

Abstract
Chronic recurrent multifocal osteomyelitis (CRMO) is a rare, pediatric, autoinflammatory disease characterized by bone pain due to sterile osteomyelitis, and is often accompanied by psoriasis or inflammatory bowel disease. There are two syndromic forms of CRMO, Majeed syndrome and DIRA, for which the genetic cause is known. However, for the majority of cases of CRMO, the genetic basis is unknown. Via whole-exome sequencing, we detected a homozygous mutation in the filamin-binding domain of FBLIM1 in an affected child with consanguineous parents. Microarray analysis of bone marrow macrophages from the CRMO murine model (cmo) determined that the Fblim1 ortholog is the most differentially expressed gene, downregulated over 20-fold in the cmo mouse. We sequenced FBLIM1 in 96 CRMO subjects and found a second proband with a novel frameshift mutation in exon 6 and a rare regulatory variant. In SaOS2 cells, overexpressing the regulatory mutation showed the flanking region acts as an enhancer, and the mutation ablates enhancer activity. Our data implicate FBLIM1 in the pathogenesis of sterile bone inflammation and our findings suggest CRMO is a disorder of chronic inflammation and imbalanced bone remodeling.
Introduction
Chronic recurrent multifocal osteomyelitis (CRMO) is a rare, autoinflammatory bone disease. Its primary symptom is bone pain resulting from sterile osteomyelitis. Laboratory tests are frequently normal but can reveal mild elevation of inflammatory markers including white blood cell count, erythrocyte sedimentation rate (ESR), C-reactive protein, and tumor necrosis alpha (TNF-α) [1]. CRMO is treated with non-steroidal anti-inflammatory drugs (NSAIDs), methotrexate, sulfasalazine, bisphosphonates, TNF-α inhibitors, or occasionally by IL-1 inhibition, on an empirical basis [1].
An additional inflammatory disorder is present in approximately 25% of individuals with CRMO. Most often, the associated disease is palmoplantar pustulosis (PPP), psoriasis, or inflammatory bowel disease (IBD); within IBD, Crohn disease is the most common. These associated conditions frequently run in families affected by CRMO, as 50% of first or second degree relatives of probands have psoriasis, IBD, or another chronic immune-mediated disorder. The high prevalence of inflammatory disease in relatives suggests a strong genetic component to CRMO.
There are two rare syndromic forms of CRMO, Majeed Syndrome and Deficiency of IL-1 Receptor Antagonist (DIRA). In general, these are disorders that affect individuals at a very young age. CRMO presents before 2 years in Majeed and in infancy in DIRA [1, 2]. Majeed Syndrome is caused by homozygous mutations in LPIN2 [3] and DIRA is caused by recessive loss-of-function mutations in IL1RN [4]. Patients with both syndromes respond well to IL-1 blocking agents [5]. However, most cases of CRMO are non-syndromic and with unknown genetic cause [1, 2].
There are several mouse models of CRMO with homozygous mutations in Pstpip2. All models have extensive multifocal osteomyelitis and inflammation of the tissues of the ears including the skin [6–8]. PSTPIP2 is a cytoskeletally associated protein involved in membrane formation that reduces inflammation and may play a role in osteoclastogenesis [9]. Recently, it was shown that the cmo disease phenotype is greatly rescued by IL-1R1 deficiency, and that the NLRP3 inflammasome plays a redundant role with caspase-8 in IL-1β mediated osteomyelitis [10, 11]. The IL-1β over-secretion appears to be neutrophil driven and can be counteracted by serine protease inhibitors in vitro [10, 12].
Monocytes from CRMO patients have decreased IL-10 expression in response to LPS stimulation [13]. There are three SNPs in the IL-10 promoter–rs1800896, rs1800871 and rs1800872 –that compose a haplotype, and some haplotypes are significantly more common in patients compared to healthy controls [13, 14]. The haplotypes ATA and ACC are associated with relatively low and medium IL-10 expression, respectively, and the haplotype GCC is associated with high expression [14].
Here, we report on a South Asian child from a consanguineous union with CRMO. Whole exome sequencing of the child and her parents revealed a list of 23 rare, protein-changing variants in 22 genes for which the child is homozygous, having inherited one copy of the mutation from each parent. Of these 22 genes, FBLIM1 stood out because it was the most differentially expressed gene from a microarray experiment examining macrophage expression in the cmo mouse. In addition, studies suggest FBLIM1 is an anti-inflammatory molecule regulated by STAT3, and one involved in bone remodeling via ERK1/2 phosphorylation and the subsequent regulation of RANKL activation. We sequenced FBLIM1 in a larger cohort of 96 patients with CRMO and found one compound heterozygote in FBLIM1, with a novel frameshift insertion in exon 6 of one allele and an enhancer variant in the third intron of the other allele. The enhancer contains binding sites for STAT3 and NR4A2. NR4A2 is a transcription factor that is also involved in regulation of the inflammatory response. Enhancer activity of a 1-kb region around the mutation was validated by luciferase assays in fluoride-treated SaOS2 cells, as was the effect of the mutation on regulatory activity. Because FBLIM1 expression is regulated by STAT3 as a function of IL-10 anti-inflammatory activity, we sequenced the SNPs in the IL-10 promoter haplotype and determined that the child with the enhancer variant is homozygous for the haplotype (ATA/ATA) that is associated with low IL-10 expression, and the child with the R38Q mutation is heterozygous (ATA/ACC). Our results implicate mutations in FBLIM1 in CRMO and autoinflammatory disease. Mutations in FBLIM1 likely contribute to CRMO pathogenesis epistatically with variants involved in the regulation of IL-10 expression.
Methods
Use of humans in research
All human research was approved by the University of Iowa Institutional Review Board (IRB). Written informed consent was obtained from all study participants and written parental or guardian consent was obtained. All participants were under the age of eighteen. Blood and saliva collected for DNA analysis were taken specifically for this study.
Use of mice in research
Mice were used as there is no adequate in vitro or invertebrate animal to model autoinflammatory bone disease. All animal care, and experiments and protocols using mice were conducted in accordance with the University of Iowa Institutional Animal Care and Use Committee (IACUC). Mice are housed in cages in a Specific-Pathogen-free (SPF) barrier facility and are not housed singly. The mice breed and move about the cage without difficulty. The mice have extra bedding to make the floor of the cage soft. To minimize pain, anesthetics are used when needed, and the mice are handled gently. Each procedure was performed in a time-efficient manner and any animals that appeared to be in discomfort or ill were euthanized. Euthanasia was performed by a trained laboratory technician under guidance by a veterinarian, and mice were euthanized by 100% carbon dioxide (C02) administered continuously until at least 1 minute after breathing stops followed by cervical dislocation. Mice were euthanized prior to harvesting tissue for gene expression analysis.
Whole exome sequencing and analysis
DNA from the child and both parents was purified from saliva and prepared for whole exome sequencing. The DNA was enriched using the Agilent SureSelectXT Human All Exon V4 (Agilent Technologies) before sequencing at Otogenetics, Inc (Atlanta, GA). The fastq files were quality-checked using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and the raw reads were aligned to the hg19 (NCBI Build 37) reference genome using the Burrows-Wheeler Alignment (BWA) software [15] and duplicates were removed using Picard Tools. GATK software [16] was used to realign indels, for base quality recalibration and to calculate coverage. Variants were called using GATK’s UnifiedGenotyper and were recalibrated using OMNI, HapMap [17] and 1000 genomes [18] data as training sets. Variants were hard-filtered in GATK based on mapping quality (MQ>40) and depth of coverage (QD>2.0) and variants were annotated with minor allele frequencies (MAFs) from EVS (Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (URL: http://evs.gs.washington.edu/EVS/)), dbSNP [19] and ExAC [20] using GATK’s VariantAnnotator. SNPSift/SNPEff [21] was used to complete annotation from dbSNP and dbNSFP. The variants were uploaded to VarSifter [22] and non-coding variants were removed, along with those not passing hard-filtering, and variants with a MAF>2% in either the global or South Asian population. The resulting list was queried for rare homozygous variants on the autosomal chromosomes where one allele was present in each parent. Variants satisfying these criteria were validated by Sanger sequencing.
FBLIM1 Sanger sequencing
The coding regions of FBLIM1 were sequenced in a cohort of 96 people with CRMO using DNA purified from either saliva or blood. The primers used to amplify each exon are listed in Table 1 below. Crude PCR products were sent to Functional Biosciences (Madison, WI) for Sanger sequencing.
