

Abstract
The mechanisms that govern node of Ranvier organization, stability, and long-term maintenance remain to be fully elucidated. One of the molecular components of the node is the cytoskeletal scaffolding protein, ankyrin G (AnkG), which interacts with multiple members of the nodal complex. The role of AnkG in nodal organization and maintenance is still not clearly defined as to whether AnkG functions as an initial nodal organizer or whether it functions as a nodal stabilizer after the nodal complex has been assembled. Using a mouse model system, we report here that perinatal and juvenile neuronal ablation of AnkG has differential consequences on nodal stability. Early loss of AnkG creates immature nodes with abnormal morphology, which undergo accelerated destabilization within a month, resulting in rapid voltage-gated sodium (NaV) channel and βIV spectrin loss with reduced effects on neurofascin 186. On the other hand, late ablation of AnkG from established nodal complexes leads to slow but progressive nodal destabilization over 10 months, primarily affecting βIV spectrin, followed by NaV channels, with modest impact on neurofascin 186. We also show that ankyrin R and βI spectrin are not sufficient to prevent nodal disorganization after AnkG ablation. Additionally, nodal disorganization in both early and late AnkG mutants is accompanied by axonal pathology and neurological dysfunction. Together, our results suggest that AnkG plays an indispensable role in the maturation and long-term stabilization of the newly assembled nodal complex, and that loss of AnkG after nodal stabilization does not lead to rapid nodal disassembly but to loss of specific nodal components in a time-dependent manner.
SIGNIFICANCE STATEMENT Nodes of Ranvier are the myelin-free gaps along myelinated axons that allow fast communication between neurons and their target cells by propagating action potentials in a saltatory manner. The cytoskeletal scaffolding protein ankyrin G (AnkG) has been thought to play an important role in node formation; however, its precise role in nodal assembly, stability, and maintenance is still not clear. By using spatiotemporal ablation of AnkG, we report its differential role in nodal maturation and stabilization. We show that early AnkG-deficient nodes fail to mature and undergo rapid destabilization. In contrast, nodes that assemble with AnkG are much more stable and undergo gradual disintegration with sequential loss of nodal components in the absence of AnkG.
Keywords: ankyrin G, axonal domains, myelin, nerve conduction, neurofascin, nodes of Ranvier
Introduction
Myelination of axons evolved to allow propagation of neuronal impulses to long distances in a saltatory manner. This instructive process also created unique anatomical domains with distinct molecular composition along the axons (Buttermore et al., 2013). These domains are nodes of Ranvier flanked by the paranodal regions, which are further bordered by juxtaparanodes followed by the internodes. The main molecular areas where the action potentials are regenerated and transmitted along the axon are the nodes of Ranvier, which are myelin-free specialized structures distributed along the axons. During early postnatal development, nodes become highly enriched in voltage-gated sodium (NaV) channels, which are externally stabilized via interactions with the extracellular matrix through cell-adhesion molecules [neurofascin 186 (NF186) and NrCAM], as well as intracellularly by scaffolding proteins [ankyrin G (AnkG) and βIV spectrin (βIV Spec)] that anchor them with the axonal actin cytoskeleton (Kordeli et al., 1995; Davis et al., 1996; Lambert et al., 1997; Komada and Soriano, 2002; Dzhashiashvili et al., 2007; Feinberg et al., 2010; Thaxton et al., 2011; Susuki et al., 2013). Even though nodal complexes in the PNS and CNS share key molecular players, mechanisms that govern nodal organization are thought to be distinct (Jenkins and Bennett, 2001; Eshed et al., 2005; Sherman et al., 2005; Salzer et al., 2008; Bekku et al., 2009). However, prenatal loss of NF186 in the PNS and CNS abolishes node formation, suggesting that NF186 plays a prominent role in nodal organization (Thaxton et al., 2011).
The cytoskeletal scaffolding protein AnkG is highly enriched at the nodes and had been suggested to play an essential role in nodal formation (Jenkins and Bennett, 2002; Dzhashiashvili et al., 2007; Jenkins et al., 2015). However, recent studies indicate that AnkG-deficient nodes are capable of clustering other nodal proteins along the axons (Ho et al., 2014). Moreover, AnkG ablation in the CNS (Ho et al., 2014) from adult myelinated axons failed to show nodal destabilization at least within a range of 2 months after AAV-Cre induced recombination. Therefore, there is still much speculation about whether this protein plays an instructive role in the initial nodal formation or is required for long-term maintenance and nodal stability.
In the current study, we have addressed differential consequences of perinatal and juvenile loss of AnkG on nodal development and maintenance. Perinatal tamoxifen-induced loss of AnkG does not affect the initial recruitment of nodal proteins but does affect the morphology of the newly assembled nodal complexes. These early AnkG-deficient nodes undergo accelerated destabilization within a month after AnkG ablation. Juvenile loss of AnkG caused slow but progressive destabilization of nodes over a period of 10 months after its ablation, with a loss of specific nodal components in a time-dependent manner. Interestingly, the loss of AnkG either during perinatal or juvenile stages altered the electrical properties and also caused ultrastructural disorganization of myelinated axons, leading to severe neurological phenotypes and eventual lethality. Together, our AnkG ablation studies reveal that the early function of AnkG is to create a proper scaffold of the newly formed nodal complex and allow its maturation. In contrast, nodes that are assembled with AnkG remain stable for extended periods of time even after AnkG loss, suggesting its role in the long-term maintenance of the node as a stabilizer by anchoring the nodal complex to the underlying axonal cytoskeleton.
Materials and Methods
Animals.
All animal experiments were performed according to the University of Texas Health Science Center Institutional Animal Care and Use Committee-approved guidelines for the ethical treatment of laboratory animals. The Ankyrin 3flox conditional knock-out mouse line (RRID:MGI:5538267) for AnkG ablation was provided by Vann Bennett (Duke University, Durham, NC; Paez-Gonzalez et al., 2011). The Single-neuron Labeling with Inducible Cre-mediated Knock-out (SLICK)-H-CreER (derivative of Thy1.2-CreER) transgenic mouse line (RRID:IMSR_JAX:012708) was generously provided by Paul Young (Young et al., 2008). The Actin-Cre transgenic mouse line (RRID:IMSR_JAX:019099) was obtained from the The Jackson Laboratory (stock #019099). Mouse lines used in these studies were on a mixed C57BL/6 and 129/Sv genetic background. To obtain Ankfx/− mice, Ankfx/fx and Actin-Cre animals were mated, which caused ubiquitous recombination of the fx allele and loss of one AnkG copy in Actin-Cre;Ank−/+ pups, where (−) represents germline recombination of the AnkG-floxed allele. Ankfx/− animals were crossed to SLICK-H-CreER;Ankfx/fx strain to obtain SLICK-H-CreER;Ankfx/− males, which were mated with Ankfx/fx females to get SLICK-H-CreER;Ankfx/− mutants and Ankfx/− as littermate controls for further experiments.
Tamoxifen injections.
For perinatal ablation of AnkG from neurons, Ankfx/fx females were bred to SLICK-H-CreER;Ankfx/− males. An initial dose of 5 mg of tamoxifen was administered to pregnant females by intraperitoneal injections at gestational day 19 [equivalent to embryonic day 19 (E19); Agarwal et al., 2012], which was followed by intraperitoneal injection of 3 mg of tamoxifen to lactating dams for 5 consecutive days starting from postnatal day 0 (P0). To study node formation at P3, we followed the same perinatal protocol where the mother received tamoxifen at E19, P0, P1, and P2 without being exposed to the final two doses at P3 and P4. For adult ablation of AnkG, mice received 1 mg of tamoxifen by intraperitoneal injection for 10 consecutive days from P23 to P32 (Pillai et al., 2009).
Antibodies.
AnkG polyclonal antibodies in rabbit and rat were generated against a polypeptide from amino acids TEDK to KKTH (Thaxton et al., 2010). Pan-NaV channel polyclonal antibodies in rabbit were made against a polypeptide from amino acids FNQQ to AFDI of the sodium channel NaV1.6. Guinea pig and rabbit anti-βIV Spec polyclonal antibodies were made against a polypeptide from amino acids ARRA to QESA. Guinea pig anti-NF186 (Thaxton et al., 2011), guinea pig and rabbit anti-Caspr (Bhat et al., 2001) antibodies were described previously. Other primary antibodies used in this study were mouse anti-α-tubulin (catalog #12G10, DSHB; RRID:AB_1157911), rabbit anti-myelin basic protein (MBP; catalog #ab40390, Abcam; RRID:AB_1141521), mouse anti-Caspr [catalog #75–001, University of California (UC) Davis/National Institutes of Health (NIH) NeuroMab Facility; RRID:AB_2083496], rabbit anti-Caspr2 (catalog #ab33994, Abcam; RRID:AB_2083506), mouse anti-Kv1.2 (catalog #75–008, UC Davis/NIH NeuroMab Facility; RRID:AB_2296313), mouse anti-AnkR (catalog #75–380, UC Davis/NIH NeuroMab Facility; RRID:AB_2491109), and rabbit anti-βI Spec (obtained from M. Stankewich, Yale University, CT). Fluorophore-conjugated secondary antibodies used for immunofluorescence and HRP-conjugated secondary antibodies used for immunoblotting were purchased from Invitrogen. Infrared (IR)-conjugated secondary antibodies used for immunoblotting were purchased from LI-COR.
Tissue preparation and immunostaining.
Animals were anesthetized with intraperitoneal injection of Avertin (400 mg/kg mouse body weight; T48402, Sigma-Aldrich) and transcardially perfused with PBS (pH 7.2–7.4) followed by ice-cold 1% paraformaldehyde (PFA) and 1% sucrose in 0.1 m pH 7.2–7.4 phosphate buffer (PB). The spinal cords (SCs) were dissected out and postfixed in the same fixative for 2 h at 4°C and then immersed in 30% sucrose in 0.1 m PB until it settled at the bottom. The tissue was rinsed several times in PBS and frozen in Tissue-Tek O.C.T. Compound (Sakura Finetek). Longitudinal 14 μm sections were cut with a cryostat (Leica), mounted on slides, and processed for immunostaining. Sciatic nerves (SNs) were dissected out from anesthetized animals, fixed in 4% PFA for 30 min, washed extensively in PBS, teased into individual nerve fibers, dried overnight at room temperature, and stored at −80°C. Immunostaining of all samples was performed as previously described (Thaxton et al., 2011).
