

Abstract
Adoptively transferred CD8+ T cells can stabilize the size of solid tumors over long periods of time by exclusively recognizing antigen cross-presented on tumor stroma. However, these tumors eventually escape T cell–mediated growth control. The aim of this study was to eradicate such persistent cancers. In our model, the SIYRYYGL antigen is expressed by cancer cells that lack the MHC-I molecule Kb needed for direct presentation, but the antigen is picked up and cross-presented by tumor stroma. A single injection of antigen-specific 2C CD8+ T cells caused long-term inhibition of tumor growth, but without further intervention, tumors started to progress after approximately 3 months. Escape was associated with reduced numbers of circulating 2C cells. Tumor-infiltrating 2C cells produced significantly less TNFα and expressed more of the “exhaustion” markers PD-1 and Tim-3 than T cells from lymphoid organs. High-dose local ionizing radiation, depletion of myeloid-derived suppressor cells, infusions of additional 2C cells, and antibodies blocking PD-L1 did not prevent tumor escape. In contrast, adoptive transfer of allogeneic CD4+ T cells restored the numbers of circulating Ag-specific CD8+ T cells and their intratumoral function, resulting in tumor eradication. These CD4+ T cells had no antitumor effects in the absence of CD8+ T cells and recognized the alloantigen cross-presented on tumor stroma. CD4+ T cells might also be effective in cancer patients when PD1/PD-L1 blockade does not rescue intratumoral CD8+ T-cell function and tumors persist.
Keywords: Adoptive therapy, T cells, exhaustion, intratumoral, tumor escape
INTRODUCTION
Various new treatments, including immunotherapy, can result in the prolonged stabilization of certain cancers in patients, i.e., the cancer does not change in extent or severity for months or even years. This outcome is often also referred to as “stable disease” (SD) (NCI Dictionary of Cancer Terms http://www.cancer.gov/publications/dictionaries/cancer-terms) and is certainly a welcome response to chemo-, immuno- or radiotherapy (1–3). The extent of which such a response may be secondary to the biology of the particular cancer being treated is poorly understood. Certainly however, as any cancer that is not cured, “stable” cancers carry the risk of relapsing.
Several years ago, we developed a cancer model in which stable disease occurs after adoptive T-cell therapy (ATT) (4, 5). The transferred CD8+ T cells do not target the cancer cells directly because they lack the appropriate MHC-I molecule to present the model neoantigen SIYRYYGL. However, the CD8+ T cells do target the stromal cells cross-presenting the Ag released by the cancer cells. This treatment achieved shrinkage of large long-established tumors, followed by the tumors persisting at a smaller size for several weeks or even months (5). This is similar to stable disease observed in some cancer patients after various types of therapies. Subsequent experiments presented here demonstrated that all of these cancers eventually relapsed and killed the hosts. Therefore, the focus of our present work was to develop approaches to prevent or revert tumor relapse and achieve eradication.
A major cause for escape from CD8+ T cell-mediated control can be loss, mutation or reduced expression of the MHC molecules presenting the targeted peptides to the T cells on the cancer cell surface (6–9). Although genetic instability is not a requirement for cancer development (10), cancers exhibit a mutator phenotype (11), which results in the often remarkable heterogeneity of cancers (12). Genetic instability allows cancer cells to develop resistance to all kind of therapies. In contrast, stroma consists mainly of leukocytes, fibroblasts and blood vessels that are genetically stable and therefore cannot easily escape destruction through mutation. Stroma provides the proper three-dimensional microenvironment, vascular supply and growth regulatory paracrine loops that are essential for cancer cells growth (for references see (13)).
Stroma also has immunological functions and can be critical for preventing or permitting T cell–mediated destruction of antigenic cancer cells (14–16). For example, highly expressed tumor-specific proteins are released by the cancer cells and load not only stromal dendritic cells but also CD11b+ macrophages. Such macrophages, when exposed to larger amounts of protein than they can break down, express the tumor-specific proteins on their cell surface (4, 17, 18). Thereby, stroma can become a target for CD8+ T cells recognizing tumor-specific Ags, as in our model of stable disease (5).
Because the aim of our study was to eradicate stable tumors, determining the mechanism of relapse was important to achieve eradication. T cells can control chronic systemic viral infection for years, but escape from this control may occur because chronic exposure to Ag can “exhaust” the CD8+ T cells (19). In persistent tumors, this problem must occur within the tumor microenvironment where antigenic cancer cells can grow progressively without evidence for systemic T-cell exhaustion or anergy (20). Monoclonal antibodies disrupting the PD1/PD-L1 axis can revive exhausted CD8+ T cells in chronic viral infection and cancer patients (21–24). However, although viral load is reduced, PD1/PD-L1 blockade is insufficient to eradicate the infection in a murine model of chronic infection by LCMV (21). PD1/PD-L1 pathway blockade can also cause durable remissions in patients with certain types of cancer, but only a fraction of the patients respond to treatment, and few become tumor-free. (25).
CD4+ T cells are required to sustain CD8+ T cells and resolve chronic viral infection (26) and the CD8+ T cells in CD4+ T cell–deficient mice have more pronounced functional defects (26). CD4+ T cells are not only required for the induction of new CD8+ T-cell responses (27), but also during the effector phase (28, 29), and for the secondary expansion and generation of memory CD8+ T lymphocytes (30–32). CD8+ T cells receive CD4+ T-cell help directly through CD40 (33). CD4+ T cells can be required (34) within the first few days after transfer (35) for optimal efficacy of ATT regimes and even have a potent antitumoral effect by themselves in some models (34, 36, 37). CD4+ T cells are also important in the maintenance of long-term protective antitumor immunity (38). Whether CD4+ T-cell transfer can rescue CD8+ T cells in chronic cancers and thereby allow tumor eradication is presently unknown. Therefore, we tested this hypothesis in our animal model of chronic cancers that escape immune control by CD8+ T cells and are resistant to treatment with PD1/PD-L1 blocking antibodies. Indeed, we found that ATT of CD4+ T cells led to eradication of cancers that relapsed after many weeks of remaining at a stable size. This eradication required the participation of Ag-specific CD8+ T cells whose function in the tumor microenvironment was rescued by the CD4+ T cells.
MATERIALS AND METHODS
Mice, cell lines, and reagents
OT1-Rag1−/− mice were provided by A. Ma (University of California San Francisco, San Francisco, CA). 2C mice were provided by J. Chen (Massachusetts Institute of Technology, Boston, MA). OT2 mice were provided by R. Shilling (University of Chicago) and crossed to C57BL/6 Rag1−/− (B6.129S7-Rag1tm1Mom/J, The Jackson Laboratory) to obtain OT2-Rag1−/−. For the generation of 2C-Prf−/−, 2C were crossed to C57BL/6-Prf tm1Sdz/J mice obtained from The Jackson Laboratory. CD8−/− mice (B6.129S2-Cd8atm1Mak/J) were from The Jackson Laboratory. MHC-II−/− mice have been described (39). All mice were maintained in a specific pathogen-free barrier facility at the University of Chicago according to the Institutional Animal Care and Use Committee guidelines. PRO4L fibrosarcoma cell line was induced by UV in C3H/HeN mice in our laboratory in the 1980s (40) and later transduced to make PRO4L-SIY-EGFP in the early 2000s (4). This cell line was tested by IDEXX-Radil and found to be negative for animal pathogens and mycoplasma. Cells were kept in culture for a maximum of 3 weeks before being used. The 2C-recognized peptide SIYRYYGL was synthesized by S. Meredith (The University of Chicago).