Table 1. Primers for FBLIM1 Sanger sequencing.
| Fblim1 exon | Forward | Reverse |
|---|---|---|
| Exon3 | ggtgctggactgagttgtt | tgtcccggaaattctgacat |
| Exon 4 | gtgcctggcctgagattaag | cctcagagctgaaggaggtg |
| Exon 5 | ctcggtacaatgcggctaat | tgcaggcttctcactgtgtc |
| Exon 6 | gtgctggcattacaggtgtg | cactgggctccttctttctg |
| Exon 6 alternative | aagcaatggtggaggttttg | cccgagtagctgggactaca |
| Exon 7 | tgcaggcctctcctgtactt | cactgcaaactccacctcct |
| Exon 8 | agcccgagttctgacagcta | accatgagccaacagattcc |
| Exon 9 | caactttccctgaaccctca | ggctcagaaggaaagtgtgc |
IL-10 promoter Sanger sequencing
The SNPs rs1800871 and rs1800872 were sequenced via Sanger sequencing using the primers previously described: forward 5’ GACAACACTACTAAGGCTTC and reverse 5’ GCTAACTTAGGCAGTCACCT [23]. For the polymorphism rs1800896, we designed and used primers with the sequences: forward 5’ CATCAAAGGATCCCCAGAGA and reverse 3’ GGCACATGTTTCCACCTCTT.
IL-10 promoter haplotype assessment in 1000 genomes participants
Genotypes for the polymorphisms rs1800896, rs1800871, and rs1800872 were downloaded for all 1000 genomes [24] participants using the 1000 genomes browser. For the 489 South Asian (SAS) 1000 genomes participants, the frequencies of the ATA/ACC and ATA/ATA haplotypes were calculated.
Gene expression microarray of cmo mouse bone marrow derived macrophages
Under aseptic conditions, the whole femur was collected from 4-month-old mice and stored in 1–2 ml α-MEM (serum free) ice-cold media. The whole marrow was flushed into 200μl α-MEM media in a microcentrifuge at 10000 rpm for 30s. The supernatant was removed. Marrow cell pellets were washed with 1ml α-MEM media and then centrifuged at 10000rpm for 2 minutes. The cell pellets were resuspended in 1ml media (α-MEM + 5% CMG) and cell suspensions were then transferred onto Petri dishes with a total volume of 20 ml media (α-MEM + 10% FBC + 5% CMG) for incubation at 37°C in 5% CO2 for 2 days. After a two-day incubation, the cells were harvested to undergo RNA extraction. The purification of RNA from the bone marrow macrophage cells was performed using Qiagen’s RNA purification kit (Cat#74104) according to the manufacturer’s instruction. RNA samples were sent to the University of Iowa DNA Core Facility for microarray analysis. Cells were cultured from 3 mice from each of the cmo, cmo IL1R+/- and cmo IL1R-/- strains.
RNA sample preparation and subsequent hybridization to the Illumina beadchips were performed at the University of Iowa DNA Facility using the manufacturer’s recommended protocol. Briefly, 100 nanograms total RNA was converted to amplified Biotin-aRNA using the Epicentre TargetAmp-Nano Labeling Kit for Illumina Expression BeadChip (Illumina, Inc., San Diego, CA, Cat. #TAN07924) according to the manufacturer’s recommended protocol. The amplified Biotin-aRNA product was purified through a QIAGEN RNeasy MinElute Cleanup column (QIAGEN Cat #74204) according to modifications from Epicentre. 750ng of this product were mixed with Illumina hybridization buffer, placed onto Illumina-Mouse WG-6 v2.0 BeadChips (Part No. BD-201-0202), and incubated at 58°C for 17h, with rocking, in an Illumina Hybridization Oven. Following hybridization, the arrays were washed, blocked, and then stained with streptavidin-Cy3 (Amersham/GE Healthcare, Piscataway, NJ) according to the Illumina Whole-Genome Gene Expression Direct Hybridization Assay protocol. Beadchips were scanned with the Illumina iScan System (ID #N0534) and data were collected using the GenomeStudio software v2011.1.
Cell culture and luciferase assays
SaOS2 cells were ordered from ATCC (Manassas, VA). Cells were maintained in McCoys 5a medium + 15% FBS and 1% P/S. SaOS2 cells were stimulated to osteoblast differentiation by the addition of NaFl at a concentration of 0.2 mM to the media immediately after transfection. The Fblim1 enhancer region was amplified by PCR and cloned into competent cells using the Gateway pENTR Directional TOPO Cloning kit from Invitrogen / Thermo Fisher Scientific (Waltham, MA). The sequences for the primers used to amplify the region are F: CTGTACCCCACTCTGTCCCT and R:GATCTGCTGAATCAGAGA. The enhancer was then recombined into the FFL Luciferase vector and amplified in Dh5α cells. The enhancer mutation rs41310367 was induced in the plasmid by site-directed mutagenesis at GenScript Inc. (Piscataway, NJ).
The Renilla and Firefly luciferase- containing plasmids were transfected into SaOS2 cells using Lipofectamine 3000 from Invitrogen / Thermo Fisher Scientific. Transfected cells were incubated for 72 hours prior to lysis. Luciferase assays were performed using the Promega (Madison, WI) Dual-Luciferase assay system. Within each experiment and treatment, there were four biological replicates. The experiment was performed three times and data from multiple experiments were combined prior to statistical analysis. Firefly to Renilla ratios were calculated for each sample and analyzed for differences between absence and presence of enhancer, and between wild-type and mutant enhancer. A two-tailed t-test was performed for each comparison.
Multiple sequence alignment
The peptide sequence for human FBLP1 was obtained from Ensembl (www.ensembl.org), March 2016) and input into NCBI protein BLAST and queried against the non-redundant protein sequence database. Fasta sequences for Mus musculus, Rattus norvegicus, Pan paniscus, Bos taurus, Equus caballus, Canis lupus familiaris and Felis catus were downloaded and input into Clustal Omega [25] for multiple sequence alignment. Default settings were used to create the alignment.
Analysis of transcription factor binding recognition sites
To predict the effect of the regulatory mutation rs41310367, we queried the sequence flanking the SNP with and without the variant for transcription factor binding site recognition motifs using the JASPAR database[26]. For the input sequences, we used the reference sequence AAGACCACGTCAC, the alternative reference sequence based on conservation AAGACCAGGTCAC, and the variant sequence AAGACCATGTCAC. Only recognition scores greater than 86 were considered for the initial analysis and then a threshold of 70 was set to determine the score after a sequence change that caused the score to be below 86.
Structural modeling and analysis of FBLIM1 (FBLP1)
Protein structural disorder predictions were performed using DISOPRED [27]. Secondary structure predictions were performed using PSI-PRED [28]. The tertiary structure of the FBLIM1 Filamin-1 binding domain (residues 1–70) was modeled using Phyre2 [29]. We next modeled the Filamin-1-FBLP1 complex by superimposing our Phyre2 model to NMR structure of Filamin-1 bound to a FBLIM1 peptide (PDB: 2K9U). Charges and hydrogen atoms were added to the wild-type and mutant FBLP1 models using PDB2PQR [30]. Electrostatic potentials were calculated using APBS [31]. Protein and solvent dielectric constants were set to 2.0 and 78.0, respectively. PyMOL generated all structural figures.
Results
Case report
An eight-year old South Asian girl was admitted to The Hospital for Sick Children (Toronto, ON) with left knee pain and an inability to bear weight. Plain x-rays were consistent with osteomyelitis of the left proximal tibia (Fig 1A). Her CBC was normal, ESR was elevated to 42 mm/hr (normal < 20), while her CRP was normal (4.2 mg/L). Cefazolin was given as therapy. An MRI of the left lower extremity showed abnormalities involving the left proximal tibial metaphysis and epiphysis and similar abnormalities in the left distal femoral metaphysis and the distal tibial metaphysis, all consistent with chronic recurrent multifocal osteomyelitis (Fig 1B). A whole body MRI showed additional lesions in the second and third right metatarsals, right cuneiform, right acetabulum, distal left humeral metaphysis and epiphysis and the left clavicle in a distribution typical of CRMO (not shown). She also had psoriasis on her right thumb and heels. Her family history was unremarkable other than that her parents were first cousins. In view of the multiple bone lesions involving metaphyses and psoriasis, a diagnosis of CRMO was made. Antibiotic therapy was discontinued. She has subsequently been treated with NSAIDs followed by two cycles of pamidronate.
Fig 1. X-ray and MRI knee images from the affected proband.

(A) X-ray showing osteomyelitis and a lesion characteristic of CRMO (red arrow) in the left proximal tibia. (B). MRI of the same knee showing inflammation and bone destruction (red arrow). Similar lesions were found in the clavicle, hip, femur, tibia, foot and toes (not shown). (C) MRI of a healthy knee for comparison.