Immunoblotting.
Mouse SCs and SNs were dissected out and homogenized with glass mortar and pestle on ice in lysis buffer (50 mm Tris-HCl, pH 7.5, 50 mm NaCl, 10 mm EDTA, 1% Triton X-100, and a protease inhibitor mixture tablet; Roche Diagnostics). Homogenized samples were incubated at 4°C followed by centrifugation at 20,000 × g at 4°C for 30 min. Supernatant was collected as a final lysate, heated for 5 min at 37°C (to detect AnkG or pan-NaV channels) or boiled (for all other proteins), loaded onto SDS-PAGE gel, transferred to nitrocellulose, and probed with primary antibodies as previously described (Thaxton et al., 2011). Afterward, membranes were incubated in HRP- or IR-conjugated secondary antibodies followed by detection of the probes. Protein concentration in samples was estimated using a Pierce BCA protein assay kit (Thermo Scientific).
Transmission electron microscopy.
Animals were anesthetized and transcardially perfused with normal saline followed by 2.5% glutaraldehyde/4% PFA (dissolved in 0.16 m NaH2PO4/0.11 m NaOH buffer, to a final of pH 7.2–7.4) EM fixative for 30 min. All solutions were freshly prepared on the day of perfusion. After perfusion, entire mouse carcasses were postfixed for 2 weeks in the same EM fixative. SNs and SCs then were dissected out and incubated overnight in 0.1 m sodium cacodylate buffer followed by incubation in a 2% OsO4 solution and gradient ethanol dehydration. Samples were incubated in propylene oxide, left in 100% PolyBed resin with constant agitation for 36 h and embedded in flat molds at 55°C for 36 h. After embedding, the molds were processed and imaged on a JEOL 1230 electron microscope at the University of Texas Health Science Center, San Antonio, Electron Microscopy Laboratory, as previously described (Green et al., 2013).
In vivo nerve conduction measurements.
All nerve conduction studies were performed under isoflurane anesthesia, and mouse external body temperature was constantly maintained between 33°C and 34°C with a warming lamp. Subdermal stainless steel needle electrodes were used for nerve stimulation with 0.02 ms impulses using the Nicolet Teca Synergy portable neurological system (Natus Neurology). Nerve conduction velocity (NCV) and amplitudes in nerves of the tail and SN in the hindlimb were measured as previously described (Oh et al., 2010; Walsh et al., 2015). Briefly, for each trace, amplitude was measured as the difference in millivolts from the onset to the peak of the compound action potential (CAP), and NCV was calculated by the distance divided by the latency. To determine the sciatic NCV, the distance between the notch and ankle divided by the difference between the notch and ankle latencies was used.
Quantification and statistics.
For each experiment, SC sections and teased SNs from at least three animals per group were immunostained. Consistently throughout the study, lateral tracts of the cervical region of the SC and distal SN were analyzed. In all experiments, the number of independent measurements (n) represents the number of animals. Images were acquired by use of Zeiss LSM 710 confocal microscope with 40× magnification objective using identical settings. All nodes/heminodes flanked by Caspr immunostaining were quantified within each field of view. For each animal, the following number of nodes was quantified in SNs and SCs: 40 nodes for P3; 60–80 nodes for P5–P30 for the early AnkG ablation; and 120–130 nodes for the late AnkG model. Data measurements and analyses were performed by one examiner in a nonblinded manner.
Protein intensities were measured over the entire nodal area, which was defined by Caspr-positive paranodal immunostaining, and were selected manually using the freeform drawing tool in ImageJ software (RRID:SCR_003070). To calculate corrected values of the nodal fluorescence, mean background readings were taken and subtracted from the integrated density values for each node. The following formula was used for calculations: corrected nodal fluorescence = integrated density − (area of selected node × mean fluorescence of background readings; Burgess et al., 2010; Gavet and Pines, 2010). Mean corrected nodal fluorescence values were averaged from three control and three AnkG knock-out animals, and the final results were standardized to control values. All animals and samples within the same age group were processed at the same time (perfused, sectioned, immunostained, and imaged), and statistically significant differences between mutant and control groups of the same time point were determined by unpaired, two-tailed Student's t test.
The percentage of positive normal (staining equally distributed over the nodal area) and positive nodes with abnormal morphology (patchy staining) were calculated over the total number of nodal gaps (defined by Caspr immunostaining) in the entire field of view, and the mean values from three animals were plotted into the graphs. Two-way ANOVA with Bonferroni's post hoc test was used for multiple comparison of groups within the same genotype, as well as between different groups of mice of various genotypes. All data are presented as the mean ± SEM using GraphPad Prism5 software (RRID:SCR_002798) and are represented as follows: *p < 0.05; **p < 0.01; ***p < 0.001.
Results
Early loss of AnkG affects nodal maturation and causes enhanced destabilization of nodal proteins
Recent studies have suggested that AnkG is not essential for node formation and that other cytoskeletal proteins at the node may compensate for the loss of AnkG (Ho et al., 2014). Ablation of AnkG during early development using pan-neuronal driver Nestin (Nes)-Cre causes lethality within a day after birth (data not shown; Ho et al., 2014). This early lethality made it difficult to establish the contribution of AnkG in nodal domain organization and other neuronal functions during postnatal development. To overcome the issue of early lethality and to generate animals that lack AnkG from late embryonic stages into postnatal development, we created an early tamoxifen-inducible AnkG model using SLICK-H-CreER mice, which express a tamoxifen-inducible Cre recombinase (CreER) under the neuronal specific promoter Thy1.2 (Young et al., 2008). Pregnant Ankfx/fx females mated with SLICK-H-CreER;Ankfx/− males were injected with an initial 5 mg dose of tamoxifen during late pregnancy (at approximately gestational day 19). After delivery, lactating mothers received injections of 3 mg of tamoxifen for 5 consecutive days for oral consumption of tamoxifen by the progeny through milk (schematized in Fig. 1A). Pups were analyzed for various phenotypes at the time points shown in Figure 1A. SLICK-H-CreER;Ankfx/− animals were significantly smaller and became distinguishable from littermate controls by P10. These animals developed tremors and ataxia and, on average, barely survived to P40 (Fig. 1B,C). SC and SN lysates from P5 SLICK-H-CreER;Ankfx/− mice showed a reduction of the 270 and 480 kDa bands of AnkG neuronal isoforms (Fig. 1D). Immunostaining of teased SNs (Fig. 1E,F) and SCs (Fig. 1G,H) from P5 SLICK-H-CreER;Ankfx/− animals using anti-AnkG (red) and anti-Caspr (green) antibodies showed an absence of AnkG at the nodes (Fig. 1F,H). Quantification of immunoreactivity for AnkG in SNs and SCs showed 83.4% and 71.3% of nodes without AnkG, respectively, in SLICK-H-CreER;Ankfx/− from different litters, compared with Ankfx/− controls (Fig. 1I,J). Even though the same amount of tamoxifen was injected into different females during their pregnancy (5 mg) and lactating period (3 mg × 5 d), there was no greater variation observed between littermates than pups from different litters of the same genotypes, which indicates similar recombination efficiencies across experiments.
Figure 1.
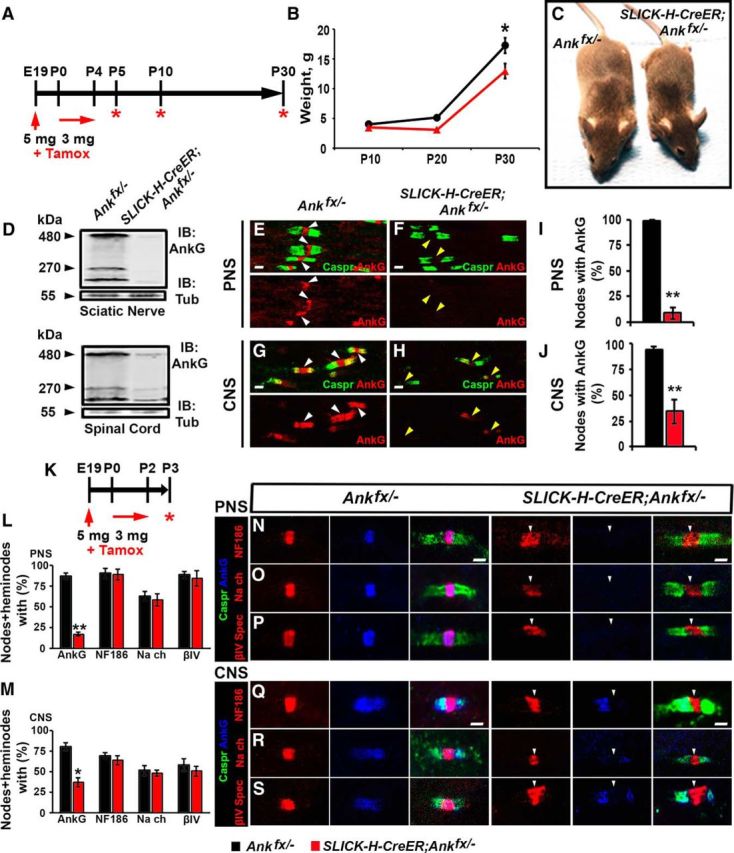
Perinatal neuron-specific AnkG ablation model. A, Schematic representation of tamoxifen injection for early AnkG ablation from neurons. Pregnant females were injected with a single dose of tamoxifen at E19, followed by daily intraperitoneal injections of tamoxifen to lactating dams for 5 consecutive days starting from the day pups were born. SLICK-H-CreER;Ankfx/− mutant and Ankfx/− control littermates were analyzed at P5, P10, and P30. B, Weight curves of Ankfx/− and SLICK-H-CreER;Ankfx/− male littermates (n = 6 mice/genotype; two-way ANOVA, Bonferroni's post hoc analysis). C, Photographic depiction of P30 SLICK-H-CreER;Ankfx/− and Ankfx/− mice after AnkG ablation by tamoxifen. D, Immunoblot analysis of SN and SC lysates from P5 Ankfx/− and SLICK-H-CreER;Ankfx/− mice with antibodies against AnkG and tubulin. E–H, Immunostaining of P5 teased SN fibers (E, F) and SCs (G, H) with antibodies against Caspr (green) and AnkG (red). White and yellow arrowheads indicate AnkG-positive and AnkG-negative nodes, respectively. I, J, Quantification of the percentage of nodes with AnkG in P5 Ankfx/− (black bar) and SLICK-H-CreER;Ankfx/− (red bar) SNs and SCs, respectively (n = 3 mice/genotype; 60–80 nodes/animal; unpaired, two-tailed Student's t test). K, Schematic of shortened tamoxifen injection protocol for perinatal AnkG ablation from neurons used for analysis of nodal formation at P3. L, M, Percentage of nodes and heminodes in SNs and SCs, respectively, with AnkG, NF186, pan-NaV, and βIV Spec at P3 in SLICK-H-CreER;Ankfx/− mice compared with Ankfx/− controls (n = 3 mice/genotype; 40 nodes/animal; unpaired, two-tailed Student's t test). N–S, Immunostaining of SN (N–P) and SC (Q–S) fibers from P3 age-matched Ankfx/− and SLICK-H-CreER;Ankfx/− littermate mice against AnkG (blue) in combination with either of the following proteins: NF186 (N, Q), pan-NaV (O, R), or βIV Spec (P, S; red), plus Caspr (green). Arrowheads indicate AnkG-negative nodes. Scale bar, 2 μm. All data are represented as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
To evaluate the earliest nodal phenotypes, we analyzed P3 pups from a perinatal tamoxifen delivery protocol (schematized in Fig. 1K). Immunostaining of SN and SC myelinated fibers revealed 70.04% and 43.25% reductions in AnkG-positive nodes in the PNS and CNS of P3 SLICK-H-CreER;Ankfx/− mutant mice, respectively, compared with Ankfx/− control littermates. There were no statistically significant differences in the percentages of NF186-, βIV Spec-, and pan-NaV-positive nodes and heminodes in SNs and SCs between P3 control and SLICK-H-CreER;Ankfx/− littermates (Fig. 1L,M). However, nodes and heminodes missing AnkG in SLICK-H-CreER;Ankfx/− myelinated axons contained NF186, βIV Spec, and pan-NaV that showed abnormal morphology at the nodal areas (Fig. 1N–S), indicating that these proteins are recruited into the node independent of AnkG in both the PNS and CNS; however, their proper assembly within the nodal area is affected.