Flow cytometry
Monoclonal antibodies (mAbs) to CD90.1 (Thy1.1, OX-7), I-Ab (AF6-1201) and IFNγ (XMG1.2) were from BD PharMingen; mAbs to CD11b (M1/70), Gr1 (RB6-8C5), MHC-II (M5/114.15.2), Vβ8.1/8.2 (KJ16), CD8α (53–6.7), PD-1 (J43), Tim-3 (RMT3-23), TNF (MP6-XT22), I-Ak (11-5.2), and I-Ek (14-4-4S) were from eBioscience. Samples were initially incubated with 2.4G2 hybridoma supernatant to block antibody binding to the Fcγ receptors. In tumor samples, dead cells were identified by 7AAD (BD Pharmingen) staining and excluded by electronic gating. Intracellular stainings were performed using the Cytofix/Cytoperm kit from BD (Cat. No. 554714). AccuCount Rainbow beads (Cat. No. ACRFP-100-3 Spherotech) were used according to the manufacturer’s instructions to determine absolute counts of 2C cells in PBL. Data were acquired on a FACSCalibur or FACSCanto and analyzed with FlowJo software (Tree Star, Ashland, OR).
Phenotypic and functional analysis of 2C cells from lymphoid organs and tumors
Tumor cell suspensions were prepared as in (5). Anti-CD45 microbeads were used (Cat. No. 130-052-301, Miltenyi Biotec, Bergisch Gladbach, Germany) for the isolation of tumor leukocytes. Tumor CD45+ cells (including unsorted APC and T cells), splenocytes, and lymph node single-cell suspensions were incubated for 4.5 h with SIY peptide (1 μM) and brefeldin A (Sigma), then surface-stained with anti-CD8 and anti-Vb8 and intracellularly with anti-TNF and anti-IFNγ. Expression of PD-1 and Tim-3 was measured in freshly isolated cells. Anti-PD-L1 (10F.9G2, Cat. No. BE0101, BioXCell) was added to tumor CD45+ cells restimulated for 3 days with SIY peptide for PD-L1 blocking experiments. In some experiments anti-Tim3 (5D12), kindly donated by A. Anderson (Harvard Medical School, Boston, MA), was added. IFNγ and TNF were measured in supernatants by ELISA (eBioscience Cat. No. 88-8314-77 and BMS607HS). For the in vivo killing experiments, OT1-Rag−/− splenocytes were pulsed with 2 concentrations of SIY peptide (SIY 5 nM and SIY 50 nM) or no peptide (No SIY). The 3 cell suspensions were labeled with different concentrations of CFSE (Sigma), mixed at 1:1:1 ratio and injected i.v. into a naïve OT1-Rag−/− mouse (control), and 2 to 3 tumor-bearing mice. After 5 h the mice were killed and the spleens were analyzed by flow cytometry.
Tumor challenge and treatment
Cancer cells lines were cultured in DMEM, 5% FCS (Gemini Bio-Products, West Sacramento, CA) at 37 °C in a 10% CO2 dry incubator. 2 × 106 PRO4L-SIY-EGFP cells were injected subcutaneously into the shaved backs of mice. Tumor volumes were measured along three orthogonal axes (a, b, and c) every 3 to 4 days and tumor volume calculated as abc/2. When tumors reached approximately 300–600 mm3 (for details, see Figure legends), mice were treated with naïve 2C or 2C-Rag−/− splenocytes (around 10 × 106 CD8+ T cells) i.v. to induce equilibrium. For the experiments testing local radiation plus 2C cells, 2C splenocytes were activated in vitro with SIY peptide before transfer. Some mice were treated with 0.4 mg purified anti-Gr1 (RB6-8C5) i.p. For treatment with CD4+ T cells, splenocytes from naïve CD8−/− (containing 10 × 106–20 × 106 CD4+ T cells) were injected at the indicated time points. Anti-PD-L1 was administered i.p. two to three times per week at 200 μg doses.
Local tumor irradiation
Mice were irradiated using an x-ray generator (PCM 1000; Pantak) at a dose of 20 Gy each day for two consecutive days (total 40 Gy). Each mouse was confined to a lead cover with its tumor-bearing flank exposed through an opening on the side, allowing the tumor to be irradiated locally.
Statistical analysis
The number of 2C/μL blood versus time and of MDSC/CD8+ T cells versus tumor size were analyzed using a random-effects regression model. The increase in number of 2C cells and their production of IFNγ in the peripheral blood after treatment of mice with CD4+ T cells was analyzed using an unpaired Student t test with unequal variances. The production of cytokines by 2C cells from different organs was compared using a paired Student t test. Tumor eradication by different treatments was compared using Fisher’s exact test. Statistical analysis was performed using Stata (Statacorp, College Station, TX).
RESULTS
Stabilized tumors caused by transferred 2C CD8+ T cells progress, but retain Ag
Experiments were designed to determine the duration of the equilibrium maintained by tumor antigen–specific 2C CD8+ T cells, under conditions in which antigen was exclusively cross-presented by tumor stroma (Fig. 1A). The C3H-derived PRO4L-SIY-EGFP cancer cells were not direct targets because they lack the H-2Kb allele needed for presenting the SIYRYYGL peptide that is recognized by the 2C TCR-transgenic CD8+ T cells (Fig. 1A) (5). In untreated mice, the tumors grew progressively for three weeks, at which time the mice were euthanized due to tumor size. Tumors in all mice treated with CD8+ T cells at 2 weeks regressed to remain almost undetectable for 7–8 weeks, when all mice developed tumors that grew slowly but progressively to become large tumors by 10 to 14 weeks (Fig. 1B). Cancer cell lines derived from escaping tumors were injected into new hosts and produced tumors that regressed when treated with CD8+ T cells (Suppl. Fig. 1A), indicating that loss of equilibrium was not due to emergence of variants.
Figure 1. Indirect antigen recognition on tumor stroma by adoptively transferred CD8+ T cells causes long-term inhibition of tumor growth followed by escape.
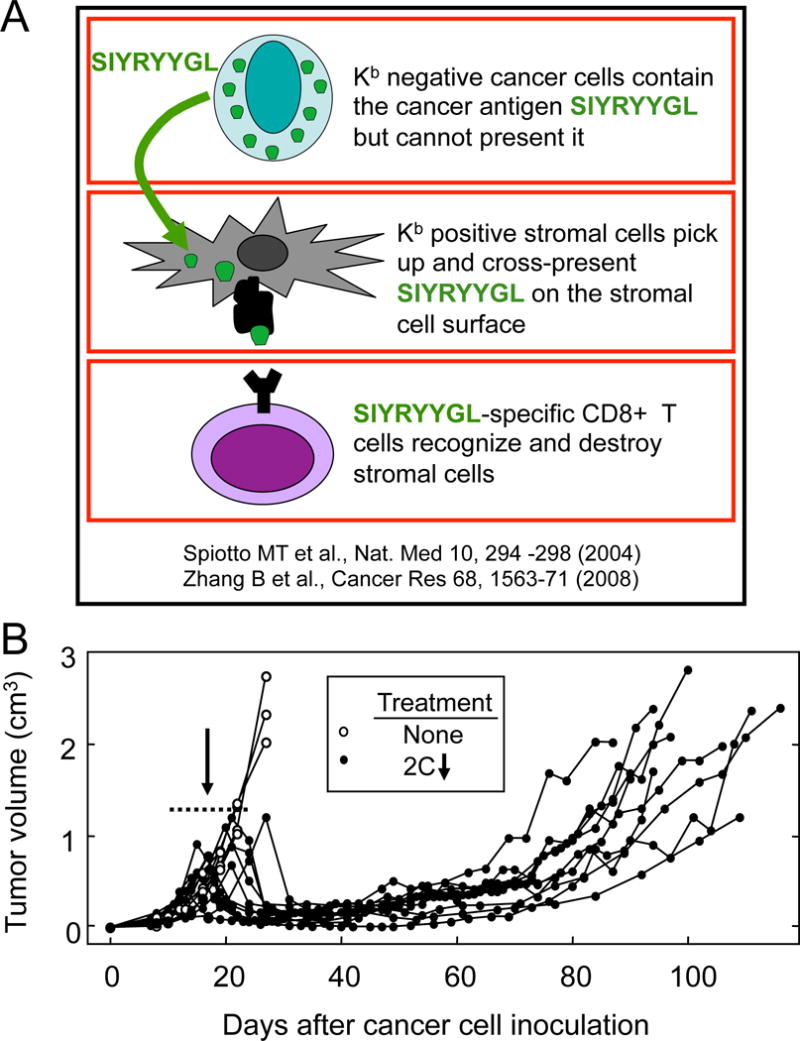
A. Diagram of the cellular interactions causing a long-term equilibrium of tumor growth through exclusive recognition of antigen on tumor stroma. B. Escape of tumors from long-term equilibrium caused by CD8+ T cells. OT1-Rag−/− B6 (H-2b) mice were injected with C3H (H-2k)-derived PRO4L-SIY-EGFP cells. When tumors reached 300–600 mm3 (average size = 425 mm3) the mice were treated with SIY-specific TCR-transgenic 2C T cells i.v. The dotted line (day 12–24) indicates the range of days after cancer cell inoculation at which the ATT took place. Data have been pooled from 9 experiments and 15 mice in total.