Affected child has a homozygous coding mutation in FBLIM1
Whole exome sequencing followed by Sanger sequencing validation revealed that the child harbored homozygous coding variants with a minor allele frequency in global or South Asian populations of less than 2% in 22 genes. The Agilent v4 platform was covered by an average number of 24.9 reads. The affected genes are listed in Table 2. Of these 22 genes, FBLIM1 was the most promising candidate based on its known function and the results of the mouse microarray experiment (see Fig 2). Her FBLIM1 variant consists of a G to A mutation that induces an arginine to glutamine amino acid change at position 38 (R38Q) in all known transcripts. The mutation is located in the filamin-binding domain and the affected residue is largely conserved in mammals (Fig 3). According to ExAC [20] (which does contain individuals with inflammatory diseases), this variant has a global MAF of 0.004193 and in the South Asian population, an MAF of 0.01417. Fig 3 shows the protein location of the FBLIM1 mutation as well as a multiple sequence alignment for the surrounding region.
Table 2. Rare homozygous coding variants in the affected child.
| Chr | bp (hg19) | Gene name | dbID | REFa | VARb | Amino Acid Changec | ExACd Global | ExAC SAsian | 1KG e Global | 1KG SAS | Child | Father | Mother |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 16069525 | TMEM182 | rs41268336 | C | T | Arg58Trp | 0.00963 | 0.01592 | 0.006 | 0.008 | TT | CT | CT |
| 1 | 16091591 | FBLIM1 | rs146575757 | G | A | Arg38Gln | 0.00419 | 0.01417 | 0.004 | 0.009 | AA | AG | AG |
| 1 | 26524551 | CATSPER4 | rs560352853 | A | G | Ile221Val | 0.00202 | 0.01429 | 0.003 | 0.016 | GG | AG | AG |
| 1 | 230824171 | COG2 | rs201551743 | C | G | Leu553Val | 9.06E-05 | 0.00061 | 0 | 0.001 | GG | CG | CG |
| 3 | 196742243 | MFI2 | rs45625439 | C | T | Arg409Gln | 0.02026 | 0.00867 | 0.005 | 0.007 | TT | CT | CT |
| 6 | 118953690 | CEP85L | rs560509236 | T | C | His56Arg | 0.00115 | 0.00806 | 0.002 | 0.008 | CC | CT | CT |
| 7 | 48317939 | ABCA13 | rs200243325 | T | G | Ile2383Arg | 0.00197 | 0.00997 | 0.001 | 0.005 | GG | GT | GT |
| 8 | 10469719 | RP1L1 | - | G | A | Thr630Ile | - | - | - | - | AA | AG | AG |
| 8 | 10480645 | RP1L1 | rs188979626 | G | A | Arg23Cys | 1.79E-05 | 0 | 0 | - | AA | AG | AG |
| 12 | 12966323 | DDX47 | rs577622688 | G | T | Asp8Tyr | 0.00158 | 0.01089 | 0.002 | 0.009 | TT | GT | GT |
| 12 | 54118405 | CALCOCO1 | rs373349679 | C | A | G95*| | 0.00322 | 0.01715 | 0.002 | 0.008 | AA | AC | AC |
| 14 | 69259681 | ZFP36L1 | rs139683444 | C | T | Arg61His | 0.00018 | 0.00097 | 0 | - | TT | CT | CT |
| 15 | 90610517 | ZNF710 | rs144139243 | G | C | Glu50Gln | 0.00239 | 0.01305 | 0.001 | 0.005 | CC | CG | CG |
| 16 | 72822645 | ZFHX3 | rs149133285 | G | A | Ser3177Leu | 0.00699 | 0.01757 | 0.005 | 0.016 | AA | AG | AG |
| 19 | 13941205 | ZSWIM4 | rs372292857 | G | A | Ala771Thr | 0.00033 | 0.00237 | 0 | 0.002 | AA | AG | AG |
| 19 | 17638144 | FAM129C | rs200304763 | A | C | Ser30Arg | 0.00532 | 0.01283 | 0.001 | 0.006 | CC | AC | AC |
| 19 | 19648290 | YJEFN3 | rs547825267 | G | A | Arg286His | 0.00105 | 0.00565 | 0.001 | 0.003 | AA | AG | AG |
| 19 | 23927685 | ZNF681 | - | C | G | Glu223Gln | - | - | - | - | GG | CG | CG |
| 20 | 57599401 | TUBB1 | rs62639974 | C | T | Arg307Cys | 0.00443 | 0.00515 | 0.011 | 0.002 | TT | CT | CT |
| 20 | 55027781 | CASS4 | - | C | T | Arg517Trp | - | - | - | - | TT | CT | CT |
| 20 | 61288068 | SLCO4A1 | - | T | C | Trp88Arg | - | - | - | - | CC | CT | CT |
| 22 | 43213187 | ARFGAP3 | rs144427016 | T | C | Asn342Ser | 0.00121 | 0.00798 | 0.001 | 0.006 | CC | CT | CT |
| 22 | 44515711 | PARVB | rs742550 | G | A | Arg140His | 0.01606 | 0.01331 | 0.008 | 0.007 | AA | AG | AG |
Fig 2. Microarray analysis of bone marrow macrophages from the cmo mouse model.

(A) Gene expression heatmap from microarray analysis of RNA from bone marrow-derived macrophages of cmo, IL-1R-/- and IL-1R+/- mice (n = 3 for each strain). Normalized expression values were analyzed for differential expression using Partek Genomics Suite software (version 6.12, Partek Inc., St. Louis, MO, USA.). The top 25 differentially expressed genes are shown. Fblim1 (red arrow) was the most differentially expressed (downregulated) gene in the cmo mouse. (B, C and D) Representative fixed decalcified tail bone sectioned and stained with H&E from cmo, IL-1R-/- and IL-1R+/- mice, respectively. The cmo mouse has extensive mixed inflammatory infiltrate with destruction of the vertebral body, the cmo.IL-1R+/- has similar but less severe inflammation and bone destruction, whereas, the cmo.IL-1R-/- mouse has normal bone with no inflammation. Representative tail kinks and foot deformities in these mice were depicted and described previously [10].
Fig 3. Coding mutations found in individuals with CRMO.

(A) Chromatogram showing the homozygous mutation in exon 3 of FBLIM1 in a child from a consanguineous union. The mutation is rs146575757, a G to A nucleotide change, causing an Arg38Gln change in the protein. (B) Chromatogram showing a novel 1-bp frameshift insertion in exon 6 of FBLIM1 in a second proband with CRMO. The mutation is one allele of a compound heterozygote and the frameshift occurs at Glu255. (C) Image of conserved domains in FBLP1 from the Protein Data Bank (PDB) (www.rcsb.org) [32]. The red arrow points to the location of the Arg38Gln position, in the filamin-binding domain. (D) Protein alignment of FBLP1 and its orthologues in other mammals. The red arrow points to the amino acid disrupted by rs146575757.
FBLIM1 is the most differentially expressed gene in BMM from cmo mice
We examined FBLIM1 expression in the Pstpip2-null cmo mouse model. These mice are protected from disease on an IL-1R-deficient genetic background, and disease is delayed in a heterozygous background (appearing at 21 weeks vs. 8 weeks in the cmo mouse) [10]. To assay gene expression in these mice, bone marrow derived macrophages from cmo, cmo IL-1R+/- and cmo IL-1R-/- four-month olds were harvested and cultured for microarray analysis. Notably, Fblim1 was the most differentially expressed gene in the cmo mouse, and was downregulated 26.3-fold (p = 7.74 x 10−9) compared to cmo IL-1R+/-, and downregulated 21.4-fold (p = 1.14x10-8) compared to cmo IL-1R-/- (Fig 2A). The top 25 differentially expressed genes are listed in Table 3.