To further establish the consequences of early loss of AnkG on nodal maturation and stability, we analyzed both Ankfx/− and SLICK-H-CreER;Ankfx/− mice at P5, P10, and P30 after the last tamoxifen injection was given at P4. We continued to analyze the localization of key nodal proteins, NF186, βIV Spec, and pan-NaV channels in teased SNs and sliced SC sections. In P5 SLICK-H-CreER;Ankfx/− SNs, AnkG is absent from the nodes (Fig. 2A,D,G). In the absence of AnkG, at this stage most nodes contained NF186, βIV Spec, and pan-NaV channels (Fig. 2A,D,G), although their clustering was disrupted and diffuse with a lack of strictly defined boundaries compared with controls. At P5 in SNs, quantification showed 16.69%, 28.11%, and 63.48% reductions, respectively, in NF186, βIV Spec, and pan-NaV channel intensities (Fig. 2J–L) at the nodal gap of SLICK-H-CreER;Ankfx/− fibers compared with Ankfx/−. At P10 in SLICK-H-CreER;Ankfx/− SNs, AnkG is absent, as expected; however, the localization of other nodal proteins remained altered in SLICK-H-CreER;Ankfx/− fibers, as the majority of the immunostaining was diffuse and redistributed into areas away from the nodal region. Remaining nodal markers NF186, βIV Spec, and pan-NaV channels showed more profound destabilization (Fig. 2B,E,H), which is reflected by significantly reduced intensity, by 34.27%, 43.75%, and 76.93%, respectively (Fig. 2J–L), compared with control values in Ankfx/− fibers. By P30, nodal proteins underwent more progressive destabilization with NF186, βIV Spec, and pan-NaV channels showing diffuse and scattered immunostaining away from the nodal areas (Fig. 2C,F,I) and significantly decreased intensity (by 44.87%, 50.7%, and 75.34%, respectively; Fig. 2J–L). In addition to the reduction in intensity, quantification analysis of P10 SLICK-H-CreER;Ankfx/− SNs showed that 87.48%, 73.1%, and 70.63% of the nodes were positive for NF186, pan-NaV channels, and βIV Spec, respectively. However, only 29.5%, 37.42%, and 14.5% of these positive nodes showed NF186, pan-NaV channels and βIV Spec, respectively, uniformly distributed over the nodal area, while the remaining percentages of the nodes showed abnormal partial or patchy staining. The percentages of nodes with normal morphology continued to decrease by P30, indicating progressive nodal destabilization (Fig. 2Y). Our data indicate that in the PNS early loss of AnkG affects proper nodal scaffolding and maturation, leading to enhanced destabilization.
Figure 2.
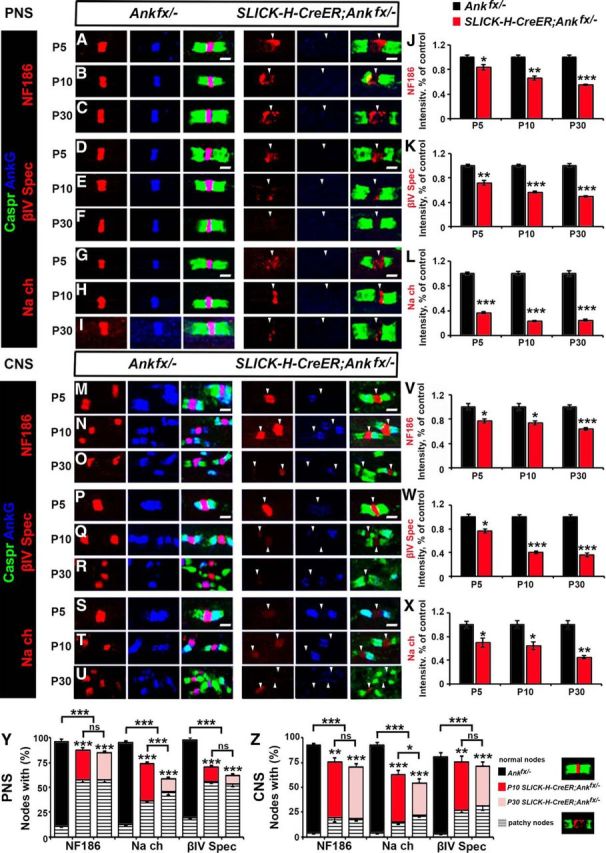
AnkG is required for nodal maturation but not initial assembly and protein clustering at the nodes. A–I, Immunostaining of SN fibers from P5, P10, and P30 age-matched Ankfx/− and SLICK-H-CreER;Ankfx/− littermate mice with antibodies against AnkG (blue) in combination with the following proteins: NF186 (A–C), βIV Spec (D–F), or pan-NaV (G–I; red) and Caspr (green). J–L, Quantification of NF186, βIV Spec, or pan-NaV intensity, respectively, in the SN nodal area standardized to the same age control values from P5, P10, and P30 Ankfx/− (black bars), and SLICK-H-CreER;Ankfx/− (red bars) mice (n = 3 mice/genotype; 60–80 nodes/animal; unpaired, two-tailed Student's t test). M–U, SCs from P5, P10, and P30 age-matched Ankfx/− and SLICK-H-CreER;Ankfx/− littermate mice were immunostained with antibodies against AnkG (blue) in combination with either of the following proteins: NF186 (M–O), βIV Spec (P–R), or pan-NaV (S–U; red), and Caspr (green). V, W, X, Intensity of NF186, βIV Spec, or pan-NaV, respectively in the SC nodal area standardized to the same age control values from P5, P10, P30 Ankfx/− (black bars), and SLICK-H-CreER;Ankfx/− (red bars) mice (n = 3 mice/genotype; 60–80 nodes/animal; unpaired, two-tailed Student's t test). Y, Z, Percentages of normal and patchy nodes in SN and SC, respectively, with remaining NF186, pan-NaV, and βIV Spec at P10 and P30 in SLICK-H-CreER;Ankfx/− mice compared with Ankfx/− controls (n = 3 mice/genotype; 60–80 nodes/animal; comparison of normal nodes performed by two-way ANOVA with Bonferroni's post hoc analysis). Arrowheads mark AnkG-negative nodes in the SLICK-H-CreER;Ankfx/− mice. Scale bar, 2 μm. All data are represented as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Next, we analyzed the localization of the nodal proteins in the SCs of SLICK-H-CreER;Ankfx/− mice. At P5 in Ankfx/− SCs, AnkG is enriched in the paranodes expressed by oligodendrocytes (Chang et al., 2014) and the nodes along myelinated nerve fibers; however, in SLICK-H-CreER;Ankfx/− mice, AnkG is absent from the nodes while its localization at the paranodes remains intact (Fig. 2M,P,S). Nodal protein immunostaining revealed that NF186, βIV Spec, and pan-NaV altered clustering at P5 (Fig. 2M,P,S), which is represented by 23.19%, 23.80%, and 29.97% reduced intensity values (Fig. 2V–X) compared with control Ankfx/− mice. At P10 in AnkG-negative SLICK-H-CreER;Ankfx/− nodes NF186, βIV Spec, and pan-NaV intensities continued to decrease (Fig. 2N,Q,T) and was reduced by 26.28%, 59.76%, and 35.93%, respectively, compared with control animals (Fig. 2V–X). By P30, prominent destabilization of all nodal proteins was observed in SLICK-H-CreER;Ankfx/− SCs, where the intensity was significantly lower compared with control Ankfx/− fibers and was even undetectable in some areas (Fig. 2O,R,U–X). Quantification analysis of nodes lacking AnkG at P10 in SC myelinated fibers revealed 17.09%, 29.2%, and 5.4%, respectively, of nodes negative for NF186, pan-NaV channels, or βIV Spec in AnkG-deficient mice. However, only 56.33%, 47.83%, and 48.69% of the remaining positive nodes were normal, showing proper localization of NF186, pan-NaV channels, or βIV Spec at the nodal area, with further progressive loss of these proteins by P30 (Fig. 2Z). Together, the developmental immunostaining in both the PNS and CNS establishes that AnkG is required for proper anchoring of the nascent nodal complex with axonal cytoskeleton for further maturation and long-term stabilization.