CD8+ T cells from tumors escaping equilibrium showed an “exhausted” phenotype
The increase in tumor volume of escaping tumors correlated with an increase in circulating myeloid-derived suppressor cells (MDSC), which was accompanied by a decrease in the percentage of CD8+ T cells (Fig. 2A). The absolute numbers of circulating 2C cells in the periphery also declined with time (Fig. 2B). In models of chronic infection (41) or cancer (42, 43), “exhausted” T cells show upregulation of certain surface markers, including PD-1 and Tim-3. Indeed, 2C cells isolated from the tumors of mice escaping equilibrium showed strong upregulation of PD-1 and Tim-3 compared to 2C cells isolated from lymphoid organs (Fig. 2C). Exhausted CD8+ T cells have less functional capacity, with decreased proliferation, cytolytic capacity, and production of cytokines (44). Here, the production of TNF by CD8+ T cells isolated from the tumors escaping equilibrium was severely reduced in comparison to T cells obtained from the lymphoid organs (Fig. 2D). CD8+ T-cell cytotoxic function in vivo was not impaired (Suppl. Fig. S1B), but equilibrium could be established by CD8+ T cells that lacked perforin (Suppl. Fig. S1C), suggesting that cytokine production rather than perforin-mediated lysis might be the dominant antitumoral mechanism of CD8+ T cells in vivo.
Figure 2. CD8+ T cells from mice with tumors relapsing after ATT showed decreased numbers in circulation and functional impairment in the tumor.
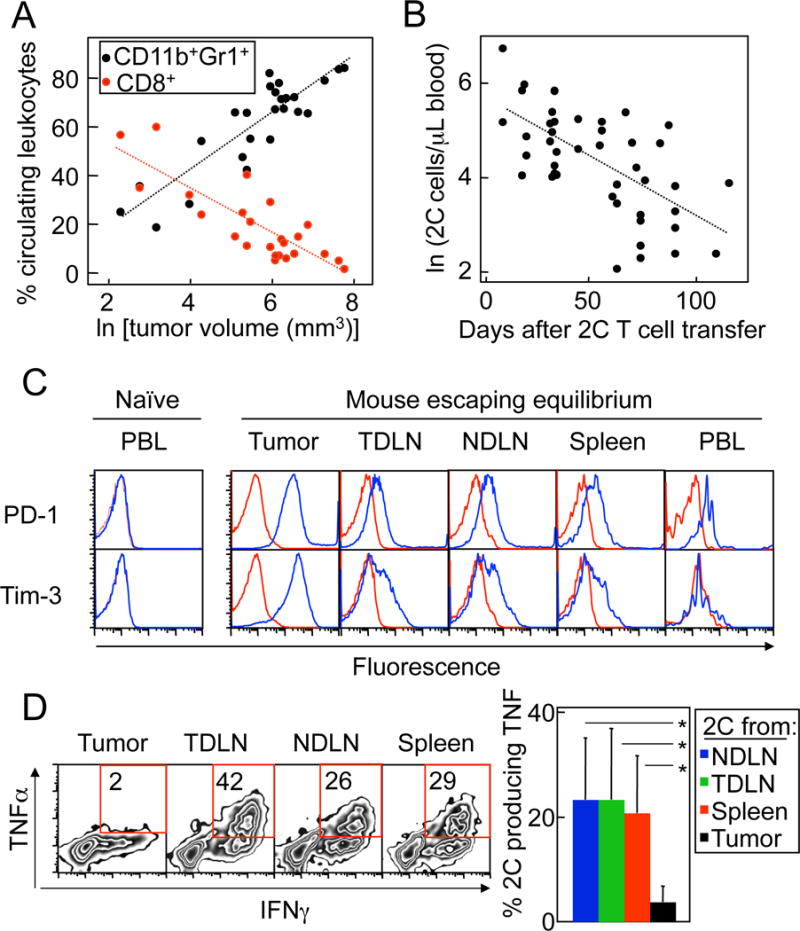
A. Decrease in CD8+ T cells and increase in MDSC in circulation with increasing tumor volumes. OT1-Rag−/− mice bearing PRO4L-SIY-EGFP tumors were treated with 2C cells on day 12–24 of tumor growth. Percentages of circulating CD11b+Gr1+ and total CD8+ T cells were plotted against the natural logarithm of tumor size in mm3. Data are from 14 mice and 8 experiments. Both the decrease in percent CD8+ and increase in percent CD11b+Gr1+ were highly statistically significant (P < 0.0001). The regression lines correspond to the following equations: for CD11b+Gr1+: y = 11.72x - 4.50, for CD8+ T cells: y = −9.16x + 71.43. B. Decrease in tumor Ag-specific CD8+ T cells with time after transfer. The absolute number of 2C cells (CD8+Vβ8+) per μL of blood was measured in mice treated as in (A). The natural logarithm of the number of 2C cells/μL of blood was calculated and plotted against the time after transfer. Data are from 23 mice and 13 experiments. The decrease in number of 2C/μL blood over time was highly statistically significant (P < 0.0001). The regression line corresponds to the equation y = −0.02x + 5.69. C. Upregulation of exhaustion markers by T cells isolated from tumors escaping equilibrium. Levels of expression of exhaustion markers PD-1 and Tim-3 were determined in 2C cells obtained from the indicated organs of a PRO4L-SIY-EGFP tumor-bearing OT1-Rag−/− mouse escaping equilibrium 63 days after ATT. Histograms are gated in 2C (CD8+Vβ8+) cells (blue: specific antibody; red: isotype control). A representative example is shown of a total of 6 repeats. D. Failure of T cells isolated from escaping tumors to produce TNF. The production of IFNγ and TNF by 2C cells from a mouse escaping equilibrium was analyzed by intracellular staining at day 91 since ATT. Gated: CD8+Vβ8+cells. Quadrants were determined based on staining with isotype control antibodies (not shown). FACS plots show a representative example out of 6 experiments performed with mice analyzed 43–98 days after 2C cell transfer. The bar graph shows pooled data on TNF production (n = 5–7 mice). The production of TNF by 2C was significantly lower in the tumor than in non-draining lymph nodes (NDLN, P = 0.028), tumor-draining lymph nodes (TDLN, P = 0.046) and spleen (P = 0.008) of the same mice.
Blockade of PD-L1 in vitro increased the production of IFNγ and TNF by peptide-stimulated tumor-infiltrating cells isolated from tumors escaping equilibrium (Suppl. Fig. S2A), suggesting that functional impairment of the CD8+ T cells in the tumor and PD-1 expression were linked. Addition of Tim-3-blocking antibodies failed to increase cytokine production any further (Suppl. Fig. S2B).