Table 3. Top 25 differentially expressed genes in the mouse cmo microarray experiment.
| CMO vs. IL1R+/- | CMO vs. IL1R-/- | |||||
|---|---|---|---|---|---|---|
| Gene | p-value | Fold-Change | Effect in CMO | p-value | Fold-Change | Effect in CMO |
| FBLIM1 | 7.74E-09 | -26.3414 | down | 1.14E-08 | -21.4121 | down |
| FCER1G | 3.76E-08 | -768.539 | down | 3.47E-08 | -840.44 | down |
| TMEM87A | 1.25E-07 | 33.9359 | up | 1.08E-07 | 37.0694 | up |
| CASP9 | 5.06E-07 | -10.0818 | down | 5.33E-07 | -9.88089 | down |
| ARL2BP | 5.93E-07 | -6.58926 | down | 6.11E-07 | -6.52675 | down |
| GUS-S | 7.42E-07 | -14.3302 | down | 1.06E-06 | -12.2753 | down |
| LOC666403 | 8.88E-07 | 40.3891 | up | 1.31E-06 | 31.8832 | up |
| TTC27 | 1.06E-06 | -15.8034 | down | 9.23E-07 | -16.8946 | down |
| BAT5 | 1.32E-06 | -21.3405 | down | 1.04E-06 | -24.2149 | down |
| MSH5 | 1.24E-06 | 4.01022 | up | 2.74E-06 | 3.36972 | up |
| 4930427A07RIK | 1.68E-06 | -3.11473 | down | 4.04E-06 | -2.6625 | down |
| WDR46 | 2.66E-06 | -11.3828 | down | 2.52E-06 | -11.6345 | down |
| RBM45 | 2.52E-06 | 30.9757 | up | 2.86E-06 | 28.8087 | up |
| CAP1 | 2.78E-06 | 19.3367 | up | 3.35E-06 | 17.6384 | up |
| PRCP | 3.23E-06 | -10.3049 | down | 5.24E-06 | -8.56969 | down |
| 9030625A04RIK | 3.65E-06 | -11.273 | down | 1.08E-05 | -7.49047 | down |
| TRIM30 | 4.63E-06 | -22.0663 | down | 5.75E-06 | -19.7381 | down |
| 2810021O14RIK | 4.91E-06 | -5.91873 | down | 6.63E-06 | -5.41564 | down |
| PSMB9 | 5.80E-06 | -10.1156 | down | 6.32E-06 | -9.78298 | down |
| CLCN7 | 5.90E-06 | -4.40784 | down | 7.57E-06 | -4.14429 | down |
| TM7SF3 | 7.55E-06 | -8.13616 | down | 4.36E-06 | -9.99593 | down |
| CTSL | 7.24E-06 | 5.25264 | up | 1.05E-05 | 4.74202 | up |
| MANBAL | 9.29E-06 | -4.12521 | down | 1.69E-05 | -3.59344 | down |
| SLC39A9 | 9.30E-06 | -2.46427 | down | 0.000132813 | -1.7638 | down |
FBLP1 structural modeling
Previous structural studies on FBLP1 using nuclear magnetic resonance (NMR) spectroscopy have determined its structure in complex with Filamin-1 [33]. However, these structures do not cover the region that is mutated in our patient. Therefore, we attempted to model the full Filamin-1 binding region of FBLIM1 using an ab initio approach in the Phyre2 program [29]. First, we submitted the primary structure to the DISOPRED and PSI-PRED servers [27, 28]. These programs predicted the Filamin-1 binding region to be highly disordered and mostly coiled (Fig 4A and 4B). Similarly, the resulting model from Phyre2 was predicted to be 86% disordered (Fig 4C). The ordered residues (positions 1–24) corresponded directly with the previously-determined NMR structure of FBLP1 (PDB: 2K9U). The model was in agreement with our primary structure analysis that predicted the Filamin-1 binding domain to be an intrinsically-disordered region. Superimposition of our model onto the previously-determined Filamin-1 complex revealed the R38Q mutation to be downstream of the Filamin-1 interaction site (Fig 4D).
Fig 4. Structural modeling of patient mutation in FBLIM1.

(A) Disorder prediction of the Filamin-1 binding region of FBLP1 (residues 1–70) was performed with DISOPRED version 3. Disorder probability scores greater than 0.5 are considered to be disordered. The R38Q mutation falls in a predicted disordered region. (B) Secondary structure prediction was performed using PSI-PRED. The confidence scores for coils, helices, and strands are shown. (C) The output model of the Filamin-1 binding region of FBLIM1 generated in Phyre2. A total of 86% of the residues are predicted to be disordered, resulting in 76% of the residues having a low confidence score (<90%). Residues 1–24 are predicted to be ordered. (D) Superimposition of the Phyre2 model onto the NMR structure of the Filamin-1-FBLIM1 complex. Modeling of the complex places the R38Q mutation downstream of the interaction site. (E) Electrostatic potential calculations in APBS reveal a loss of positive charge in FBLP1 as a result of the CRMO mutation.
Intrinsically-disordered regions (IDRs) in proteins are known to be important for interactions with other proteins and binding partners [34]. Since the Filamin-1-binding domain was predicted to be intrinsically-disordered, we sought to determine other potential deleterious effects of the R38Q mutation on FBLP1 function other than conformational changes. The substitution from an arginine to a glutamine removes a positive charge downstream of the Filamin-1 interaction site. This resulting change in electrostatic potential was modeled using APBS (Fig 4E) [31]. Taken together, our structural modeling results place the patient mutation downstream of the Filamin-1 interaction site on an intrinsically-disordered region and predict a loss of positive charge, which may alter FBLP1’s interaction with Filamin-1. Since the Filamin-1-binding region of FBLP1 is predicted to be an IDR, the results of our structural modeling are limited and require further biophysical studies to determine the effects of CRMO mutations on FBLP1 structure and function.
FBLIM1 sequencing in our CRMO cohort
FBLIM1 was sequenced in a cohort of 96 unrelated individuals with CRMO. All rare (global MAF < 2%), coding variants detected are summarized in Table 4. Among this group was a second child, also of South Asian ancestry, who harbored two FBLIM1 mutations: a novel 1-bp frameshift insertion in exon 6, (the fourth coding exon, Fig 2B) and a variant (rs41310367) in an intron, 32 nucleotides 3’ of the end of the first coding exon (Fig 5A). Sequencing the parents verified that the child inherited one mutation from each parent. According to data from the ENCODE project, the intronic variant (rs41310367) is centrally located in the middle of an enhancer, in a STAT3 binding region and in an NR4A2 recognition site (Fig 5B and 5C). The enhancer was reported to be active in several cell lines, including osteoblasts, since it was covered by H3K27ac and H3K4me1 peaks, the two marks of an active enhancer [35] (Fig 5C). Chromatograms for the FBLIM1 mutations found in the family with the R38Q mutations and the family with the frameshift and rs41310367 mutations are provided in the S1 Chromatograms.
Table 4. Rare (<2% GMAFa) mutationsb found in FBLIM1 in a cohort of 96 subjects with CRMO.
| Subject (total number) | exon3c | rs41310367 | exon4 | exon5 | exon6 | exon7 | exon8 | Reported Ancestry |
|---|---|---|---|---|---|---|---|---|
| 1–72d (72) | xe | X | x | x | x | x | x | European |
| 73–76 (4) | X | yesf | x | x | x | x | x | European |
| 77 (1) | X | X | x | x | x | n/a g | x | European |
| 78 (1) | X | X | x | n/a | x | x | x | European |
| 79–88 (10) | X | X | x | x | x | rs114077715 | x | European |
| 89 (1) | X | X | x | x | x | x | x | Latin American |
| 90 (1) | X | Yes | x | x | Glu225fs | x | x | South Asian |
| 91–94 (4) | X | X | x | x | x | x | x | East Asian |
| 95–96 (2) | X | X | x | x | x | x | x | Ashkenazi Jewish |
aGMAF = global minor allele frequency.
bAll mutations in the table listed are non-synonymous except for rs41310367.
cExons 1 and 3 are 5’UTR, so they were not sequenced.
dSubjects pooled together are identical in FBLIM1 genotype and reported ancestry.
ex = successful Sanger sequencing, no rare coding variants.
fyes = presence of mutation.
gn/a = Sanger sequencing for the exon in that individual failed.
Fig 5. FBLIM1 enhancer variant in 2nd proband.

(A) Chromatogram from the second proband. Red arrow indicates rs41310367 (C→T) in the third intron of FBLIM1. (B) From JASPAR [26], the human NR4A2 consensus transcription factor binding site (TFBS) motif. The red arrow shows where rs41310367 is located within the binding site and that the TFBS is disrupted. (C) Screen capture from UCSC genome browser (hg19) showing the regulatory region that includes rs41310367 (red arrow) and a STAT3 binding region. H3K27ac and H3K4me1 peaks from ENCODE are shown, including in osteoblasts. The red box indicates the region that was amplified and cloned into the Firefly reporter vector.