Juvenile loss of AnkG causes slow destabilization of the nodes
To determine the consequences of AnkG loss at a later postnatal stage on nodal stability and whether later loss affects long-term maintenance of the node differently, at P23, when myelination is complete, SLICK-H-CreER;Ankfx/− and Ankfx/− mice were given tamoxifen injections for 10 consecutive days and examined for various phenotypes 1, 3, and 10 months later (Fig. 3A). As early as 3 months postinjection (mpi), SLICK-H-CreER;Ankfx/− mice developed a foot-clasping phenotype and slight tremor; by 8 mpi, more profound tremor, abnormal gait, significant body weight reduction, and severe spine deformation were observed (Fig. 3B,C). On average, SLICK-H-CreER;Ankfx/− animals survived until 11 months of age (10 mpi), while their tamoxifen-injected Ankfx/− littermates maintained a normal lifespan. To confirm AnkG ablation by tamoxifen, SN fibers and SC slices were examined 1 mpi with immunostaining, which revealed a loss of AnkG from nodes in SLICK-H-CreER;Ankfx/− myelinated fibers (Fig. 3E–H). Quantification analysis of nodes without AnkG in SLICK-H-CreER;Ankfx/− SCs and SNs compared with Ankfx/− controls showed 82.2% and 81.0% reduction of AnkG-positive nodes, respectively (Fig. 3I,J). Further, immunoblotting from 1 mpi SLICK-H-CreER;Ankfx/− mice also showed reduction in the total amount of AnkG 270 and 480 kDa neuronal isoforms present in whole SN and SC lysates (Fig. 3D). Immunoblot quantification analysis normalized to tubulin showed 87.8%/33.0% intensity reduction of 270 kDa and 83.43% decrease/absence of 480 kDa bands in SLICK-H-CreER;Ankfx/− SN/SC lysates, respectively, compared with Ankfx/− control values. It should be noted that the discrepancy in the percentage of each isoform that is lost likely reflects the presence of the 270 kDa isoform of AnkG in oligodendrocytes reported previously (Chang et al., 2014). Overall, these data reveal that within 1 mpi, AnkG protein was undetectable at the nodes, and that the loss of AnkG leads to a progressive weakening of the animals, resulting in their demise within a year.
Figure 3.

Juvenile neuron-specific AnkG ablation model. A, Schematic representation of tamoxifen injections for AnkG ablation from adult myelinated axons. Tamoxifen was injected starting at P23 for 10 consecutive days to SLICK-H-CreER;Ankfx/− and Ankfx/− control mice, which were analyzed 1, 3, and 10 mpi. B, Graph representing the weight of Ankfx/− and SLICK-H-CreER;Ankfx/− mice (n = 6 mice/genotype; two-way ANOVA, Bonferroni's post hoc analysis). C, Photographic depiction of 8 mpi SLICK-H-CreER;Ankfx/− mutant and Ankfx/− control mice after AnkG ablation by tamoxifen. D, Immunoblot analysis of SN and SC lysates from 1 mpi Ankfx/− and SLICK-H-CreER;Ankfx/− mice with antibodies against AnkG and α-tubulin (Tub). Asterisk in the SLICK-H-CreER;Ankfx/− lane indicates a 270 kDa AnkG isoform expressed by oligodendrocytes. E–H, Immunostaining of 1 mpi teased SN fibers (E, F) and SCs (G, H) with antibodies against Caspr (green) and AnkG (red) from Ankfx/− and SLICK-H-CreER;Ankfx/− mice. White and yellow arrowheads indicate AnkG-positive and AnkG-negative nodes, respectively. I, J, Quantification of the percentage of nodes with AnkG in 1 mpi Ankfx/− and SLICK-H-CreER;Ankfx/− SNs and SCs, respectively (n = 3 mice/genotype; 120–130 nodes/animal; unpaired, two-tailed Student's t test). Scale bar, 2 μm. All data are represented as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
To determine the consequences of AnkG loss on nodal long-term stability, SNs and SCs from 1, 3, and 10 mpi Ankfx/− and SLICK-H-CreER;Ankfx/− were triple immunostained with antibodies against AnkG, paranodal Caspr, and NF186, βIV Spec, or pan-NaV channels (Fig. 4A–I,M–U). Quantifying nodes in SN that had no detectable AnkG remaining revealed that by 3 mpi 1.5%, 29.57%, and 9.8% of nodes, respectively, were lacking NF186, pan-NaV, and βIV Spec, with only 68.72%, 53.32%, and 45.33% of positive nodes showing normal clustering (Fig. 4Y). In addition, progressive reduction in intensity staining of the remaining nodal proteins were apparent in SLICK-H-CreER;Ankfx/− compared with Ankfx/− SNs. At 1 mpi in SLICK-H-CreER;Ankfx/− SNs, NF186 and pan-NaV channels intensity is not altered (Fig. 4A,G,J,L); however, reduction of 29.9% was observed for βIV Spec intensity (Fig. 4D,K) compared with control values of Ankfx/−. By 3 mpi in SLICK-H-CreER;Ankfx/− nodes, NF186, βIV Spec, and pan-NaV channel clusters were reduced in size and their intensity decreased to 31.01%, 58.07%, and 54%, respectively, compared with control levels (Fig. 4B,E,H,J–L). At 10 mpi, SLICK-H-CreER;Ankfx/− SNs showed more profound destabilization of the remaining nodal proteins with 34%, 60%, and 62.01%, respectively, intensity reduction of NF186, βIV Spec, and pan-NaV channels (Fig. 4C,F,I–L). These data suggest that the loss of AnkG has a direct effect on the stability of pan-NaV channels and the cytoskeletal βIV Spec with relatively less effect on the transmembrane NF186 at the nodal region.
Figure 4.
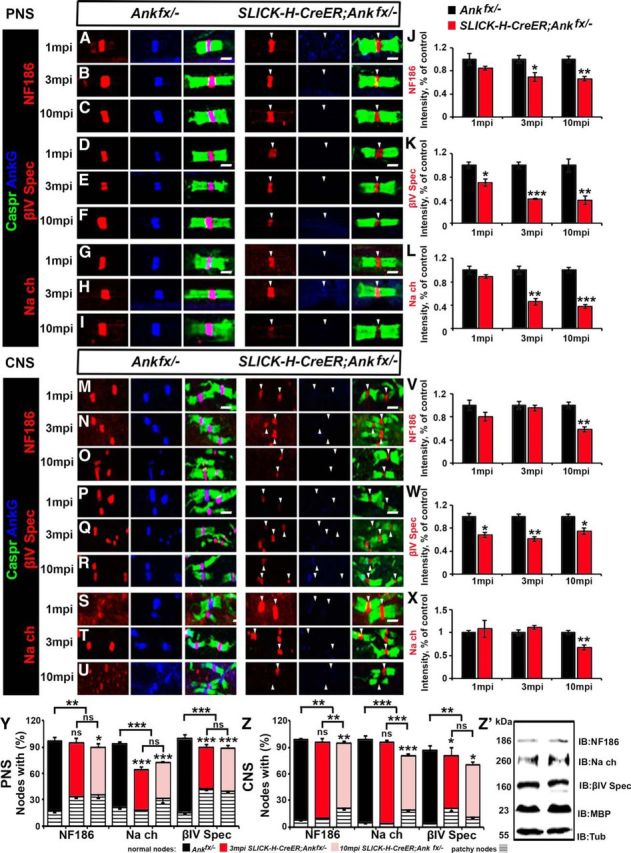
Late loss of AnkG causes prolonged nodal destabilization compared with early loss of AnkG. A–I, SN fibers from 1, 3, and 10 mpi age-matched Ankfx/− and SLICK-H-CreER;Ankfx/− littermate mice were immunostained with antibodies against AnkG (blue) and Caspr (green) in combination with any of the following proteins: NF186 (A–C), βIV Spec (D–F), or pan-NaV (G–I; red). J–L, Graphs representing intensity quantification of NF186, βIV Spec, or pan-NaV, respectively, in the SN nodal area of SLICK-H-CreER;Ankfx/− (red bars) standardized to the same age control values from 1, 3, and 10 mpi Ankfx/− mice (black bars; n = 3 mice/genotype; 120–130 nodes/animal; unpaired, two-tailed Student's t test). M–U, Immunostaining of SC sections from 1, 3, and 10 mpi age-matched Ankfx/− and SLICK-H-CreER;Ankfx/− littermate mice with antibodies against AnkG (blue) and Caspr (green) in combination with any of the following proteins: NF186 (M–O), βIV Spec (P–R), or pan-NaV (S–U; red). V, W, X, Graphs representing intensity quantification of NF186, βIV Spec, or pan-NaV, respectively, in the SC nodal area of SLICK-H-CreER;Ankfx/− (red bars) standardized to the same age control values from Ankfx/− mice (black bars; n = 3 mice/genotype; 120–130 nodes/animal; unpaired, two-tailed Student's t test). Y, Z, Percentage of normal and patchy nodes in SNs and SCs, respectively, with the remaining NF186, pan-NaV, and βIV Spec at 3 and 10 mpi in SLICK-H-CreER;Ankfx/− mice compared with Ankfx/− controls (n = 3 mice/genotype; 120–130 nodes/animal; comparison of normal nodes performed by two-way ANOVA with Bonferroni's post hoc analysis). Z′, Immunoblot analysis of SC lysates from 3 mpi Ankfx/− and SLICK-H-CreER;Ankfx/− mice with antibodies against NF186, pan-NaV, βIV Spec, MBP, and tubulin (Tub; as a loading control). Arrowheads mark the nodes without AnkG in the SLICK-H-CreER;Ankfx/− mice. Scale bar, 2 μm. All data are represented as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
In the SC, AnkG was undetectable at most of the nodes after 1 mpi in SLICK-H-CreER;Ankfx/− mice (Fig. 4M,P,S). Despite the lack of AnkG, the localization and intensity of NF186 and pan-NaV channel staining was not altered in SLICK-H-CreER;Ankfx/− SCs compared with control Ankfx/− fibers (Fig. 4M,S,V,X). However, similar to what was found in SNs, there was a 32% reduction in the intensity of βIV Spec staining in the SCs of 1 mpi SLICK-H-CreER;Ankfx/− animals compared with controls (Fig. 4P,W). At 3 mpi, no changes in NF186 (Fig. 4N,V) and pan-NaV channel intensity (Fig. 4T,X) were observed; however, βIV Spec intensity progressively decreased (Fig. 4Q,W) in SLICK-H-CreER;Ankfx/− SCs. By 10 mpi, quantification analysis revealed that all three nodal proteins, NF186, βIV Spec, and pan-NaV channels, were significantly reduced in intensity at the nodal area in SLICK-H-CreER;Ankfx/− SC (Fig. 4O,R,U–X). Scoring for nodes in SC lacking AnkG revealed that only 58.96% of βIV Spec-positive nodes had preserved morphology with no significant changes in NF186 and pan-NaV-positive nodes in SLICK-H-CreER;Ankfx/− at 3 mpi. However, at 10 mpi, the percentages of normal nodes with NF186, βIV Spec, and pan-NaV were reduced by 17.45%, 32.33%, and 24.02%, respectively, compared Ankfx/− SCs (Fig. 4Z).