Transferred CD4+ T cells allowed eradication of escaped tumors
We tested several therapeutic approaches to prevent tumor escape. Additional late injections of activated CD8+ T cells preceded by local irradiation of the tumors with the purpose of loading stroma more efficiently (17) did not work (Suppl. Fig. S3A). Similarly, depletion of MDSCs using a mAb to Gr1, followed by new administration of CD8+ T cells, was not effective (Suppl. Fig. S3B). Continuous treatment of mice with tumors in equilibrium with anti-PD-L1 was also ineffective to prevent tumor relapse in the majority of the animals (7 relapses in 8 mice vs. 7 relapses in 7 mice, P = 1.0) (Fig. 3). However, we found that ATT of allogeneic B6 CD4+ T cells not only prevented tumor escape, but also eradicated these chronic tumors (Fig. 4A). Splenocytes of CD8−/− B6 mice were used as source of CD4+ T cells to bypass the requirement of CD4+ T-cell purification/sorting. CD8−/− splenocytes and sorted CD4+ T cells from B6 mice were equally effective in helping CD8+ T cells eliminate tumors in a different model (K. Schreiber and B. Engels, unpublished observations). Tumor-bearing mice received the CD4+ T cells at different times (varying between 21–95 days) after the initial CD8+ transfer, and some mice rejected their cancers even at very late times when their tumors were relapsing (Fig. 4A). Sometimes rejection was preceded by a transient increase in tumor volume, probably due to inflammation. Rejection took place also if CD8+ and CD4+ T cells were given together as first treatment (Fig 4B). It has been shown that non-Ag-specific CD4+ T cells are required for the maintenance phase of long-lived CD8+ T-cell memory (32). In our model, Ag-specific CD4+ T cells were required in the effector phase, because transferred OT2 CD4+ T cells from TCR-transgenic OT2 Rag−/− mice, specific for the irrelevant Ag ovalbumin, did not cause rejection (Fig. 4C). Since the PRO4L-SIY-EGFP cancer cells are of C3H (H-2k) origin, the transferred H-2b-derived splenic CD4+ T cells likely recognized H-2k-encoded alloantigens expressed by the PRO4L cancer cells. Alloantigens are known to generate potent CD4+ T-cell responses (45), therefore we sought to determine whether the CD4+ T cells could destroy the tumors without the participation of the CD8+ 2C T cells. However, CD4+ T cells had no antitumoral effects by themselves, as seen in Fig. 4D, supporting the requirement of CD8+ and CD4+ T-cell collaboration for the rejection of tumors (Table 1).
Figure 3. Treatment with anti-PD-L1 antibodies fails to prevent tumor relapse in most mice with tumors in equilibrium.
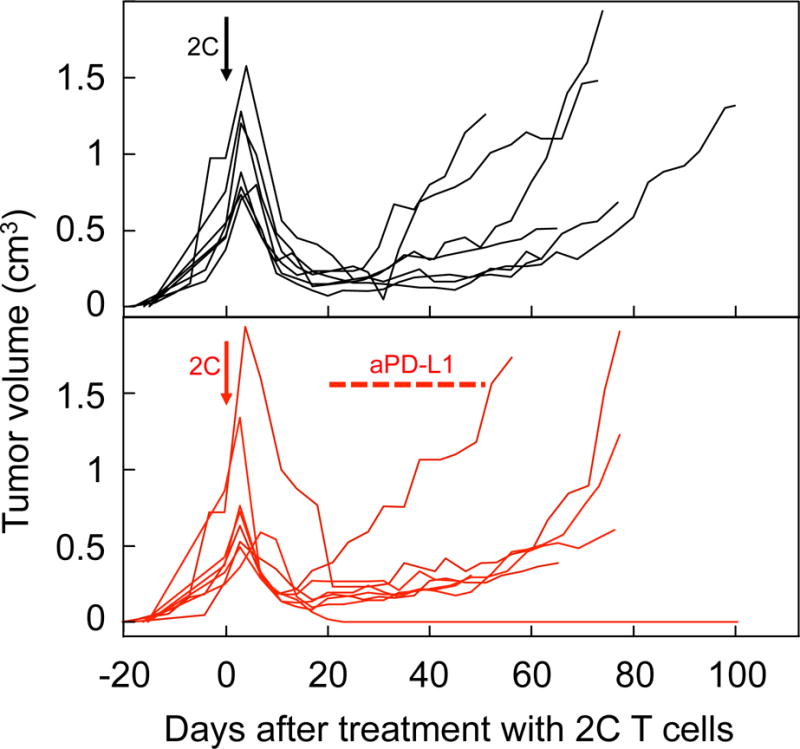
PRO4L-SIY-EGFP tumor-bearing OT1-Rag−/− mice were treated with 2C CD8+ T cells (arrows). When tumor “equilibrium” was established, defined here by at least two consecutive tumor measurements rendering sizes smaller than the size at the time of treatment, some mice started receiving 200 μg anti-PD-L1 i.p. twice weekly, until tumor rejection or escape were observed (average start day and duration of anti-PD-L1 treatment indicated in the figure). Data are pooled from 5 experiments with a total of 8 mice treated with anti-PD-L1 (red lines), and 7 concurrent control mice that received no anti-PD-L1 (black lines). One out of eight mice treated with anti-PD-L1 rejected the tumor.
Figure 4. Allogeneic CD4+ T cells help CD8+ T cells to eradicate tumors.
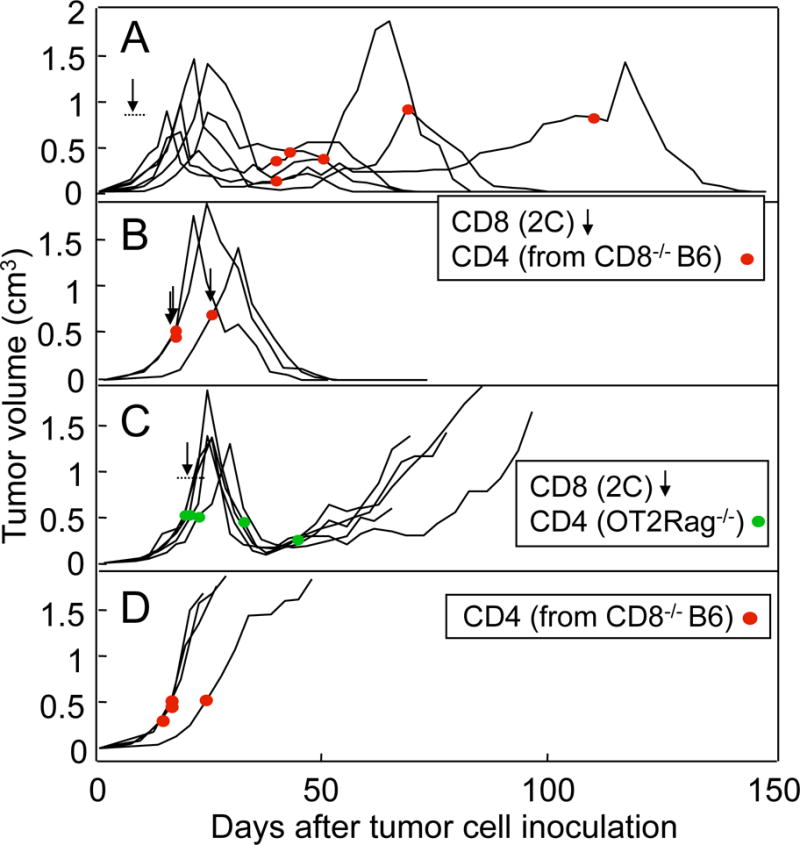
PRO4L-SIY-EGFP tumor-bearing OT1-Rag−/− mice were treated with 2C CD8+ T cells (arrows) and/or with CD4+ T cells from CD8−/− B6 mice (red dots). (A) Transfer of CD4+ T cells from CD8−/− B6 mice eradicates tumors previously treated with 2C cells and established for months. The CD4+ T cells were given at different times after the 2C cells as indicated (n = 6). (B) Tumors were eradicated when CD8+ and CD4+ T cells were given simultaneously (n = 3). (C) OT2-Rag−/− CD4+ T cells, specific for an irrelevant antigen, were ineffective (n = 4). (D) Transfer of CD4+ T cells from CD8−/− B6 mice alone did not eradicate tumors (n = 4).
Table 1.