While we did not find more individuals with homozygous or compound heterozygous mutations in FBLIM1 in the cohort of 96 patients, there was significant enrichment for rs114077715, a non-synonymous SNP in exon 7 (Table 4). The Gly311Arg polymorphism has a global and European MAF of 0.01977 and 0.02638, respectively (ExAC). Ten of the 88 European-American individuals with CRMO carried the minor allele (MAF = 0.057), indicating two-fold enrichment compared to the European MAF (p = 0.0120). rs114077715 was not detected as part of a compound heterozygote–however, its enrichment suggests that there could be other non-coding regulatory variants like rs41310367 in FBLIM1 contributing to the pathogenesis of CRMO.
rs41310367 disrupts an NR4A2 recognition site
The sequence surrounding variant rs#41310367, located in the third intron of FBLIM1 was analyzed using the JASPAR transcription factor (TF) database (JASPAR CORE Vertebrate data set) [26]. Only four candidate TFs scored higher than the 86% score threshold, including NR4A2, a C4 zinc finger class nuclear receptor. Analysis of this sequence window featuring the rs#41310367 (T) variant resulted in reduced scores for three of these TFs except for MEIS1, which was substantially increased. This same sequence window features a G nucleotide in many other species based on conservation of the regulatory region surrounding and including the base impacted by rs#41310367, as viewed in UCSC Genome Browser’s Vertebrate Multiz Alignment and Conservation track [36]. Analysis of this sequence showed that only NR4A2 was predicted to retain a high quality binding site. The scores for each of the five TFs are listed in Table 5.
Table 5. NR4A2 is a candidate transcription factor affected by the variant rs#.
| Factor | AAGACCACGTCAC | AAGACCATGTCAC | AAGACCAGGTCAC |
|---|---|---|---|
| NR4A2 | 0.876 | 0.762 | 0.820 (0.866) |
| ATF1 | 0.910 | 0.779 | 0.759 |
| ZNF354C | 0.887 | 0.712 | 0.755 |
| MEIS1 | 0.906 | 0.970 | 0.757 (0.702) |
| RORA | 0.743 | 0.743 | 0.865 |
The five transcription factors (TFs) that recognize the region surrounding rs#41310367 are in the first column. The second column lists the JASPAR [26] scores for the reference sequence in humans. The third column lists the scores for the sequence with the minor T allele, and the last column lists the scores with the G variant at the same position, which is the consensus sequence in many vertebrates.
IL-10 promoter haplotype investigation
The IL-10 promoter haplotype associated with differential IL-10 expression contains the polymorphisms rs1800896 (G/A), rs1800871 (C/T) and rs1800872 (C/A). We sequenced the SNPs in the two families with the FBLIM1 mutations. Phasing of each variant was validated by sequencing the SNPs in the parents. The child with the R38Q mutation is heterozygous for the low and medium expressing haplotypes ATA/ACC and the child with rs114077715 is homozygous for the lowest-expressing ATA haplotype. We investigated the frequency of the ATA/ACC and ATA/ATA haplotypes in the South Asian 1000 genomes participants and determined that the two genotypes occur at a frequency of 24.5% and 22.1% respectively. The genotypes in both children are supportive of additive effects of the IL-10 regulating promoter haplotype and the FBLIM1 mutations. The chromatograms for the IL-10 promoter haplotype sequencing are provided in the S2 Chromatograms.
Functional validation of rs41301367
The 1000 genomes global and South Asian minor allele frequencies for rs41310367 are 0.014 and 0.040, respectively, so the variant is relatively common. However, since the second proband also harbored a novel frameshift on the other FBLIM1 allele, we functionally characterized the intronic variant in a firefly luciferase reporter construct. The 1138 base pairs flanking the SNP was cloned upstream of the cfos promoter, in an enhancer position (Fig 6A). Transfected cells were assayed in the presence of added fluoride, since supplemental fluoride can differentiate SaOS2 cells into osteoblasts and increase expression of osteoblast marker genes [37]. In its normal form, the 1138 bp sequence showed robust enhancer activity in the NaFl-treated SaOS2 cells, and introducing the mutation ablated this activity (Fig 6B). The raw luciferase readings are provided in S1 Table.
Fig 6. Functional validation of rs41310367 by luciferase assay.

(A) Diagram of the firefly luciferase reporter driven by the cfos promoter plus the putative FBLIM1 enhancer region (hg19:chr1:16,091,437–16,092,574). attB1 and attB2 are the recombination sites for the Gateway recombination system. The diagram shows recombination inserted the regulatory region into the construct backwards. The red arrow shows the position of rs41310367. (B) Firefly to Renilla luciferase ratios in transfected SaOS2 cells (72-hours after transfection). The number of replicates for each experiment is indicated in parentheses. The presence of the enhancer (red) increased luciferase activity compared to the empty vector (blue), and the mutation (gray) significantly reduced enhancer activity, both with and without fluoride treatment; however, the effects were greater and more significant in NaFl-treated SaOS2 cells. In all experiments, luciferase activity was normalized to the co-transfected Renilla luciferase. Data from individual experiments were combined prior to statistical analysis and differences in relative luciferase activity between experimental scenarios were determined using a two-tailed t-test.
Discussion
CRMO is a rare pediatric autoinflammatory bone disease and family pedigrees suggest a strong genetic component. Via whole-exome sequencing, a child from a consanguineous union affected by CRMO was found to harbor a rare, homozygous coding mutation in FBLIM1, and another child with CRMO was found to be a compound heterozygote for mutations in FBLIM1, with one frameshift mutation and one expression-altering enhancer mutation.
FBLIM1 codes for Filamin-binding LIM protein 1 (FBLP1) or migfilin, a filamin-binding protein involved in the regulation of bone remodeling [38]. The protein has an N-terminal filamin-binding domain that binds to Filamin A/B/C, a central proline-rich domain that interacts with vasodilator-stimulated phosphoprotein (VASP), and a C-terminal LIM domain that interacts with the transcription factor CSX/NKX2-5 or mitogen-inducible gene 2 (MIG2), depending on the splice variant [39].
FBLP1 is a key regulator of the cytoskeleton, where it anchors cell-extracellular matrix adhesion proteins and filamin-containing actin filaments [39, 40]. FBLP1 binds filamin at cell-cell and cell-ECM contacts [41]. There, it competes with integrin β for filamin binding to promote integrin activation [33, 42]. This mechanism was recently demonstrated in neutrophils and vascular and endothelial cells [42]. In relation to the development of CRMO, this is very interesting in light of the recent work demonstrating macrophage-1 (mac-1) activation in neutrophils during sterile inflammation [43]. Mac-1 is composed of both integrin α and β, the latter of which interacts directly with filamin [44]. Notably, the mutation found in the child from the consanguineous family is in the filamin-binding domain of FBLP1 and so may disrupt FBLP1-FLN binding resulting in aberrant integrin activation in neutrophils. In the cmo mouse, the phenotype is driven by neutrophils that hyperexcrete IL-1β [10, 11].
There is a structural model of the FBLP1-Filamin1 complex based on NMR spectroscopy [33]. Unfortunately, because the R38Q mutation is outside the region included in the model, our in silico predictive capacity of the mutation’s effect on Filamin binding is limited. However, the R38Q mutation induces a loss of positive charge in an intrinsically disordered region; such regions are known to be involved in interacting with and/or binding to other proteins [34]. Future experiments include those that test the hypothesis that the R38Q disrupts filamin binding and subsequent integrin β activation, thereby leading to sterile inflammation.
Mice null for FBLIM1 have severe osteopenia and increased osteoclast differentiation marked by increased RANKL expression in bone marrow stromal cells [38]. RANK/RANKL/OPG signaling was identified in the late 1990s, and in the last fifteen years, much has been discovered regarding its involvement in bone remodeling and its dysregulation in immune-mediated diseases [45–47]. RANKL/RANK binding triggers osteoclast differentiation and activation and too much of either results in excess bone resorption leading to osteoporosis and other bone disorders [46, 47]. In rheumatoid arthritis, RANKL overexpression in the synovial fluid is induced by Th17 signaling [48]. In Fblim1-null mice, increased RANKL is mediated by ERK1/2 phosphorylation and blocking ERK1/2 phosphorylation in the mice restored lower RANKL levels [38]. ERK phosphorylation is crucial for priming of the inflammasome by lipopolysaccharide (LPS) [49], suggesting that FBLP1 may act as an inhibitor of inflammasome activation.
As part of the anti-inflammatory response, FBLIM1 is regulated by STAT3 upon macrophage stimulation by IL-10 [50]. A 2012 study identified the genomic regions bound by STAT3 and the nearby (within 20 kb) genes up- or down-regulated in macrophages treated with IL-10, an anti-inflammatory cytokine [50]. FBLIM1 was one of the most significantly upregulated genes providing additional evidence that FBLP1 is a molecule with anti-inflammatory activity.
