wick W, Steinman L, Platten M. Tryptophan degradation in autoimmune diseases. Cell Mol Life Sci 2007; 64:2542-63; PMID:17611712; http://dx.doi.org/ 10.1007/s00018-007-7140-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, Sorenson EC, Popow R, Ariyan C, Rossi F et al.. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med 2011; 17:1094-100; PMID:21873989; http://dx.doi.org/ 10.1038/nm.2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Litzenburger UM, Opitz CA, Sahm F, Rauschenbach KJ, Trump S, Winter M, Ott M, Ochs K, Lutz C, Liu X et al.. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014; 5:1038-51; PMID:24657910; http://dx.doi.org/ 10.18632/oncotarget.1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z et al.. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009; 27:1160-7; PMID:19204204; http://dx.doi.org/ 10.1200/JCO.2008.18.1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stavrum AK, Heiland I, Schuster S, Puntervoll P, Ziegler M. Model of tryptophan metabolism, readily scalable using tissue-specific gene expression data. J Biol Chem 2013; 288:34555-66; PMID:24129579; http://dx.doi.org/ 10.1074/jbc.M113.474908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J et al.. Integrative analysis of 111 reference human epigenomes. Nature 2015; 518:317-30; PMID:25693563; http://dx.doi.org/ 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M et al.. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res 2015; 43:D670-81; PMID:25428374; http://dx.doi.org/ 10.1093/nar/gku1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest 2006; 116:561-70; PMID:16511588; http://dx.doi.org/ 10.1172/JCI27987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med 2013; 210:1389-402; PMID:23752227; http://dx.doi.org/ 10.1084/jem.20130066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B, Lepage P, Boneca IG, Chamaillard M, Kroemer G et al.. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity 2016; 44:1255-69; PMID:27332730; http://dx.doi.org/ 10.1016/j.immuni.2016.06.001 [DOI] [PubMed] [Google Scholar]
- 20.Rose DP. The influence of sex, age and breast cancer on tryptophan metabolism. Clin Chim Acta 1967; 18:221-5; PMID:5624543; http://dx.doi.org/ 10.1016/0009-8981(67)90161-1 [DOI] [PubMed] [Google Scholar]
- 21.Lyon DE, Walter JM, Starkweather AR, Schubert CM, McCain NL. Tryptophan degradation in women with breast cancer: a pilot study. BMC Res Notes 2011; 4:156; PMID:21615916; http://dx.doi.org/ 10.1186/1756-0500-4-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Juhasz C, Nahleh Z, Zitron I, Chugani DC, Janabi MZ, Bandyopadhyay S, Ali-Fehmi R, Mangner TJ, Chakraborty PK, Mittal S et al.. Tryptophan metabolism in breast cancers: molecular imaging and immunohistochemistry studies. Nucl Med Biol 2012; 39:926-32; PMID:22444239; http://dx.doi.org/ 10.1016/j.nucmedbio.2012.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soliman H, Rawal B, Fulp J, Lee JH, Lopez A, Bui MM, Khalil F, Antonia S, Yfantis HG, Lee DH et al.. Analysis of indoleamine 2-3 dioxygenase (IDO1) expression in breast cancer tissue by immunohistochemistry. Cancer Immunol Immunother 2013; 62:829-37; PMID:23344392; http://dx.doi.org/ 10.1007/s00262-013-1393-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isla Larrain MT, Rabassa ME, Lacunza E, Barbera A, Creton A, Segal-Eiras A, Croce MV. IDO is highly expressed in breast cancer and breast cancer-derived circulating microvesicles and associated to aggressive types of tumors by in silico analysis. Tumour Biol 2014; 35:6511-9; PMID:24687552; http://dx.doi.org/ 10.1007/s13277-014-1859-3 [DOI] [PubMed] [Google Scholar]
- 25.Jacquemier J, Bertucci F, Finetti P, Esterni B, Charafe-Jauffret E, Thibult ML, Houvenaeghel G, Van den Eynde B, Birnbaum D, Olive D et al.. High expression of indoleamine 2,3-dioxygenase in the tumour is associated with medullary features and favourable outcome in basal-like breast carcinoma. Int J Cancer 2012; 130:96-104; PMID:21328335; http://dx.doi.org/ 10.1002/ijc.25979 [DOI] [PubMed] [Google Scholar]