Adoptive transfer of both CD8+ and CD4+ T cells is required for tumor eradication
Transferred CD4+ T cells improved CD8+ T cell cytokine production and numbers
2C cells recovered from tumors and tumor-draining lymph nodes from mice treated with CD4+ T cells produced more IFNγ and TNF than those from control mice not treated with CD4+ T cells, as percentage of cells producing cytokines and amount of cytokine produced per cell (indicated by mean fluorescence intensity, Fig. 5A, and Suppl. Table S1). Tumor-infiltrating 2C cells obtained from mice treated with CD4+ T cells also expressed less PD-1 (Fig. 5B), whereas Tim-3 was not affected (unpublished data). The beneficial effects of CD4+ treatment were also reflected in the periphery: CD4+ T cell–treated mice had more circulating 2C cells than mice that were not treated with CD4+ T cells (Fig. 5C), and a higher percentage of circulating 2C cells produced IFNγ (Fig. 5D), although TNF production by circulating 2C cells was unchanged (unpublished data). Thus, CD4+ T cells rescued or prevented exhaustion of CD8+ T cells and cooperated with them to cause tumor eradication.
Figure 5. CD4+ T cells restored the function of exhausted CD8+ T cells.
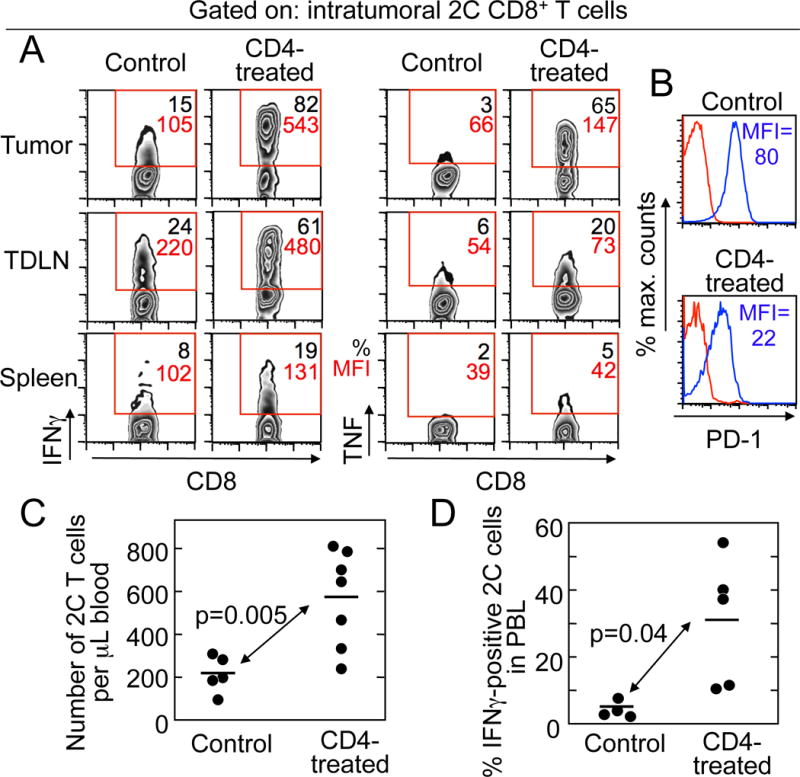
A. CD4+ T cells increased IFNγ and TNF production by tumor-infiltrating CD8+ T cells. IFNγ and TNF were measured in 2C cells recovered from lymphoid organs and PRO4L-SIY-EGFP tumors from OT1-Rag−/− mice with tumors in equilibrium treated or not with allogeneic CD4+ T cells (day 24 after CD4+ T-cell transfer). For each plot the percentage of 2C cells (CD8+Vb8+) producing cytokines is shown in black and the mean fluorescence intensity (MFI) in red. B. CD4+ T-cell transfer reduced the amount of PD-1 on 2C cells (CD8+Vb8+) from tumors. Red: Isotype control, Blue: anti-PD-1. C. CD4+ T cells increased the absolute numbers of 2C CD8+ T cells in the peripheral blood of mice bearing PRO4L-SIY-EGFP tumors. D. CD4+ T-cell treatment increased the production of IFNγ by circulating 2C cells. Mice received CD4+ T cells at days 29–76 after receiving 2C cells, and were bled 13–49 days after CD4+ T-cell transfer. For (A) and (B), 1/3 experiments with consistent results are shown. Data shown in (C) are from 3 independent experiments and 12 mice, and in (D) from 3 independent experiments and 9 mice.
Help by allogeneic CD4+ T cells required an MHC Class II-positive host
As for most non-hematopoietic cancer cells, PRO4L-SIY-EGFP cells in culture are MHC-II-negative (Suppl. Fig. S4A). However, as reported for other models (37, 46), we found that PRO4L-SIY-EGFP cancer cells freshly isolated from tumors escaping equilibrium had upregulated MHC-II expression (Suppl. Fig. S4B), implying that allogeneic CD4+ T cells might recognize the alloantigens directly on the cancer cells. However, combined treatment of PRO4L-SIY-EGFP tumors with CD4+ and CD8+ T cells in MHCII−/−C57BL/6 (B6) Rag−/− mice failed to eradicate the tumors in most animals (Fig. 6). Therefore, expression of MHC-II by cancer cells in vivo was insufficient, consistent with the notion that recognition of C3H-tumor-derived alloantigen on host cells was required for the CD4+ T cells to help tumor eradication.
Figure 6. Allogeneic CD4+ T cells could not help CD8+ T cells eradicate tumors in the absence of MHCII expression by the host.
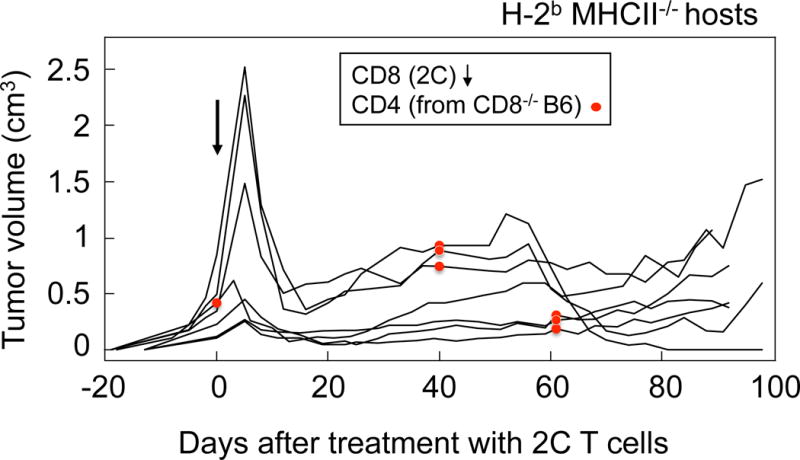
PRO4L-SIY-EGFP tumor-bearing MHCII−/− Rag−/− B6 mice were treated with sorted 2C CD8+ T cells (arrows) and sorted CD4+ T cells from CD8−/− B6 mice at the indicated times (red dots). Data were pooled from 3 independent experiments. Only 1/7 total mice rejected the tumor.
DISCUSSION
Here we show that alloantigen-specific CD4+ T cells can rescue exhausted neoantigen-specific CD8+ T cells to eradicate relapsing established solid tumors. These CD8+ T cells controlled the growth of aggressive cancers for weeks to months, but eventually failed. Tumor regrowth correlated with a decrease in the number of circulating tumor-reactive CD8+ T cells and the decreased ability of T cells to produce cytokines, in particular TNF, specifically inside tumors (Fig. 2D). T-cell dysfunction could well have been caused by chronic exposure of CD8+ T cells to Ag presented on tumor stromal cells. Ag persisted throughout the entire course of the experiment in this and previous studies using our model of exclusive stromal targeting (Suppl. Fig. S1A and (5)). A probable reason for the lack of Ag-loss variants may be the lack of the Ag-presenting MHC-I molecules on the cancer cells, because selection might be difficult in the absence of direct recognition and killing of cancer cells. Other epigenetic or genetic changes of the cancer cells were not the mechanism for escape, since re-isolates of the escaped cancers readapted to culture and where re-injected into mice were as effectively forced into equilibrium as the original cancers (Suppl. Fig. S1A). Thus, the escape tumors had retained sensitivity to T-cell killing, and the main mechanism for escape was CD8+ T-cell exhaustion or dysfunction (Fig. 2).