Given its documented role as an anti-inflammatory molecule involved in bone remodeling, FBLIM1 was the most robust candidate gene found in the consanguineous family. Further support for FBLIM1 came from a microarray experiment with the cmo mouse model of CRMO. Differential expression analysis among bone-marrow derived macrophages from the cmo mouse, a cmo IL-1R-/- knockout and a cmo IL-1R -/+ mouse found Fblim1 to be the most significantly differentially expressed gene, downregulated over 20-fold. Sequencing the coding portion of FBLIM1 in a larger cohort of CRMO subjects found a second proband with a novel frameshift (and likely loss-of-function) insertion in the sixth exon of FBLIM1, along with a variant in a putative enhancer. While we did not find more than two individuals harboring homozygous or compound heterozygous FBLIM1 mutations, we did find two-fold enrichment for rs114077715, a coding mutation in the 7th exon. This significant association coupled with our functional validation of the regulatory mutation rs41310367 suggests that there may be other non-coding mutations in linkage disequilibrium with rs114077715 contributing to CRMO disease.
FBLIM1 is crucial for bone remodeling, which requires a balance between osteoclast and osteoblast activity. Our experimental results suggest rs41310367 disrupts this balance. More specifically, the region flanking the mutation acts as an enhancer in osteoblasts and our experiment shows that its regulatory activity is markedly ablated when the mutation is present. This implies that the mutation would enhance osteoclastogenesis and diminish osteoblastogenesis, resulting in excessive bone resorption. Our interpretation is supported by findings in the Fblim1-null mouse, which has decreased osteoblast and increased osteoclast differentiation [38].
The STAT3 transcription factor participates in the anti-inflammatory response mediated by IL-10 in macrophages, and in MCF10A cells, ENCODE ChIP-Seq experiments identified a STAT3 binding region at rs41310367 [51–53]. Such binding would likely enhance FBLIM1 expression in osteoblasts. Although the transcription factor binding profile database JASPAR [26] indicates the mutation is not in the sequence recognized by STAT3, it could nevertheless disrupt binding and regulation by other transcription factors that cooperate with STAT3. That other factor may be NR4A2, since the mutation rs41310367 disrupts an NR4A2 binding site.
We tested in silico the effect of the regulatory variant on transcription factor (TF) binding recognition as predicted using JASPAR [26]. Recognition scores were determined using the two reference alleles for rs41310367 (C or G) among vertebrates as well as the variant allele (T). Only NR4A2 recognized the sequences featuring both the C or G nucleotides, suggesting that in the most conserved model, NR4A2 is the TF that is most likely impacted by the variant. Notably, the MEIS1 recognition score increased with the T allele. A second non-exclusive interpretation is that the variant nucleotide creates a MEIS1 binding site that competes with NR4A2 binding. However, given that NR4A2 is directly involved in the differentiation of osteoblasts [54, 55], we believe that it is a more likely candidate for involvement of the regulation of FBLIM1 expression in the fluoride-treated SaOS2 cells.
NR4A2 is an orphan nuclear receptor and transcription factor well characterized in its involvement in dopaminergic neuron signaling [56]. NR4A2 is also implicated in the inflammatory response in relation to the development of arthritis, atherosclerosis and psoriasis [56]. Its expression is significantly higher in the synovial fluid of patients with rheumatoid arthritis and in psoriatic skin [56, 57] and this overexpression is abrogated by treatment with dexamethasone, methotrexate, or by TNF-α inhibition [56, 57]. In synovial tissue, NR4A2 activates the expression of the inflammatory chemokine IL-8, [57] and NR4A2 expression and correlated TNF-α levels are modulated by thrombospondin-1 (TSP-1), [58] an ECM glycoprotein that binds to integrins and integrin-associated proteins in its role as a regulator of cell-cell and cell-ECM interaction [59]. In human chondrocytes, histamine (which induces cartilage destruction) increases RANKL activity via regulation by NR4A2 [60]. Recently, NR4A2 was identified as an NLRP3 inflammasome activation-responsive “hub” gene in human monocytes, as a transcription factor that activates the expression of anti-inflammatory genes [61]. Based on NR4A2’s documented involvement in the inflammatory response in the skin, joints, and cartilage, rs41310367 may disrupt FBLIM1’s regulation by NR4A2 as part of the inflammatory or anti-inflammatory response, depending on the cell type.
It is notable that aside from the novel frameshift mutation detected in the second family, the other mutations described in this report are not extremely rare. The homozygous mutation rs146575757 found in the child with consanguineous parents has a South Asian minor allele frequency of 0.01417 (ExAC) and there are two homozygotes in the ExAC database. The regulatory variant rs41310367 is even more common, with a minor allele frequency of 0.04 in the South Asian individuals in 1000 genomes, and there is one homozygote for this variant in the database. Of the 97 CRMO subjects for which FBLIM1 was sequenced (including the child from the first family), only two harbored rare, recessive mutations, and they were the only two individuals with South Asian ancestry. It is possible that there are additional variants contributing to disease pathogenesis; in the two South Asian individuals, those variants could be common, and possibly ancestry-specific.
Given that FBLIM1 expression is regulated by STAT3 in the context of IL-10 signaling and the IL-10 promoter haplotypes are associated with variable IL-10 expression [13, 14], we sequenced the haplotypes in the two families described in this report. Both children have haplotypes associated with relatively low IL-10 expression; the child with the R38Q mutation is heterozygous for the ATA and ACC haplotypes and the child with the novel frameshift mutation in trans with the regulatory variant rs41310367 is homozygous for the lowest IL-10 expressing ATA haplotype. The ATA/ACC and ATA/ATA genotypes occur at a frequency of 24.5% and 22.1% respectively in the 1000 genomes South Asian (SAS) participants, so these probabilities multiplied by the probabilities of the FBLIM1 mutations found in the two families result in combined genotype frequencies that are more consistent with the incidence of CRMO. The low IL-10 expression likely contributes to loss of FBLIM1 function in an additive manner, especially in the child with the rs41310367 variant, as low IL-10 expression likely induces low FBLIM1 expression synergistically with the reduced enhancer activity caused by the regulatory variant. In trans with the frameshift mutation, this would result in a complete loss of function in FBLIM1.
We report here that recessive mutations in FBLIM1 contribute to the pathogenesis of chronic recurrent multifocal osteomyelitis. Previous experiments support FBLP1’s function as an anti-inflammatory molecule, and one involved in the regulation of integrin activation in neutrophils as well as the maintenance of bone homeostasis. In this study, we identified two families affected by CRMO with likely pathogenic mutations in FBLIM1. The results of the cmo mouse microarray experiment strongly support FBLIM1 as the most likely candidate gene underlying CRMO in these two families. This is the first report of FBLIM1 as a gene mutated in human disease. Although further experimentation is necessary to determine the exact mechanism by which aberrant FBLIM1 function leads to the CRMO phenotype, we hypothesize that FBLIM1 expression is regulated by STAT3 and NR4A2 in immune and bone cells and that it maintains the balance between osteoclast bone resorption and osteoblast bone formation in healthy individuals via regulation of ERK1/2 phosphorylation and subsequent RANKL expression (Fig 7). FBLP1 activity is part of the anti-inflammatory response and mutations in FBLIM1 lead to the chronic inflammation and bone lesions characteristic of CRMO.
Fig 7. Proposed model for FBLIM1’s involvement in CRMO.
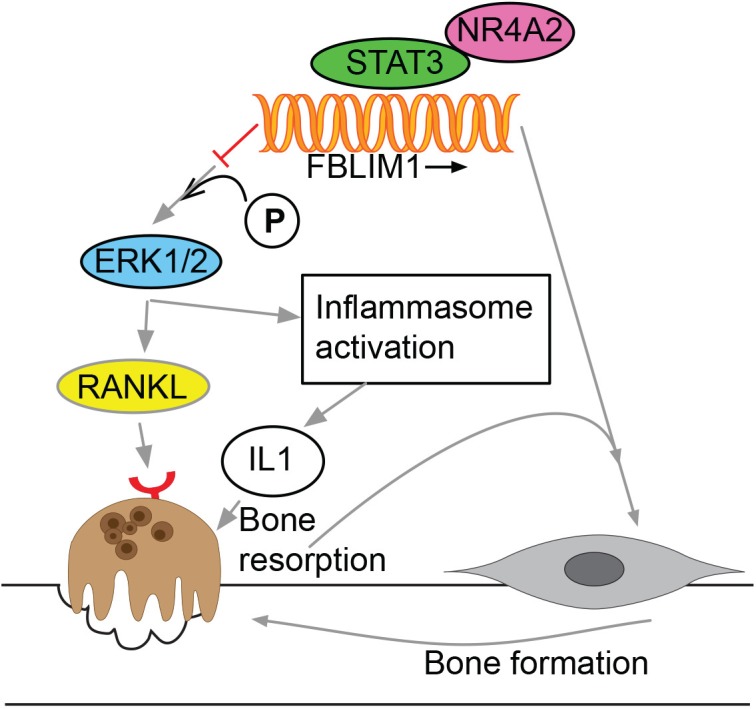
FBLIM1 expression is regulated by STAT3 and NR4A2 binding. FBLP1 blocks the phosphorylation of ERK1/2; in its absence or when dysfunctional, the increased phosphorylation of ERK1/2 leads to increased RANKL production, subsequent osteoclast activation, and bone resorption, as well as inflammasome activation. Mutations like rs41310367 disrupt regulation and expression of FBLIM1, resulting in decreased osteoblast activity and subsequent bone loss.