To further address whether the absence of a single nodal protein altered the total levels of proteins of interest, we performed immunoblot analyses on 10 mpi Ankfx/− and SLICK-H-CreER;Ankfx/− SC lysates. Although in SLICK-H-CreER;Ankfx/− mice, AnkG 270 and 480 kDa bands showed significant reduction, no differences were observed in NF186, βIV Spec, pan-NaV channels, or MBP in response to AnkG loss (Fig. 4Z′), suggesting that AnkG loss alters proteins stability at the node, leading to their mislocalization along the axon. Overall, our data indicate that nodal destabilization occurs more slowly when the cytoskeletal component is ablated after nodal maturation is complete, indicating that AnkG is critical both for the early stability and lifelong maintenance of the nodes in PNS and CNS.
Perinatal or juvenile loss of ankyrin G leads to elevated intensities of ankyrin R and βI spectrin at the node
To determine whether perinatal ablation of AnkG and sequential destabilization of βIV Spec is compensated by ankyrin R (AnkR) and βI Spec, as proposed previously by Ho et al., 2014, we analyzed PNS and CNS myelinated fibers for AnkR and βI Spec (Fig. 5A–J). Immunostaining for AnkR at P30 in the model of early AnkG ablation revealed an equal presence of AnkR-positive nodes in both the control and SLICK-H-CreER;Ankfx/− animals in the PNS (Fig. 5A,B,J). However, at the CNS by P30, a reduction of AnkR-positive nodes was observed in SLICK-H-CreER;Ankfx/− mice (Fig. 5C,D,J), suggesting AnkG-mediated AnkR destabilization. AnkR intensity quantifications revealed significantly elevated levels at the nodes of PNS-(3.2-fold change) and CNS-myelinated fibers (1.4-fold change; Fig. 5I). Similar to AnkR, βI Spec-positive nodes were equally detected in control (82.1 ± 1.9) and mutant (68.7 ± 12.3) animals in the PNS at P30 (Fig. 5E,F,J), with its 3.35-fold increased intensity in SLICK-H-CreER;Ankfx/− animals (Fig. 5I). In the CNS, only 5% of the nodes contained βI Spec in both control and mutant animals by P30, with no statistical difference in intensity (Fig. 5G–J). These data indicate that an early loss of AnkG leads to elevated intensities but not to an increased percentage of AnkR or βI Spec-positive nodes, and most importantly these two resident cytoskeletal proteins fail to protect nodes from destabilization.
Figure 5.
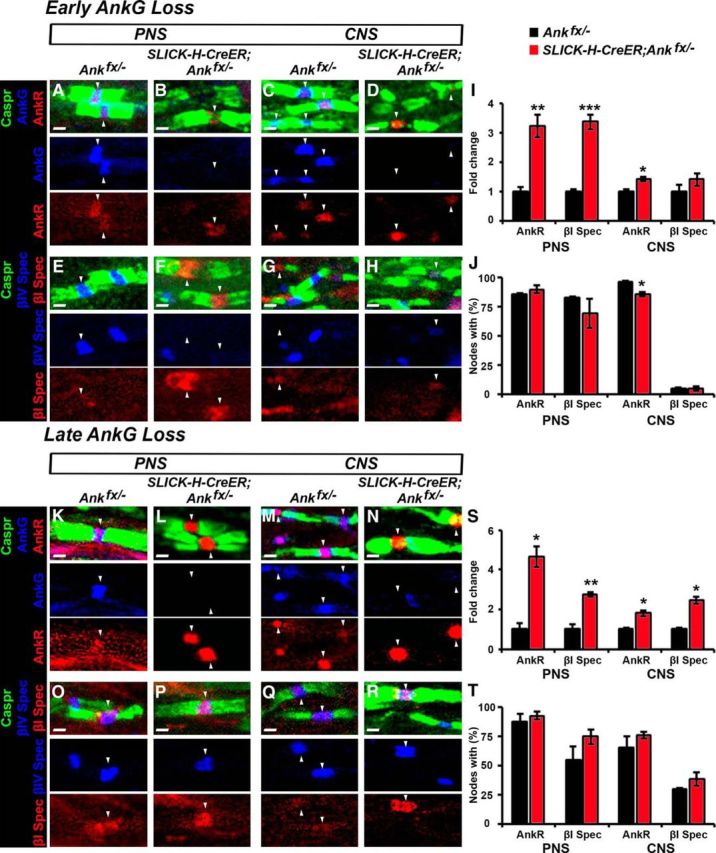
AnkR and βI spectrin intensities are elevated at the node after perinatal and juvenile loss of AnkG. A–D, Immunostaining of SN fibers (A, B) or SC slices (C, D) from P30 age-matched Ankfx/− and SLICK-H-CreER;Ankfx/− littermate mice with antibodies against Caspr (green), AnkG (blue), and AnkR (red). E–H, Immunostaining of SN (E, F) or SC (G, H) myelinated axons from P30 age-matched Ankfx/− and SLICK-H-CreER;Ankfx/− littermate mice with antibodies against Caspr (green), βIV Spec (blue), and βI Spec (red). I, Intensity fold change of AnkR and βI Spec in the SN and SC nodal areas standardized to the same age control values from P30 Ankfx/− (black bars) and SLICK-H-CreER;Ankfx/− (red bars) mice (n = 3 mice/genotype; 60–80 nodes/animal; unpaired, two-tailed Student's t test). J, Percentage of nodes with AnkR and βI Spec in P30 Ankfx/− (black bar) and SLICK-H-CreER;Ankfx/− (red bar) SNs and SCs, respectively (n = 3 mice/genotype; 60–80 nodes/animal; unpaired, two-tailed Student's t test). K–N, Immunostaining of SN fibers (K, L) or SC slices (M, N) from 10 mpi age-matched Ankfx/− and SLICK-H-CreER;Ankfx/− littermate mice with antibodies against Caspr (green), AnkG (blue) and AnkR (red). O–R, Immunostaining of SN (O, P) or SC (Q, R) myelinated axons from 10 mpi age-matched Ankfx/− and SLICK-H-CreER;Ankfx/− littermate mice with antibodies against Caspr (green), βIV Spec (blue), and βI Spec (red). S, Intensity fold change of AnkR and βI Spec in the SN and SC nodal areas standardized to the same age control values from 10 mpi Ankfx/− (black bars) and SLICK-H-CreER;Ankfx/− (red bars) mice (n = 3 mice/genotype; 120–130 nodes/animal; unpaired, two-tailed Student's t test). T, Percentage of nodes with AnkR and βI Spec in 10 mpi Ankfx/− (black bar) and SLICK-H-CreER;Ankfx/− (red bar) SNs and SCs, respectively (n = 3 mice/genotype; 120–130 nodes/animal; unpaired, two-tailed Student's t test). Arrowheads indicate AnkR or βI Spec-positive nodes. Scale bar, 1 μm. All data are represented the as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Further, to address whether AnkR and βI Spec compensate and protect the node from destabilization after juvenile AnkG ablation, we performed immunostaining with quantificational analysis of AnkR and βI Spec in the PNS and CNS myelinated fibers of 10 mpi control and mutant mice (Fig. 5K–T). While the total percentage of the nodes containing AnkR or βI Spec did not change between Ankfx/− and SLICK-H-CreER;Ankfx/− fibers at 10 mpi (Fig. 5T), their intensities at the node were significantly elevated (Fig. 5S). In contrast to previous published studies by Ho et al., 2014, we did not observe a natural decline of AnkR-positive nodes in control adult animals; instead, a majority of the control nodes in both PNS and CNS contained AnkR (Fig. 5K,M,T). Our data reveal that both AnkR and βI Spec are resident nodal proteins, as they are seen in control animals at least up to 11 months of age, and late AnkG loss does not change the total number of AnkR/βI Spec-positive nodes exclusively, leading to their increased intensities, while other nodal proteins (NF186, βIV Spec, and NaV channels) undergo progressive destabilization.