It is remarkable that the adoptively transferred CD8+ T cells were amenable to rescue by the CD4+ T cells even though the CD8+ T cells had been exposed to numerous immunosuppressive factors in the tumor environment for many weeks. Both locally derived stromal fibroblasts (47) and tumor-associated macrophages and neutrophils originated from circulating CD11b+Gr1+ cells (48), can be immunosuppressive (49–52), and their modulation frequently increases immune rejection of tumors (53–55). CD11b+Gr1+ cells increased in the circulation with progressive tumor growth (Fig. 2A), as expected from studies published decades ago (56–59). The adoptively transferred CD4+ T cells together with the rescued CD8+ T cells eradicated the long-established cancers despite the prominence of myeloid-derived suppressor cells (MDSCs) in the tumor-bearing hosts, in agreement with our recent findings in a different tumor model of ATT (60). Here, in contrast to CD4+ T-cell transfer, a single or repeated treatment of the tumor-bearing mice with anti-Gr1 antibody not only failed to prevent relapse, but even failed to slow down the growth of the relapsing tumors (Suppl. Fig. S3B).
A single dose of adoptively transferred CD4+ T cells was more effective than other strategies including checkpoint inhibition by anti-PD-L1 blockade, depletion of immunosuppressive MDSC or immunostimulatory local high-dose ionizing radiation (17, 61), to help existing dysfunctional CD8+ T cells eradicate the tumor. CD4+ T-cell transfer led to tumor eradication at any time point tested, contrary to other studies where CD4+ T cells needed to be present early after adoptive transfer of CD8+ T cells to be effective (35). This suggests that in our model, CD4+ T cells were not just preventing exhaustion, but reversing it. It may seem surprising that blocking the PD1/PD-L1 pathway did not prevent the persistent cancers from escaping growth control even though anti–PD-L1 increased the production of TNF by T cells re-isolated from relapsing tumors in vitro (Suppl. Fig. 2). Blocking the PD1/PD-L1 pathway as the only treatment can also fail in experimental advanced melanoma (62) and experimental ovarian cancers (63), similarly to what was observed in chronic infectious disease (21). Additional treatments may be required to make PD1/PD-L1 pathway blocking effective in advanced tumors, such as simultaneous intravenous infusion of attenuated Salmonella typhimurium, which will localize to tumor tissue (62). Furthermore, blockade of TIM-3, that we found to be ineffective in restoring cytokine production in exhausted T cells in vitro, may nevertheless lead to tumor control and restore cytokine production in vivo particularly when combined with PD1/PD-L1 blockade. Alternatively, local high-dose ionizing radiation can be combined with PD1/PD-L1 blockade to achieve greater therapeutic effects (17, 61). We also do not know whether the CD4+ T cells would require PD1/PD-L1 blockade as additional treatment if these T cells were not alloantigen-reactive, for example if a single mutant tumor-specific Ag was targeted. This is suggested by studies on a murine model of chronic infection by LCMV with “helpless” CD8 T cells (64). In such model, PD1/PD-L1 blockade by itself failed to rescue CD8+ T cells at late stages of infection (41, 64), but in combination with monoclonal naïve virus-specific CD4+ T cells, anti-PD-L1 achieved the greatest improvement of CD8+ T-cell function and viral load control, including the elimination of the virus in a fraction of mice. Therefore it would be possible that our tumor model reflects the severely exhausted/late stage CD8+ T cells, and rescue requires either monoclonal CD4+ T cells with checkpoint inhibitor adjuvant therapy or a polyclonal CD4+ T-cell response like ours.
Lack of “help” at the site of cancer growth may be an important reason for the inability of CD8+ T cells to reject established tumors. This situation might be somewhat analogous to transgenic mice that express allogeneic MHC class I molecules as self-antigen on islet cells and have autoreactive T cells that infiltrate the islets (65). In these mice, even after priming, the autoreactive T cells cannot destroy the islet cells unless local help is provided in the form of IL2 (65). Local help is also essential for breaking immune privilege in the eye (66). In our model, the mechanism of CD4+ T cell-mediated rejection functioned at least in part by providing help to the exhausted/dysfunctional tumor-infiltrating CD8+ T cells which increased their production of TNF and IFNγ several fold. Help of CD4+ to CD8+ T cells could be taking place through an improved APC function mediated by CD40/CD40L (67, 68) or the production of IL2 (29). Previous studies using early/developing tumors have reported increased numbers of tumor-specific CD8+ T cells in the periphery (35, 69) and in the tumor (29, 68, 69) if CD4+ T cells are co-transferred with CD8+ T cells. However, when established tumors were treated, increased numbers of intratumoral CD8+ T cells did not result in tumor regression (69), therefore, functional enhancement and not only an increase in the number of tumor-infiltrating CD8+ T cells might be required for elimination of established tumors.
Our proof-of-concept model targeted alloantigens on MHC-II+ tumor stroma while avoiding graft-versus-host disease because the alloantigen was only expressed by the C3H-derived cancer cells. Although alloantigens usually generate strong immune responses, tumor elimination upon transfer of CD4 T cells required the cooperation of tumor-specific CD8+ T cells, indicating that the alloantigen recognition by the CD4+ T cells alone was not sufficient for any therapeutic effects. We cannot exclude that the CD4+ T cells directly recognized (70) and killed MHC class II+ cancer cells (Suppl. Fig. 4B). However, such direct cancer cell recognition was insufficient since CD4+ T-cell transfer failed to achieve tumor eradication in MHC class II–negative tumor-bearing hosts (Fig. 6). Therefore, the B6 CD4+ T cells needed to recognize H-2k alloantigens released from the PRO4L-SIY-EGFP cancer cells and cross-presented on B6 MHC class II-positive stromal cells to rescue the CD8+ T cells and eradicate the cancers. Although the identity of the antigens recognized by CD4+ allogeneic T cells is still unclear, it has been shown that the indirect pathway of CD4+ T-cell allorecognition can provide sufficient help for effective cytotoxic CD8+ T-cell responses (71). We found that CD4+ and CD8+ epitopes must come from the same cancer cell for synergy of CD4+ T cells and CD8+ T cells at the effector phase and successful elimination of cancer inocula (28). This suggests that a very close proximity between APC and cancer cells expressing both CD4+ and CD8+ antigens might be required. In our model, cancer cells also express both the CD8+ and CD4+ T cell-recognized antigens. Therefore, CD4+ T cells and CD8+ T cells may also need to recognize epitopes presented by the same cell.
Our results are consistent with the findings from a clinical trial in which a patient first treated with TILs containing both CD8+ and CD4+ T cells needed a later infusion of tumor-specific CD4+ T cells to achieve the maximal reduction of tumor burden (2). Our observations suggest that the transferred CD4+ T cells possibly cooperated with the CD8+ T cells and could have rescued their function in the patient. Our results are also interesting with regards to findings in patients with hematopoietic cancers receiving allogeneic stem cell transplants, in whom relapse appeared to be prevented by donor CD4+ T cells responding to the mismatched alloantigens (72). Remarkably, the beneficial effect occurred in the absence or presence of GVHD (72). The danger of GVHD might be reduced or avoided by targeting minor histocompatibility Ags that are more highly expressed on the target cancer (73). Nevertheless, the best way to achieve tumor-specific targeting would be using truly cancer-specific Ags not expressed by normal cells of the patient. Proof that such Ags indeed exist initially came from studies demonstrating that tumor-specific Ags resulted from single amino acid substitutions caused by non-synonymous nucleotide substitutions in the genes of the cancer cells, absent from autologous normal tissues of the host (74). These finding were rapidly confirmed in human cancers (75, 76) and used as the basis of a clinical trial testing ATT of ex vivo–expanded mutation-specific CD4+ TILs (2). The mutant gene originally described by us (74) was a driver mutation (77) and could not be silenced with tumor progression because expression of the mutant Ag was essential for survival and the normal allele was lost (77). Such ideal targets may not be uncommon (77–80). Ongoing studies will determine whether the TCRs of CD4+ T cells with such specificities could be cloned, transduced into T cells, and re-infused into the tumor-bearing hosts to rescue exhausted CD8+ T cells in the tumor and eradicate the cancers.
Supplementary Material
Acknowledgments
We thank Donald A. Rowley, Ainhoa Pérez-Díez, Boris Engels, Matthias Leisegang, and Yang-Xin Fu for helpful discussion, and Justin Kline and Ana Anderson for reagents.