Supporting information
Sanger sequencing chromatograms showing that in the family (FamA) with rs146575757, the affected child is homozygous and both parents are heterozygous. For the second family (FamB), the chromatograms show the novel frameshift mutation and rs41310367, and that the mutations are in trans. C, D, M = child, dad, mom
(ZIP)
Sanger sequencing chromatograms showing values for rs1800896 (G/A), rs1800871 (C/T) and rs1800872 (C/A) in families A and B. C, D, M = child, dad, mom.
(ZIP)
Luciferase readings from the experiments functionally validating the regulatory effect of rs41310367. The readings are shown as three separate experiments, and combined for statistical analysis.
(XLSX)
Acknowledgments
Drs. Huan Liu and Robert Cornell at the University of Iowa for providing us with plasmids, and Melinda Smits for assistance in figure design.
Data Availability
The Sanger sequencing results for the mutations are described in the paper and the chromatograms are included as supplementary data. The gene expression microarray data are available in the Gene Expression Omnibus (accession number: GSE94112). The whole exome sequence data (fastq files) cannot be made publicly available as data sharing was not included in the consent. Requests for these data can be sent to the corresponding author.
Funding Statement
This work was supported by NIH/NIAMS R01AR059703 (PF, AB, VM), by the NIH Predoctoral Training Grants in Genetics T32GM008629 (AC), and Bioinformatics T32GM082729-01 (AC), as well as National Institutes of Health grant T32GM007337 to Gabriel Velez.
References
- 1.Ferguson PJ, Sandu M. Current understanding of the pathogenesis and management of chronic recurrent multifocal osteomyelitis. Current rheumatology reports. 2012;14(2):130–41. PubMed Central PMCID: PMC3656493. 10.1007/s11926-012-0239-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hedrich CM, Hofmann SR, Pablik J, Morbach H, Girschick HJ. Autoinflammatory bone disorders with special focus on chronic recurrent multifocal osteomyelitis (CRMO). Pediatric rheumatology online journal. 2013;11(1):47 PubMed Central PMCID: PMC3881012. 10.1186/1546-0096-11-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferguson PJ, Chen S, Tayeh MK, Ochoa L, Leal SM, Pelet A, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). Journal of medical genetics. 2005;42(7):551–7. PubMed Central PMCID: PMC1736104. 10.1136/jmg.2005.030759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. The New England journal of medicine. 2009;360(23):2426–37. PubMed Central PMCID: PMC2876877. 10.1056/NEJMoa0807865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herlin T, Fiirgaard B, Bjerre M, Kerndrup G, Hasle H, Bing X, et al. Efficacy of anti-IL-1 treatment in Majeed syndrome. Annals of the rheumatic diseases. 2013;72(3):410–3. PubMed Central PMCID: PMC3660147. 10.1136/annrheumdis-2012-201818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferguson PJ, Bing X, Vasef MA, Ochoa LA, Mahgoub A, Waldschmidt TJ, et al. A missense mutation in pstpip2 is associated with the murine autoinflammatory disorder chronic multifocal osteomyelitis. Bone. 2006;38(1):41–7. PubMed Central PMCID: PMC3726202. 10.1016/j.bone.2005.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chitu V, Ferguson PJ, de Bruijn R, Schlueter AJ, Ochoa LA, Waldschmidt TJ, et al. Primed innate immunity leads to autoinflammatory disease in PSTPIP2-deficient cmo mice. Blood. 2009;114(12):2497–505. PubMed Central PMCID: PMC2746474. 10.1182/blood-2009-02-204925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grosse J, Chitu V, Marquardt A, Hanke P, Schmittwolf C, Zeitlmann L, et al. Mutation of mouse Mayp/Pstpip2 causes a macrophage autoinflammatory disease. Blood. 2006;107(8):3350–8. PubMed Central PMCID: PMC1895761. 10.1182/blood-2005-09-3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chitu V, Nacu V, Charles JF, Henne WM, McMahon HT, Nandi S, et al. PSTPIP2 deficiency in mice causes osteopenia and increased differentiation of multipotent myeloid precursors into osteoclasts. Blood. 2012;120(15):3126–35. PubMed Central PMCID: PMC3471520. 10.1182/blood-2012-04-425595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cassel SL, Janczy JR, Bing X, Wilson SP, Olivier AK, Otero JE, et al. Inflammasome-independent IL-1beta mediates autoinflammatory disease in Pstpip2-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(3):1072–7. PubMed Central PMCID: PMC3903222. 10.1073/pnas.1318685111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lukens JR, Gross JM, Calabrese C, Iwakura Y, Lamkanfi M, Vogel P, et al. Critical role for inflammasome-independent IL-1beta production in osteomyelitis. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(3):1066–71. PubMed Central PMCID: PMC3903206. 10.1073/pnas.1318688111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gurung P, Burton A, Kanneganti TD. NLRP3 inflammasome plays a redundant role with caspase 8 to promote IL-1beta-mediated osteomyelitis. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(16):4452–7. 10.1073/pnas.1601636113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hofmann SR, Schwarz T Fau—Moller JC, Moller Jc Fau—Morbach H, Morbach H Fau—Schnabel A, Schnabel A Fau—Rosen-Wolff A, Rosen-Wolff A Fau—Girschick HJ, et al. Chronic non-bacterial osteomyelitis is associated with impaired Sp1 signaling, reduced IL10 promoter phosphorylation, and reduced myeloid IL-10 expression. (1521–7035 (Electronic)). [DOI] [PubMed]
- 14.Hamel J Fau—Paul D, Paul D Fau—Gahr M, Gahr M Fau—Hedrich CM, Hedrich CM. Pilot study: possible association of IL10 promoter polymorphisms with CRMO. (1437-160X (Electronic)). [DOI] [PubMed]
- 15.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–95. PubMed Central PMCID: PMC2828108. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20(9):1297–303. PubMed Central PMCID: PMC2928508. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.International HapMap C. A haplotype map of the human genome. Nature. 2005;437(7063):1299–320. PubMed Central PMCID: PMC1880871. 10.1038/nature04226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73. PubMed Central PMCID: PMC3042601. 10.1038/nature09534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11. PubMed Central PMCID: PMC29783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92. PubMed Central PMCID: PMC3679285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teer JK, Green ED, Mullikin JC, Biesecker LG. VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 2012;28(4):599–600. PubMed Central PMCID: PMC3278764. 10.1093/bioinformatics/btr711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moller JC, Paul D Fau—Ganser G, Ganser G Fau—Range U, Range U Fau—Gahr M, Gahr M Fau—Kelsch R, Kelsch R Fau—Rosen-Wolff A, et al. IL10 promoter polymorphisms are associated with systemic onset juvenile idiopathic arthritis (SoJIA). (0392-856X (Print)). [PubMed]
- 24.Auton A Fau—Brooks LD, Brooks Ld Fau—Durbin RM, Durbin Rm Fau—Garrison EP, Garrison Ep Fau—Kang HM, Kang Hm Fau—Korbel JO, Korbel Jo Fau—Marchini JL, et al. A global reference for human genetic variation. (1476–4687 (Electronic)).