Perinatal and juvenile loss of AnkG leads to altered nerve conduction in myelinated axons
To determine whether AnkG loss and nodal complex destabilization led to altered electrophysiological activity of myelinated nerves in our early and late AnkG knock-out models, we performed in vivo recordings from the tail and sciatic nerves of age-matched SLICK-H-CreER;Ankfx/− and Ankfx/− mice. Our studies in a perinatal AnkG ablation model revealed no changes in the NCV between P30 Ankfx/− and SLICK-H-CreER;Ankfx/− nerve fibers [22.64 ± 1.09 vs 21.1 ± 0.96 for the SN (Fig. 6A,B) and 8.3 ± 0.26 vs 8.1 ± 0.18 for the tail (Fig. 6D,E)]; however, a significant reduction in the amplitude was observed in P30 SLICK-H-CreER;Ankfx/− nerves compared with controls [0.9 ± 0.14 vs 0.5 ± 0.10 for the SNs (Fig. 6A,C); 1.5 ± 0.26 vs 0.95 ± 0.10 for the tail (Fig. 6D,F)]. Similarly, in a juvenile AnkG ablation model, no significant differences were observed in conduction velocity between age-matched Ankfx/− and SLICK-H-CreER;Ankfx/− mice at 1, 3, or even 10 mpi in SNs (Fig. 6G,H) or tail (Fig. 6J,K) during in vivo recordings. However, a significant reduction in CAP amplitude was observed in SNs by 10 mpi (Fig. 6G,I), as well as the tail nerves starting from 3 mpi in SLICK-H-CreER;Ankfx/− mice (Fig. 6J,L). Overall, these electrophysiological measurements indicate that axons, which underwent degeneration or possess a low density of NaV channels at nodes after AnkG ablation, most likely fail to propagate action potentials. This results in the decrease of measured CAP amplitude, while conduction velocity remains unaltered due to the remaining axons having enough residual nodal NaV channels for action potential propagation. In support of the above observations, immunostaining of SCs from 10 mpi animals revealed the presence of pan-NaV staining at the axon initial segment (AIS), while AnkG was undetectable in SLICK-H-CreER;Ankfx/− animals (data not shown), suggesting that action potential generation may not be affected in late ablated AnkG-deficient mice. However, when AnkG is ablated at early developmental stages, AIS formation is compromised and may severely impact the generation of action potentials (Zhou et al., 1998; Ho et al., 2014).
Figure 6.

Early and late ablation of AnkG modestly affects nerve conduction. A–C, Representative electrophysiological profiles of CAPs from SNs of P30 Ankfx/− (black line) and SLICK-H-CreER;Ankfx/− (red line) mice (A) with quantification of NCV (B) and amplitude (C; n = 5 mice/genotype; two-tailed Student's t test). D–E. Representative traces of CAPs from tails of P30 Ankfx/− (black line) and SLICK-H-CreER;Ankfx/− (red line) animals (D) with quantification of NCV (E), and amplitude (F). G–I, Representative electrophysiological profiles of CAPs from SNs of 1, 3, and 10 mpi Ankfx/− (black line) and SLICK-H-CreER;Ankfx/− (red line) mice (G) with quantification of NCV (H) and amplitude (I; n = 5 mice/genotype; two-way ANOVA, Bonferroni's post hoc analysis). J–L, Depiction of representative CAP traces from tails of 1, 3, and 10 mpi Ankfx/− (black line) and SLICK-H-CreER;Ankfx/− (red line) animals (J) with quantification of NCV (K) and amplitude (L). All data are represented as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
We also addressed whether the loss of AnkG led to altered expression of NaV channel isoforms, which undergo developmental transition from immature NaV1.2 to mature NaV1.6 isoform, and in some pathological conditions, such as multiple sclerosis, a reversion of isoforms occurs from NaV1.6 to NaV1.2 (Boiko et al., 2001; Craner et al., 2004). To test this possibility, we performed immunostaining of 10 mpi SC and SN myelinated fibers for isotype-specific NaV1.2 and NaV1.6 antibodies. Both in control and in AnkG-deficient animals, the NaV1.6 isoform was present at the nodes in SCs and SNs, and no NaV1.2 isoform was detected. Moreover, as an additional conformation of nodal destabilization after AnkG ablation, NaV1.6 channels were either significantly decreased in intensity or missing from the nodes in SLICK-H-CreER;Ankfx/− mice (data not shown), suggesting that changes in conduction properties in mutants are due to NaV1.6 disruption and not to the presence of NaV1.2 isoforms at the nodes. Together, our studies indicate an important role of AnkG as a key nodal cytoskeletal stabilizer protein to maintain proper architecture and electrical properties of myelinated axons.
Loss of AnkG leads to ultrastructural defects in myelinated axons
Since nodal disorganization was evident in both early and late AnkG ablation models, we wanted to examine the myelinated axons in SLICK-H-CreER;Ankfx/− and Ankfx/− animals for any ultrastructural changes that might have possibly occurred over time as a result of the loss of AnkG at the nodes. At P30 and 4 mpi, for early and late AnkG ablation models, respectively, SNs and SCs from control and mutant mice were processed for electron microscopy. To capture the intermediate stage and to show the progression of various axonal degenerative phenotypes after adult AnkG ablation, we used a 4 mpi time point for phenotypic analysis. Representative images in Figure 7 from the cross-sections of control P30 and 4 mpi Ankfx/− SNs and SCs show normal axons surrounded by tight intact myelin ensheathment (Fig. 7A,E,I,M). In contrast, at P30 and 4 mpi myelinated fibers with signs of axonal degeneration and disorganized myelin sheath were observed in SNs and SCs of SLICK-H-CreER;Ankfx/− animals (Fig. 7B–D,F–H,J–L,N–P). In both models of AnkG ablation, nerve fibers from mutant mice revealed shrunken axons, fragmented myelin, and axoplasmic accumulation of membranous debris. Quantification analysis in control versus early and late AnkG-ablated animals showed a tendency toward an increased percentage of myelinated fibers with signs of pathology in both SNs and SCs (data not shown). Moreover, longitudinal sections from control SNs and SCs showed a clear nodal gap surrounded by paranodal axo–glial junctions. However, accumulations of organelles were often observed at the nodal area of P30 and 4 mpi SLICK-H-CreER;Ankfx/− animals, suggesting that AnkG loss affects local axonal cytoskeleton and accordingly axonal transport along the axons (data not shown). Together, our data reveal that the nodal cytoskeletal protein AnkG is crucial to maintain proper nodal structure and that AnkG-deficient nodes undergo nodal destabilization that alters their electrical properties, which may over time contribute to axonal ultrastructural changes and degeneration.
Figure 7.
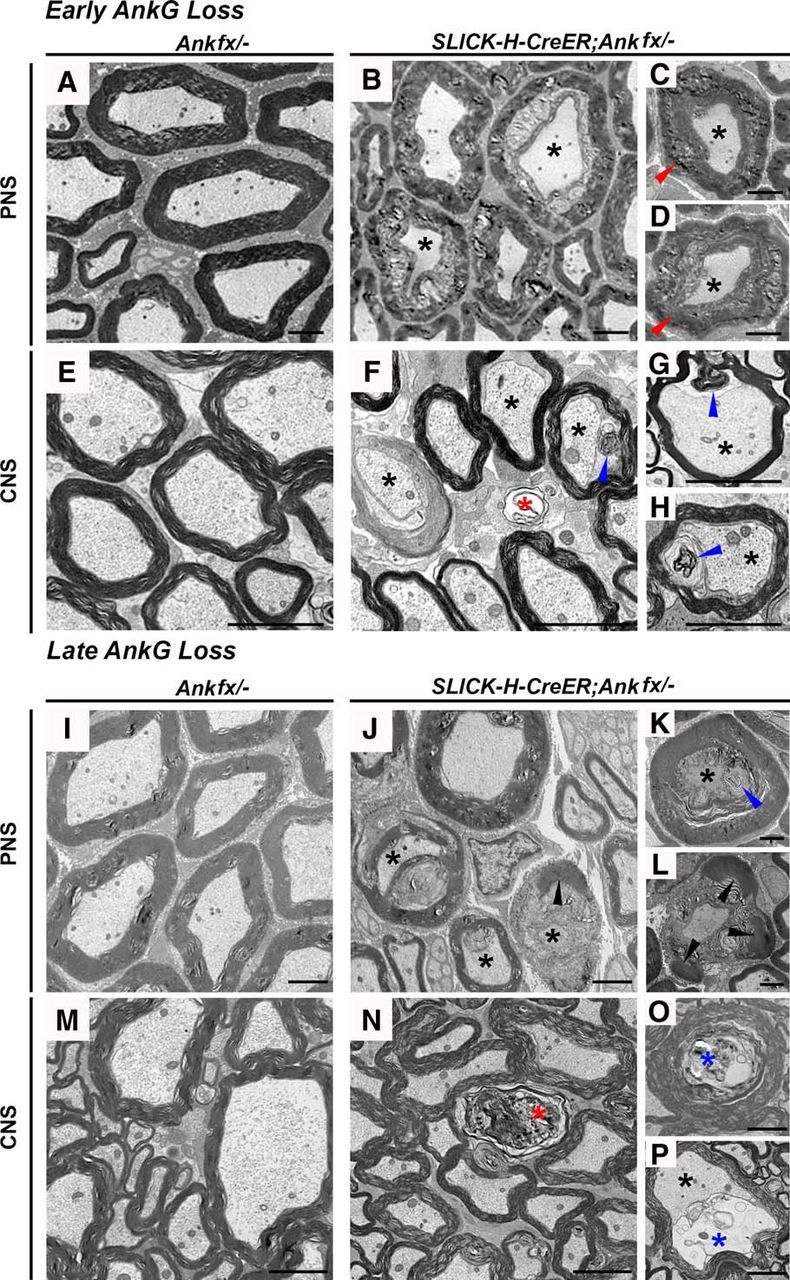
Loss of AnkG leads to degeneration of myelinated axons. A–D, Transmission electron microscopy (TEM) of cross-sections from SNs of P30 Ankfx/− (A) and SLICK-H-CreER;Ankfx/− (B–D) mice. E–H, TEM of cross-sections from SCs of P30 Ankfx/− (E) and SLICK-H-CreER;Ankfx/− (F–H) mice. I–L, TEM of cross-sections from SNs of 4 mpi control (I) and SLICK-H-CreER;Ankfx/− (J–L) mice. M–P, TEM of cross-sections from SCs of 4 mpi control (M) and SLICK-H-CreER;Ankfx/− (N–P) mice. The following types of axonal degeneration are marked: shrunken axons (black asterisks); swollen myelin sheath (red arrowheads); invasion of myelin into the former axonal space (blue arrowheads); decomposed myelin debris (black arrowhead); completely degenerated axons without microfilaments and microtubules (red asterisks); and accumulation of axonal membranous debris (blue asterisks). Scale bar, 2 μm.