This work was supported by National Institute of Health grants R01-CA037156, R01-CA22677 and P01-CA74182, The Berlin Institute of Health, The Einstein Foundation and The University of Chicago Cancer Center Grant CA-14599 to HS, and in part by the Ludwig Foundation for Cancer Research at the University of Chicago to R.R.W. A.A. had a fellowship from Fundación Alfonso Martín Escudero (Spain).
Abbreviations
- B6
C57BL/6
- ATT
adoptive T-cell therapy
Footnotes
The authors have no conflicting financial interests.
References
- 1.Prasad V, Vandross A. Characteristics of Exceptional or Super Responders to Cancer Drugs. Mayo Clin Proc. 2015;90:1639–49. doi: 10.1016/j.mayocp.2015.08.017. [DOI] [PubMed] [Google Scholar]
- 2.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344:641–5. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seyal AR, Parekh K, Velichko YS, Salem R, Yaghmai V. Tumor growth kinetics versus RECIST to assess response to locoregional therapy in breast cancer liver metastases. Acad Radiol. 2014;21:950–7. doi: 10.1016/j.acra.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 4.Spiotto MT, Rowley DA, Schreiber H. Bystander elimination of antigen loss variants in established tumors. Nat Med. 2004;10:294–8. doi: 10.1038/nm999. [DOI] [PubMed] [Google Scholar]
- 5.Zhang B, Zhang Y, Bowerman NA, Schietinger A, Fu YX, Kranz DM, et al. Equilibrium between host and cancer caused by effector T cells killing tumor stroma. Cancer Res. 2008;68:1563–71. doi: 10.1158/0008-5472.CAN-07-5324. [DOI] [PubMed] [Google Scholar]
- 6.Ruiter DJ, Bergman W, Welvaart K, Scheffer E, van Vloten WA, Russo C, et al. Immunohistochemical analysis of malignant melanomas and nevocellular nevi with monoclonal antibodies to distinct monomorphic determinants of HLA antigens. Cancer Res. 1984;44:3930–5. [PubMed] [Google Scholar]
- 7.Smith ME, Marsh SG, Bodmer JG, Gelsthorpe K, Bodmer WF. Loss of HLA-A,B,C allele products and lymphocyte function-associated antigen 3 in colorectal neoplasia. Proc Natl Acad Sci U S A. 1989;86:5557–61. doi: 10.1073/pnas.86.14.5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Surmann EM, Voigt AY, Michel S, Bauer K, Reuschenbach M, Ferrone S, et al. Association of high CD4-positive T cell infiltration with mutations in HLA class II-regulatory genes in microsatellite-unstable colorectal cancer. Cancer Immunol Immunother. 2015;64:357–66. doi: 10.1007/s00262-014-1638-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33:1152–8. doi: 10.1038/nbt.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bodmer W, Bielas JH, Beckman RA. Genetic instability is not a requirement for tumor development. Cancer Res. 2008;68:3558–60. doi: 10.1158/0008-5472.CAN-07-6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loeb LA, Bielas JH, Beckman RA. Cancers exhibit a mutator phenotype: clinical implications. Cancer Res. 2008;68:3551–7. doi: 10.1158/0008-5472.CAN-07-5835. [DOI] [PubMed] [Google Scholar]
- 12.Wu C-I, Wang H-Y, Ling S, Lu X. The Ecology and Evolution of Cancer—The Ultra-Microevolutionary Process. Annu Rev Genet. 2016;50:347–69. doi: 10.1146/annurev-genet-112414-054842. [DOI] [PubMed] [Google Scholar]
- 13.Schreiber H. Cancer Immunology. In: Paul WE, editor. Fundamental Immunology. 7th. Philadelphia, PA: Lippincott-Williams & Wilkins; 2013. pp. 1200–34. [Google Scholar]
- 14.Singh S, Ross SR, Acena M, Rowley DA, Schreiber H. Stroma is critical for preventing or permitting immunological destruction of antigenic cancer cells. J Exp Med. 1992;175:139–46. doi: 10.1084/jem.175.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ochsenbein AF, Klenerman P, Karrer U, Ludewig B, Pericin M, Hengartner H, et al. Immune surveillance against a solid tumor fails because of immunological ignorance. Proc Natl Acad Sci U S A. 1999;96:2233–8. doi: 10.1073/pnas.96.5.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spiotto MT, Yu P, Rowley DA, Nishimura MI, Meredith SC, Gajewski TF, et al. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity. 2002;17:737–47. doi: 10.1016/s1074-7613(02)00480-6. [DOI] [PubMed] [Google Scholar]
- 17.Zhang B, Bowerman NA, Salama JK, Schmidt H, Spiotto MT, Schietinger A, et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med. 2007;204:49–55. doi: 10.1084/jem.20062056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang B, Karrison T, Rowley DA, Schreiber H. IFN-gamma- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J Clin Invest. 2008;118:1398–404. doi: 10.1172/JCI33522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 1993;362:758–61. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- 20.Wick M, Dubey P, Koeppen H, Siegel CT, Fields PE, Chen L, et al. Antigenic cancer cells grow progressively in immune hosts without evidence for T cell exhaustion or systemic anergy. J Exp Med. 1997;186:229–38. doi: 10.1084/jem.186.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 22.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–75. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol. 1994;68:8056–63. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keene JA, Forman J. Helper activity is required for the in vivo generation of cytotoxic T lymphocytes. J Exp Med. 1982;155:768–82. doi: 10.1084/jem.155.3.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schietinger A, Philip M, Liu RB, Schreiber K, Schreiber H. Bystander killing of cancer requires the cooperation of CD4(+) and CD8(+) T cells during the effector phase. J Exp Med. 2010;207:2469–77. doi: 10.1084/jem.20092450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res. 2010;70:8368–77. doi: 10.1158/0008-5472.CAN-10-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–6. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 31.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–9. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 32.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol. 2004;5:927–33. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bourgeois C, Rocha B, Tanchot C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science. 2002;297:2060–3. doi: 10.1126/science.1072615. [DOI] [PubMed] [Google Scholar]
- 34.Greenberg PD, Cheever MA, Fefer A. Eradication of disseminated murine leukemia by chemoimmunotherapy with cyclophosphamide and adoptively transferred immune syngeneic Lyt-1+2- lymphocytes. J Exp Med. 1981;154:952–63. doi: 10.1084/jem.154.3.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Church SE, Jensen SM, Antony PA, Restifo NP, Fox BA. Tumor-specific CD4+ T cells maintain effector and memory tumor-specific CD8+ T cells. Eur J Immunol. 2014;44:69–79. doi: 10.1002/eji.201343718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greenberg PD, Kern DE, Cheever MA. Therapy of disseminated murine leukemia with cyclophosphamide and immune Lyt-1+,2- T cells. Tumor eradication does not require participation of cytotoxic T cells. J Exp Med. 1985;161:1122–34. doi: 10.1084/jem.161.5.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perez-Diez A, Joncker NT, Choi K, Chan WF, Anderson CC, Lantz O, et al. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood. 2007;109:5346–54. doi: 10.1182/blood-2006-10-051318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu HM, Winter H, Urba WJ, Fox BA. Divergent roles for CD4+ T cells in the priming and effector/memory phases of adoptive immunotherapy. J Immunol. 2000;165:4246–53. doi: 10.4049/jimmunol.165.8.4246. [DOI] [PubMed] [Google Scholar]
- 39.Grusby MJ, Johnson RS, Papaioannou VE, Glimcher LH. Depletion of CD4+ T cells in major histocompatibility complex class II-deficient mice. Science. 1991;253:1417–20. doi: 10.1126/science.1910207. [DOI] [PubMed] [Google Scholar]