- 25.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539 PubMed Central PMCID: PMC3261699. 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mathelier A, Fornes O, Arenillas DJ, Chen CY, Denay G, Lee J, et al. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016;44(D1):D110–5. PubMed Central PMCID: PMC4702842. 10.1093/nar/gkv1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ward JJ, McGuffin Lj Fau—Bryson K, Bryson K Fau—Buxton BF, Buxton Bf Fau—Jones DT, Jones DT. The DISOPRED server for the prediction of protein disorder. (1367–4803 (Print)). [DOI] [PubMed]
- 28.McGuffin LJ, Bryson K Fau—Jones DT, Jones DT. The PSIPRED protein structure prediction server. (1367–4803 (Print)). [DOI] [PubMed]
- 29.Kelley LA-O, Mezulis S, Yates CM, Wass MN, Sternberg MA-O. The Phyre2 web portal for protein modeling, prediction and analysis. (1750–2799 (Electronic)). [DOI] [PMC free article] [PubMed]
- 30.Dolinsky TJ, Nielsen Je Fau—McCammon JA, McCammon Ja Fau—Baker NA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. (1362–4962 (Electronic)). [DOI] [PMC free article] [PubMed]
- 31.Baker NA, Sept D Fau—Joseph S, Joseph S Fau—Holst MJ, Holst Mj Fau—McCammon JA, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. (0027–8424 (Print)). [DOI] [PMC free article] [PubMed]
- 32.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–42. PubMed Central PMCID: PMC102472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ithychanda SS, Das M, Ma YQ, Ding K, Wang X, Gupta S, et al. Migfilin, a molecular switch in regulation of integrin activation. J Biol Chem. 2009;284(7):4713–22. PubMed Central PMCID: PMC2640964. 10.1074/jbc.M807719200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tompa P. Intrinsically disordered proteins: a 10-year recap. (0968–0004 (Print)). [DOI] [PubMed]
- 35.Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;15(4):272–86. 10.1038/nrg3682 [DOI] [PubMed] [Google Scholar]
- 36.Blanchette M, Kent Wj Fau—Riemer C, Riemer C Fau—Elnitski L, Elnitski L Fau—Smit AFA, Smit Af Fau—Roskin KM, Roskin Km Fau—Baertsch R, et al. Aligning multiple genomic sequences with the threaded blockset aligner. (1088–9051 (Print)). [DOI] [PMC free article] [PubMed]
- 37.Huo L, Liu K, Pei J, Yang Y, Ye Y, Liu Y, et al. Fluoride promotes viability and differentiation of osteoblast-like Saos-2 cells via BMP/Smads signaling pathway. Biol Trace Elem Res. 2013;155(1):142–9. PubMed Central PMCID: PMC3763164. 10.1007/s12011-013-9770-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao G, Cheng H, Cao H, Chen K, Tu Y, Yu S, et al. Critical role of filamin-binding LIM protein 1 (FBLP-1)/migfilin in regulation of bone remodeling. J Biol Chem. 2012;287(25):21450–60. PubMed Central PMCID: PMC3375566. 10.1074/jbc.M111.331249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu C. Migfilin and its binding partners: from cell biology to human diseases. J Cell Sci. 2005;118(Pt 4):659–64. 10.1242/jcs.01639 [DOI] [PubMed] [Google Scholar]
- 40.Tu Y, Wu S, Shi X, Chen K, Wu C. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell. 2003;113(1):37–47. [DOI] [PubMed] [Google Scholar]
- 41.Gkretsi V, Zhang Y, Tu Y, Chen K, Stolz DB, Yang Y, et al. Physical and functional association of migfilin with cell-cell adhesions. J Cell Sci. 2005;118(Pt 4):697–710. 10.1242/jcs.01638 [DOI] [PubMed] [Google Scholar]
- 42.Das M, Ithychanda SS, Qin J, Plow EF. Migfilin and filamin as regulators of integrin activation in endothelial cells and neutrophils. PloS one. 2011;6(10):e26355 PubMed Central PMCID: PMC3197140. 10.1371/journal.pone.0026355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Volmering S, Block H, Boras M, Lowell CA, Zarbock A. The Neutrophil Btk Signalosome Regulates Integrin Activation during Sterile Inflammation. Immunity. 2016;44(1):73–87. 10.1016/j.immuni.2015.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma CP, Ezzell RM, Arnaout MA. Direct interaction of filamin (ABP-280) with the beta 2-integrin subunit CD18. J Immunol. 1995;154(7):3461–70. [PubMed] [Google Scholar]
- 45.Walsh MC, Choi Y. Biology of the RANKL-RANK-OPG System in Immunity, Bone, and Beyond. Frontiers in immunology. 2014;5:511 PubMed Central PMCID: PMC4202272. 10.3389/fimmu.2014.00511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337–42. 10.1038/nature01658 [DOI] [PubMed] [Google Scholar]
- 47.Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4(8):638–49. 10.1038/nrg1122 [DOI] [PubMed] [Google Scholar]
- 48.Takayanagi H. New developments in osteoimmunology. Nature reviews Rheumatology. 2012;8(11):684–9. 10.1038/nrrheum.2012.167 [DOI] [PubMed] [Google Scholar]
- 49.Ghonime MG, Shamaa OR, Das S, Eldomany RA, Fernandes-Alnemri T, Alnemri ES, et al. Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J Immunol. 2014;192(8):3881–8. PubMed Central PMCID: PMC3980013. 10.4049/jimmunol.1301974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hutchins AP, Poulain S, Miranda-Saavedra D. Genome-wide analysis of STAT3 binding in vivo predicts effectors of the anti-inflammatory response in macrophages. Blood. 2012;119(13):e110–9. 10.1182/blood-2011-09-381483 [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Zhuang J, Iyer S, Lin XY, Greven MC, Kim BH, et al. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013;41(Database issue):D171–6. PubMed Central PMCID: PMC3531197. 10.1093/nar/gks1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome research. 2012;22(9):1798–812. PubMed Central PMCID: PMC3431495. 10.1101/gr.139105.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489(7414):91–100. PubMed Central PMCID: PMC4154057. 10.1038/nature11245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pirih FQ, Tang A Fau—Ozkurt IC, Ozkurt Ic Fau—Nervina JM, Nervina Jm Fau—Tetradis S, Tetradis S. Nuclear orphan receptor Nurr1 directly transactivates the osteocalcin gene in osteoblasts. (0021–9258 (Print)). [DOI] [PubMed]
- 55.Lee MK, Choi H Fau—Gil M, Gil M Fau—Nikodem VM, Nikodem VM, Pirih FQ, Tang A Fau—Ozkurt IC, et al. Regulation of osteoblast differentiation by Nurr1 in MC3T3-E1 cell line and mouse calvarial osteoblasts Nuclear orphan receptor Nurr1 directly transactivates the osteocalcin gene in osteoblasts. (0730–2312 (Print)).
- 56.McMorrow JP, Murphy EP. Inflammation: a role for NR4A orphan nuclear receptors? Biochem Soc Trans. 2011;39(2):688–93. 10.1042/BST0390688 [DOI] [PubMed] [Google Scholar]
- 57.Aherne CM, McMorrow J, Kane D, FitzGerald O, Mix KS, Murphy EP. Identification of NR4A2 as a transcriptional activator of IL-8 expression in human inflammatory arthritis. Mol Immunol. 2009;46(16):3345–57. 10.1016/j.molimm.2009.07.019 [DOI] [PubMed] [Google Scholar]
- 58.McMorrow JP, Crean D, Gogarty M, Smyth A, Connolly M, Cummins E, et al. Tumor necrosis factor inhibition modulates thrombospondin-1 expression in human inflammatory joint disease through altered NR4A2 activity. Am J Pathol. 2013;183(4):1243–57. 10.1016/j.ajpath.2013.06.029 [DOI] [PubMed] [Google Scholar]
- 59.Lopez-Dee Z, Pidcock K, Gutierrez LS. Thrombospondin-1: multiple paths to inflammation. Mediators Inflamm. 2011;2011:296069 PubMed Central PMCID: PMC3134184. 10.1155/2011/296069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marzaioli V, McMorrow JP, Angerer H, Gilmore A, Crean D, Zocco D, et al. Histamine contributes to increased RANKL to osteoprotegerin ratio through altered nuclear receptor 4A activity in human chondrocytes. Arthritis Rheum. 2012;64(10):3290–301. 10.1002/art.34554 [DOI] [PubMed] [Google Scholar]
- 61.Nea Kawana. Molecular Network of NLRP3 Inflammasome Activation-Responsive Genes in a Human Monocyte Cell Line. Austin Journal of Clinical Immunology. 2014;1(4). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sanger sequencing chromatograms showing that in the family (FamA) with rs146575757, the affected child is homozygous and both parents are heterozygous. For the second family (FamB), the chromatograms show the novel frameshift mutation and rs41310367, and that the mutations are in trans. C, D, M = child, dad, mom
(ZIP)
Sanger sequencing chromatograms showing values for rs1800896 (G/A), rs1800871 (C/T) and rs1800872 (C/A) in families A and B. C, D, M = child, dad, mom.
(ZIP)
Luciferase readings from the experiments functionally validating the regulatory effect of rs41310367. The readings are shown as three separate experiments, and combined for statistical analysis.
(XLSX)
Data Availability Statement
The Sanger sequencing results for the mutations are described in the paper and the chromatograms are included as supplementary data. The gene expression microarray data are available in the Gene Expression Omnibus (accession number: GSE94112). The whole exome sequence data (fastq files) cannot be made publicly available as data sharing was not included in the consent. Requests for these data can be sent to the corresponding author.