Discussion
The integrity of the molecular complex at the nodes of Ranvier is crucial for the fast action potential propagation by myelinated axons, and hence neuronal function. The development of the nodes occurs in the following three stages: (1) initial organization (accompanied by onset of myelination and sequential recruitment of nodal proteins into the nodal gap; (2) maturation (establishment of stable interactions between nodal components and substitution of the NaV1.2 by NaV1.6 subset of channels); and (3) maintenance of the nodal complex throughout adult life. As a result of this developmental process, nodes become highly enriched in NaV channels, and their stability at the node is ensured by highly coordinated external interactions (NF186-dependent interactions with the extracellular matrix), as well as internal interactions (AnkG- and βIV Spec-mediated anchoring to the actin cytoskeleton; Eshed et al., 2005; Koticha et al., 2006; Thaxton et al., 2011; Susuki et al., 2013). In the current study, we addressed the role of the key cytoskeletal scaffolding protein AnkG during early nodal development and also after the nodal complex had been assembled. Here we report that the initial nodal assembly does not require AnkG, instead AnkG allows maturation and stabilization of the nodal complex, and, once assembled, AnkG is crucial for the long-term maintenance of the nodal region in myelinated axons.
Ankyrin G-dependent nodal assembly and integrity
Previous developmental analysis of myelinated fibers suggested that nodal organization in the CNS is defined by AnkG (Jenkins and Bennett, 2002), as other proteins such as βIV Spec, NaV channels, and NF186 were subsequently recruited to the sites of AnkG-positive clusters. In vitro studies using shRNA knockdown of AnkG in DRG neurons led to a significant elimination of other proteins from the nodes, suggesting that AnkG plays an important role in the PNS node formation (Dzhashiashvili et al., 2007). Using isotype-specific AnkG mutant mice, it was recently shown that the 480 kDa AnkG isoform is critical for the node formation in the corpus callosum, since its absence leads to a significant reduction in the nodal clusters, elongation of the nodal gap, and loss of NF186 (Jenkins et al., 2015). In vivo studies on cerebellar isoform-specific AnkG knock-out mice revealed nodes with missing NaV channels together with their overall diminished levels along the axon (Zhou et al., 1998; Barry et al., 2014). However, concurrent studies revealed the following surprising finding: AnkG may not be required for nodal organization (Ho et al., 2014). Since the clustering of other nodal components occurred normally in both PNS and CNS, Ho et al. (2014) suggested that, in the absence of AnkG, AnkR was able to substitute to allow node formation. In our studies, perinatal loss of AnkG led to progressive destabilization of NaV channels at the AnkG-deficient nodes, which was not observed by Ho et al. (2014). We addressed both the intensity changes and the number of nodes that retained normal levels of NaV channels. Our data consistently showed progressive loss of NaV channels at the nodes when AnkG was ablated. Whether these differences are due to differences in procedures or differences in the genetic backgrounds of the strains used remains to be addressed. Both studies, however, revealed a decrease in βIV Spec levels at the AnkG-deficient nodes. The many studies described above have led to more controversy regarding the role of AnkG in nodal development and have raised additional questions about whether nodes lacking AnkG but containing AnkR behaved as normal nodes and remain stable, like the nodes that contained a full complement of all nodal proteins. When AnkG was ablated during perinatal (i.e., late embryonic/early postnatal) stages, nodes were formed with abnormal morphology, did not become stable, and disorganized rapidly (Fig. 8A). This rapid nodal disintegration, when AnkG is ablated before myelination, defines its role in early maturation and its requirement for the establishment of stable interactions between nodal proteins. Interestingly, recent studies have shown that AnkG functions as an adaptor protein that links NaV1.2 subset of ion channels to kinesin-1 and transports them along the axon (Barry et al., 2014). This particular subset of sodium channels is exclusively expressed in immature nodes of Ranvier during myelination, which later are replaced by NaV1.6 channels (Boiko et al., 2001; Kaplan et al., 2001), which differ not only in molecular structure and affinity to their binding partners, but also in electrophysiological properties as well (Rush et al., 2005). Early loss of AnkG potentially might disturb such a transition during nodal maturation and result in unstable nodal architecture subjected to accelerated disorganization.
Figure 8.

AnkyrinG-dependent nodal organization, maturation, and long-term stability. A, Perinatal ablation of AnkG results in nodes that lack AnkG but assemble NF186, NaV channels, and βIV Spec. Without AnkG, nodes fail to mature and lose NaV channels and βIV Spec with only minor effects of NF186. Mice die at approximately P40. B, Juvenile ablation of AnkG results in AnkG loss from the majority of nodes within 1 mpi. In the absence of AnkG, βIV Spec stability is primarily affected, followed by NaV channels, with only minor effects on NF186. Mice die at ∼10 mpi.
A completely different situation might be expected when AnkG is lost from already mature nodes with strengthened interactions between constituent proteins. Using the adult tamoxifen-inducible AnkG ablation model, we were able to track the consequences of AnkG loss on the maintenance of the mature nodes for up to 10 months. Although AnkG loss was complete by 1 mpi, the most prominent reductions in pan-NaV channels, βIV Spec, and NF186 were observed as late as 10 mpi (Fig. 8B). In contrast, Ho et al. (2014) analyzed nodes up to two months after AnkG ablation and did not observe much nodal destabilization. The discrepancy between the two studies could be explained by a significantly prolonged timeline of our studies (10 mpi vs 2 month post viral Cre injection).
Interestingly, AnkR/βI Spec did not rescue AnkG-deficient nodes from destabilization in either model. While there is an increase in the intensities of AnkR and βI Spec at the nodes in the absence of AnkG, AnkR and βI Spec were consistently present at the control wild-type nodes up to 11 months, the last time point analyzed. One can speculate that AnkR from pre-existing axonal pools occupies molecular areas that had been previously occupied by AnkG because AnkR interacts with other nodal proteins (Ho et al., 2014), thus resulting in elevated intensities but not an increased number of AnkR-positive nodes when AnkG is lost from the nodes. Similarly, βI Spec may occupy βIV Spec-free positions in destabilizing nodes. Since AnkR has lower binding affinity with NF186 and NaV channels compared with AnkG (Ho et al., 2014), it most likely fails to maintain stability at the AnkG-deficient nodes. These data suggest that AnkR together with βI Spec are not sufficient to compensate for the loss of AnkG at the nodes and that their presence fails to maintain a fully functional nodal complex, leading to progressive nodal disorganization, severe neurological disabilities, and lethality. Thus, AnkG is necessary for both early maturation and long-term stabilization of the nodal domain, and, more importantly, nodes that form in the absence of AnkG fail to mature and undergo enhanced destabilization. On the other hand, nodes that form with AnkG mature normally and establish proper stoichiometry and interactions that guarantee their long-term maintenance. While AnkG is undetectable at the nodes within a month of its ablation, nodal components become unstable and are lost in a sequential manner, with βIV spectrin disappearing first followed by NaV channel and NF186, which is the last to disappear. This sequence of disappearance of nodal proteins may also reflect the nature and placement of these molecules within the nodal complex. Furthermore, the process of nodal destabilization is more progressive in the PNS compared with the CNS, which could be attributed to different mechanisms that organize/stabilize the nodal complex. However, these differences do not affect the outcome, as in both the PNS and CNS, the ablation of AnkG leads to compromised nodal integrity, indicating a general role of AnkG at the nodes of Ranvier.
Ankyrin/spectrin cytoskeletal scaffolding proteins in nodal development
Ankyrin and spectrin family members organize plasma membrane complexes in a majority of animal cells (Bennett and Lorenzo, 2016). Since myelinated axons are polarized structures and distinctly divided into domains, each of the domains has unique ankyrin/spectrin-associated complexes (Susuki and Rasband, 2008; Buttermore et al., 2013). The cytoskeletal scaffolding proteins AnkG and βIV Spec show highly enriched localization at the nodal domain; yet, there has been much speculation about whether these two proteins play an instructive role in node formation or in creating scaffolds that maintain nodal organization. Studies using βIV Spec mutant animals revealed that, in its absence, other nodal proteins were able to cluster but over time these clusters began to destabilize, suggesting that βIV Spec was required more for the long-term nodal stability than for initial organization and maturation (Komada and Soriano, 2002). Interestingly, disintegration of the node in the absence of βIV Spec occurs within 8–10 months, where AnkG is the first protein that gets destabilized in the absence of βIV Spec, followed by NaV channels and finally NF186 (unpublished data). On the other hand, the early loss of AnkG reported here leads to rapid nodal destabilization within a month, with βIV Spec impaired first, followed by NaV channels, and last by NF186. Thus, a comparison of the individual contributions of AnkG and βIV Spec to nodal organization and stability reveals that AnkG provides greater stability at the node due to its binding with both NF186 and NaV channels and that βIV spectrin plays an additional stabilization and scaffolding function in long-term nodal maintenance.
Ankyrin G-mediated nodal destabilization and myelinated axon pathology
Numerous experimental studies have shown that changes in nodal organization dramatically affect nerve activity, leading to sensory and motor dysfunction (Lacas-Gervais et al., 2004; Sherman et al., 2005; Susuki et al., 2012). However, AnkG-mediated axonal pathology has not been addressed before. In both our models of AnkG ablation, there was a reduction in CAP amplitude without significant changes in NCV. Interestingly, such a phenomenon has been observed previously in patients with axonal neuropathies, who displayed primary pathology of axonal loss (Kimura, 1993; Li, 2015) with normal myelination. Similarly, the examination of myelinated fibers in our early and late AnkG ablation mouse models confirmed ultrastructural changes in the myelinated axons associated with axonal degeneration. Moreover, longitudinal sections (data not shown) revealed a significant accumulation of organelles in the nodal area, suggesting potential axonal cytoskeletal disorganization leading to axonal transport defects and ultimately to axonal degeneration. These data support the idea that perturbation in the axonal domains, and specifically the nodal architecture, has dramatic consequences on axonal health and changes in nerve conduction. Furthermore, the disrupted nodal cytoskeleton potentially contributes to altered axonal transport and ultimately to axonal pathology (Ballin and Thomas, 1969; Lappe-Siefke et al., 2003; Garcia-Fresco et al., 2006; Fabrizi et al., 2007). Together, our studies establish that AnkG is not essential for the initial nodal assembly but is required for nodal maturation and the establishment of a stoichiometrically stable nodal complex; once the nodal complex is fully established with AnkG, the loss of AnkG leads to a sequential disorganization of the nodal components over time, which is associated with changes in axonal electrical properties and ultrastructure.