- 26.Tang X, Lin CC, Spasojevic I, Iversen ES, Chi JT, Marks JR. A joint analysis of metabolomics and genetics of breast cancer. Breast Cancer Res 2014; 16:415; PMID:25091696; http://dx.doi.org/ 10.1186/s13058-014-0415-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan H, Kopelovich L, Yin Y, Lu J, Glazer RI. Drug-targeted inhibition of peroxisome proliferator-activated receptor-gamma enhances the chemopreventive effect of anti-estrogen therapy. Oncotarget 2012; 3:345-56; PMID:22538444; http://dx.doi.org/ 10.18632/oncotarget.457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Amato NC, Rogers TJ, Gordon MA, Greene LI, Cochrane DR, Spoelstra NS, Nemkov TG, D'Alessandro A, Hansen KC, Richer JK. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res 2015; 75:4651-64; PMID:26363006; http://dx.doi.org/ 10.1158/0008-5472.CAN-15-2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyan M, Schmidt-Mende J, Kiessling R, Poschke I, de Boniface J. Differential tumor infiltration by T-cells characterizes intrinsic molecular subtypes in breast cancer. J Transl Med 2016; 14:227; PMID:27473163; http://dx.doi.org/ 10.1186/s12967-016-0983-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stossi F, Madak-Erdogan Z, Katzenellenbogen BS. Estrogen receptor alpha represses transcription of early target genes via p300 and CtBP1. Mol Cell Biol 2009; 29:1749-59; PMID:19188451; http://dx.doi.org/ 10.1128/MCB.01476-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dedeurwaerder S, Desmedt C, Calonne E, Singhal SK, Haibe-Kains B, Defrance M, Michiels S, Volkmar M, Deplus R, Luciani J et al.. DNA methylation profiling reveals a predominant immune component in breast cancers. EMBO Mol Med 2011; 3:726-41; PMID:21910250; http://dx.doi.org/ 10.1002/emmm.201100801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fackler MJ, Umbricht CB, Williams D, Argani P, Cruz LA, Merino VF, Teo WW, Zhang Z, Huang P, Visvananthan K et al.. Genome-wide methylation analysis identifies genes specific to breast cancer hormone receptor status and risk of recurrence. Cancer Res 2011; 71:6195-207; PMID:21825015; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi JF, Li XJ, Si XX, Li AD, Ding HJ, Han X, Sun YJ. ERalpha positively regulated DNMT1 expression by binding to the gene promoter region in human breast cancer MCF-7 cells. Biochem Biophys Res Commun 2012; 427:47-53; PMID:22975348; http://dx.doi.org/ 10.1016/j.bbrc.2012.08.144 [DOI] [PubMed] [Google Scholar]
- 34.Xue ZT, Sjogren HO, Salford LG, Widegren B. An epigenetic mechanism for high, synergistic expression of indoleamine 2,3-dioxygenase 1 (IDO1) by combined treatment with zebularine and IFN-gamma: potential therapeutic use in autoimmune diseases. Mol Immunol 2012; 51:101-11; PMID:22424783; http://dx.doi.org/ 10.1016/j.molimm.2012.01.006 [DOI] [PubMed] [Google Scholar]
- 35.Vigneron N, van Baren N, Van den Eynde BJ. Expression profile of the human IDO1 protein, a cancer drug target involved in tumoral immune resistance. Oncoimmunology 2015; 4:e1003012; PMID:26155395; http://dx.doi.org/ 10.1080/2162402X.2014.1003012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung SH, Franceschi S, Lambert PF. Estrogen and ERalpha: culprits in cervical cancer? Trends Endocrinol Metab 2010; 21:504-11; PMID:20456973; http://dx.doi.org/ 10.1016/j.tem.2010.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Srijaipracharoen S, Tangjitgamol S, Tanvanich S, Manusirivithaya S, Khunnarong J, Thavaramara T, Leelahakorn S, Pataradool K. Expression of ER, PR, and Her-2/neu in endometrial cancer: a clinicopathological study. Asian Pac J Cancer Prev 2010; 11:215-20; PMID:20593959 [PubMed] [Google Scholar]
- 38.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al.. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2:401-4; PMID:22588877; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6:pl1; PMID:23550210; http://dx.doi.org/ 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cline MS, Craft B, Swatloski T, Goldman M, Ma S, Haussler D, Zhu J. Exploring TCGA pan-cancer data at the UCSC cancer genomics browser. Sci Rep 2013; 3:2652; PMID:24084870; http://dx.doi.org/ 10.1038/srep02652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res 2002; 12:996-1006.; PMID:12045153; http://dx.doi.org/ 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trump S, Bieg M, Gu Z, Thurmann L, Bauer T, Bauer M, Ishaque N, Röder S, Gu L, Herberth G et al.. Prenatal maternal stress and wheeze in children: novel insights into epigenetic regulation. Sci Rep 2016; 6:28616; PMID:27349968; http://dx.doi.org/ 10.1038/srep28616 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.