- 40.Stauss HJ, Van Waes C, Fink MA, Starr B, Schreiber H. Identification of a unique tumor antigen as rejection antigen by molecular cloning and gene transfer. J Exp Med. 1986;164:1516–30. doi: 10.1084/jem.164.5.1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2010;107:14733–8. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–94. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207:2175–86. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–27. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teshima T, Ordemann R, Reddy P, Gagin S, Liu C, Cooke KR, et al. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med. 2002;8:575–81. doi: 10.1038/nm0602-575. [DOI] [PubMed] [Google Scholar]
- 46.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207:637–50. doi: 10.1084/jem.20091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arina A, Idel C, Hyjek EM, Alegre ML, Wang Y, Bindokas VP, et al. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc Natl Acad Sci U S A. 2016;113:7551–6. doi: 10.1073/pnas.1600363113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A. 2012;109:2491–6. doi: 10.1073/pnas.1113744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–30. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 50.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–94. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu YX, Watson G, Jimenez JJ, Wang Y, Lopez DM. Expansion of immunoregulatory macrophages by granulocyte-macrophage colony-stimulating factor derived from a murine mammary tumor. Cancer Res. 1990;50:227–34. [PubMed] [Google Scholar]
- 52.Torroella-Kouri M, Silvera R, Rodriguez D, Caso R, Shatry A, Opiela S, et al. Identification of a subpopulation of macrophages in mammary tumor-bearing mice that are neither M1 nor M2 and are less differentiated. Cancer Res. 2009;69:4800–9. doi: 10.1158/0008-5472.CAN-08-3427. [DOI] [PubMed] [Google Scholar]
- 53.Ibe S, Qin Z, Schuler T, Preiss S, Blankenstein T. Tumor rejection by disturbing tumor stroma cell interactions. J Exp Med. 2001;194:1549–59. doi: 10.1084/jem.194.11.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schuler T, Kornig S, Blankenstein T. Tumor rejection by modulation of tumor stromal fibroblasts. J Exp Med. 2003;198:1487–93. doi: 10.1084/jem.20030849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Medina-Echeverz J, Fioravanti J, Zabala M, Ardaiz N, Prieto J, Berraondo P. Successful colon cancer eradication after chemoimmunotherapy is associated with profound phenotypic change of intratumoral myeloid cells. J Immunol. 2011;186:807–15. doi: 10.4049/jimmunol.1001483. [DOI] [PubMed] [Google Scholar]
- 56.Milas L, Basic I. Stimulated granulocytopoiesis in mice bearing fibrosarcoma. Eur J Cancer. 1972;8:309–13. doi: 10.1016/0014-2964(72)90026-6. [DOI] [PubMed] [Google Scholar]
- 57.Delmonte L, Liebelt RA. Granulocytosis-Promoting Extract of Mouse Tumor Tissue: Partial Purification. Science. 1965;148:521–3. doi: 10.1126/science.148.3669.521. [DOI] [PubMed] [Google Scholar]
- 58.Hibberd AD, Metcalf D. Proliferation of macrophage and granulocyte precursors in response to primary and transplanted tumors. Isr J Med Sci. 1971;7:202–10. [PubMed] [Google Scholar]
- 59.Balducci L, Hardy C. High proliferation of granulocyte-macrophage progenitors in tumor-bearing mice. Cancer Res. 1983;43:4643–7. [PubMed] [Google Scholar]
- 60.Arina A, Schreiber K, Binder DC, Karrison TG, Liu RB, Schreiber H. Adoptively transferred immune T cells eradicate established tumors despite cancer-induced immune suppression. J Immunol. 2014;192:1286–93. doi: 10.4049/jimmunol.1202498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deng L, Liang H, Burnette B, Weicheslbaum RR, Fu YX. Radiation and anti-PD-L1 antibody combinatorial therapy induces T cell-mediated depletion of myeloid-derived suppressor cells and tumor regression. Oncoimmunology. 2014;3:e28499. doi: 10.4161/onci.28499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Binder DC, Engels B, Arina A, Yu P, Slauch JM, Fu YX, et al. Antigen-specific bacterial vaccine combined with anti-PD-L1 rescues dysfunctional endogenous T cells to reject long-established cancer. Cancer Immunol Res. 2013;1:123–33. doi: 10.1158/2326-6066.CIR-13-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Binder DC, Schreiber H. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors–letter. Cancer Res. 2014;74:632. doi: 10.1158/0008-5472.CAN-13-2216. discussion 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aubert RD, Kamphorst AO, Sarkar S, Vezys V, Ha SJ, Barber DL, et al. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc Natl Acad Sci U S A. 2011;108:21182–7. doi: 10.1073/pnas.1118450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heath WR, Allison J, Hoffmann MW, Schonrich G, Hammerling G, Arnold B, et al. Autoimmune diabetes as a consequence of locally produced interleukin-2. Nature. 1992;359:547–9. doi: 10.1038/359547a0. [DOI] [PubMed] [Google Scholar]
- 66.Streilein JW, Ksander BR, Taylor AW. Immune deviation in relation to ocular immune privilege. J Immunol. 1997;158:3557–60. [PubMed] [Google Scholar]
- 67.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–3. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 68.Shafer-Weaver KA, Watkins SK, Anderson MJ, Draper LJ, Malyguine A, Alvord WG, et al. Immunity to murine prostatic tumors: continuous provision of T-cell help prevents CD8 T-cell tolerance and activates tumor-infiltrating dendritic cells. Cancer Res. 2009;69:6256–64. doi: 10.1158/0008-5472.CAN-08-4516. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Marzo AL, Kinnear BF, Lake RA, Frelinger JJ, Collins EJ, Robinson BW, et al. Tumor-specific CD4+ T cells have a major “post-licensing” role in CTL mediated anti-tumor immunity. J Immunol. 2000;165:6047–55. doi: 10.4049/jimmunol.165.11.6047. [DOI] [PubMed] [Google Scholar]
- 70.Armstrong TD, Clements VK, Ostrand-Rosenberg S. MHC class II-transfected tumor cells directly present antigen to tumor-specific CD4+ T lymphocytes. J Immunol. 1998;160:661–6. [PubMed] [Google Scholar]
- 71.Lee RS, Grusby MJ, Glimcher LH, Winn HJ, Auchincloss H., Jr Indirect recognition by helper cells can induce donor-specific cytotoxic T lymphocytes in vivo. J Exp Med. 1994;179:865–72. doi: 10.1084/jem.179.3.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rutten CE, van Luxemburg-Heijs SA, Halkes CJ, van Bergen CA, Marijt EW, Oudshoorn M, et al. Patient HLA-DP-specific CD4+ T cells from HLA-DPB1-mismatched donor lymphocyte infusion can induce graft-versus-leukemia reactivity in the presence or absence of graft-versus-host disease. Biol Blood Marrow Transplant. 2013;19:40–8. doi: 10.1016/j.bbmt.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 73.Griffioen M, van Bergen CA, Falkenburg JH. Autosomal Minor Histocompatibility Antigens: How Genetic Variants Create Diversity in Immune Targets. Front Immunol. 2016;7:100. doi: 10.3389/fimmu.2016.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Monach PA, Meredith SC, Siegel CT, Schreiber H. A unique tumor antigen produced by a single amino acid substitution. Immunity. 1995;2:45–59. doi: 10.1016/1074-7613(95)90078-0. [DOI] [PubMed] [Google Scholar]
- 75.Coulie PG, Lehmann F, Lethe B, Herman J, Lurquin C, Andrawiss M, et al. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc Natl Acad Sci U S A. 1995;92:7976–80. doi: 10.1073/pnas.92.17.7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–4. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- 77.Beck-Engeser GB, Monach PA, Mumberg D, Yang F, Wanderling S, Schreiber K, et al. Point mutation in essential genes with loss or mutation of the second allele: relevance to the retention of tumor-specific antigens. J Exp Med. 2001;194:285–300. doi: 10.1084/jem.194.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. 2005;123:49–63. doi: 10.1016/j.cell.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 79.Chen J, Guo K, Kastan MB. Interactions of nucleolin and ribosomal protein L26 (RPL26) in translational control of human p53 mRNA. J Biol Chem. 2012;287:16467–76. doi: 10.1074/jbc.M112.349274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim TH, Leslie P, Zhang Y. Ribosomal proteins as unrevealed caretakers for cellular stress and genomic instability. Oncotarget. 2014;5:860–71. doi: 10.18632/oncotarget.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.