Footnotes
This work was supported by National Institutes of Health/National Institute of General Medical Sciences Grant GM063074, the National Multiple Sclerosis Society, and the Zachry Foundation. We thank Van Bennett (Duke University, Durham, NC) for providing Ankyrin 3 (Ank3) floxed mice, Matt Rasband (Baylor College of Medicine, Houston, TX) for providing ankyrin R antibodies, Michael Stankewich (Yale University, New Haven, CT) for providing βI spectrin antibodies, and Jeff Dupree (Virginia Commonwealth University, Richmond, VA) for his advice on electron microscopy, which was performed at the University of Texas Health Science Center San Antonio facility.
The authors declare no competing financial interests.
References
- Agarwal A, Dibaj P, Kassmann CM, Goebbels S, Nave KA, Schwab MH (2012) In vivo imaging and noninvasive ablation of pyramidal neurons in adult NEX-CreERT2 mice. Cereb Cortex 22:1473–1486. 10.1093/cercor/bhr214 [DOI] [PubMed] [Google Scholar]
- Ballin RH, Thomas PK (1969) Changes at the nodes of ranvier during wallerian degeneration: an electron microscope study. Acta Neuropathol 14:237–249. 10.1007/BF00685303 [DOI] [PubMed] [Google Scholar]
- Barry J, Gu Y, Jukkola P, O'Neill B, Gu H, Mohler PJ, Rajamani KT, Gu C (2014) Ankyrin-G directly binds to kinesin-1 to transport voltage-gated Na+ channels into axons. Dev Cell 28:117–131. 10.1016/j.devcel.2013.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekku Y, Rauch U, Ninomiya Y, Oohashi T (2009) Brevican distinctively assembles extracellular components at the large diameter nodes of Ranvier in the CNS. J Neurochem 108:1266–1276. 10.1111/j.1471-4159.2009.05873.x [DOI] [PubMed] [Google Scholar]
- Bennett V, Lorenzo DN (2016) An adaptable spectrin/ankyrin-based mechanism for long-range organization of plasma membranes in vertebrate tissues. Curr Top Membr 77:143–184. 10.1016/bs.ctm.2015.10.001 [DOI] [PubMed] [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St Martin M, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ (2001) Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron 30:369–383. 10.1016/S0896-6273(01)00294-X [DOI] [PubMed] [Google Scholar]
- Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, Matthews G (2001) Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron 30:91–104. 10.1016/S0896-6273(01)00265-3 [DOI] [PubMed] [Google Scholar]
- Burgess A, Vigneron S, Brioudes E, Labbé JC, Lorca T, Castro A (2010) Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci U S A 107:12564–12569. 10.1073/pnas.0914191107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttermore ED, Thaxton CL, Bhat MA (2013) Organization and maintenance of molecular domains in myelinated axons. J Neurosci Res 91:603–622. 10.1002/jnr.23197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KJ, Zollinger DR, Susuki K, Sherman DL, Makara MA, Brophy PJ, Cooper EC, Bennett V, Mohler PJ, Rasband MN (2014) Glial ankyrins facilitate paranodal axoglial junction assembly. Nat Neurosci 17:1673–1681. 10.1038/nn.3858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG (2004) Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A 101:8168–8173. 10.1073/pnas.0402765101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JQ, Lambert S, Bennett V (1996) Molecular composition of the node of Ranvier: identification of ankyrin-binding cell adhesion molecules neurofascin (mucin+/third FNIII domain-) and NrCAM at nodal axon segments. J Cell Biol 135:1355–1367. 10.1083/jcb.135.5.1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhashiashvili Y, Zhang Y, Galinska J, Lam I, Grumet M, Salzer JL (2007) Nodes of Ranvier and axon initial segments are ankyrin G-dependent domains that assemble by distinct mechanisms. J Cell Biol 177:857–870. 10.1083/jcb.200612012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshed Y, Feinberg K, Poliak S, Sabanay H, Sarig-Nadir O, Spiegel I, Bermingham JR Jr, Peles E (2005) Gliomedin mediates Schwann cell-axon interaction and the molecular assembly of the nodes of Ranvier. Neuron 47:215–229. 10.1016/j.neuron.2005.06.026 [DOI] [PubMed] [Google Scholar]
- Fabrizi GM, Cavallaro T, Angiari C, Cabrini I, Taioli F, Malerba G, Bertolasi L, Rizzuto N (2007) Charcot-Marie-Tooth disease type 2E, a disorder of the cytoskeleton. Brain 130:394–403. 10.1093/brain/awl284 [DOI] [PubMed] [Google Scholar]
- Feinberg K, Eshed-Eisenbach Y, Frechter S, Amor V, Salomon D, Sabanay H, Dupree JL, Grumet M, Brophy PJ, Shrager P, Peles E (2010) A glial signal consisting of gliomedin and NrCAM clusters axonal Na+ channels during the formation of nodes of Ranvier. Neuron 65:490–502. 10.1016/j.neuron.2010.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Fresco GP, Sousa AD, Pillai AM, Moy SS, Crawley JN, Tessarollo L, Dupree JL, Bhat MA (2006) Disruption of axo-glial junctions causes cytoskeletal disorganization and degeneration of Purkinje neuron axons. Proc Natl Acad Sci U S A 103:5137–5142. 10.1073/pnas.0601082103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavet O, Pines J (2010) Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell 18:533–543. 10.1016/j.devcel.2010.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JA, Yang J, Grati M, Kachar B, Bhat MA (2013) Whirlin, a cytoskeletal scaffolding protein, stabilizes the paranodal region and axonal cytoskeleton in myelinated axons. BMC Neurosci 14:96. 10.1186/1471-2202-14-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TS, Zollinger DR, Chang KJ, Xu M, Cooper EC, Stankewich MC, Bennett V, Rasband MN (2014) A hierarchy of ankyrin-spectrin complexes clusters sodium channels at nodes of Ranvier. Nat Neurosci 17:1664–1672. 10.1038/nn.3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins PM, Kim N, Jones SL, Tseng WC, Svitkina TM, Yin HH, Bennett V (2015) Giant ankyrin-G: a critical innovation in vertebrate evolution of fast and integrated neuronal signaling. Proc Natl Acad Sci U S A 112:957–964. 10.1073/pnas.1416544112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V (2001) Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol 155:739–746. 10.1083/jcb.200109026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V (2002) Developing nodes of Ranvier are defined by ankyrin-G clustering and are independent of paranodal axoglial adhesion. Proc Natl Acad Sci U S A 99:2303–2308. 10.1073/pnas.042601799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MR, Cho MH, Ullian EM, Isom LL, Levinson SR, Barres BA (2001) Differential control of clustering of the sodium channels Na(v)1.2 and Na(v)1.6 at developing CNS nodes of Ranvier. Neuron 30:105–119. 10.1016/S0896-6273(01)00266-5 [DOI] [PubMed] [Google Scholar]
- Kimura J. (1993) Consequences of peripheral nerve demyelination: basic and clinical aspects. Can J Neurol Sci 20:263–270. 10.1017/S0317167100048162 [DOI] [PubMed] [Google Scholar]
- Komada M, Soriano P (2002) [Beta]IV-spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J Cell Biol 156:337–348. 10.1083/jcb.200110003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordeli E, Lambert S, Bennett V (1995) AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem 270:2352–2359. 10.1074/jbc.270.5.2352 [DOI] [PubMed] [Google Scholar]
- Koticha D, Maurel P, Zanazzi G, Kane-Goldsmith N, Basak S, Babiarz J, Salzer J, Grumet M (2006) Neurofascin interactions play a critical role in clustering sodium channels, ankyrin G and beta IV spectrin at peripheral nodes of Ranvier. Dev Biol 293:1–12. 10.1016/j.ydbio.2005.05.028 [DOI] [PubMed] [Google Scholar]
- Lacas-Gervais S, Guo J, Strenzke N, Scarfone E, Kolpe M, Jahkel M, De Camilli P, Moser T, Rasband MN, Solimena M (2004) BetaIVSigma1 spectrin stabilizes the nodes of Ranvier and axon initial segments. J Cell Biol 166:983–990. 10.1083/jcb.200408007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S, Davis JQ, Bennett V (1997) Morphogenesis of the node of Ranvier: co-clusters of ankyrin and ankyrin-binding integral proteins define early developmental intermediates. J Neurosci 17:7025–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA (2003) Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33:366–374. 10.1038/ng1095 [DOI] [PubMed] [Google Scholar]
- Li J. (2015) Molecular regulators of nerve conduction—lessons from inherited neuropathies and rodent genetic models. Exp Neurol 267:209–218. 10.1016/j.expneurol.2015.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SS, Hayes JM, Sims-Robinson C, Sullivan KA, Feldman EL (2010) The effects of anesthesia on measures of nerve conduction velocity in male C57Bl6/J mice. Neurosci Lett 483:127–131. 10.1016/j.neulet.2010.07.076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Gonzalez P, Abdi K, Luciano D, Liu Y, Soriano-Navarro M, Rawlins E, Bennett V, Garcia-Verdugo JM, Kuo CT (2011) Ank3-dependent SVZ niche assembly is required for the continued production of new neurons. Neuron 71:61–75. 10.1016/j.neuron.2011.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai AM, Thaxton C, Pribisko AL, Cheng JG, Dupree JL, Bhat MA (2009) Spatiotemporal ablation of myelinating glia-specific neurofascin (Nfasc NF155) in mice reveals gradual loss of paranodal axoglial junctions and concomitant disorganization of axonal domains. J Neurosci Res 87:1773–1793. 10.1002/jnr.22015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD, Waxman SG (2005) Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J Physiol 564:803–815. 10.1113/jphysiol.2005.083089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer JL, Brophy PJ, Peles E (2008) Molecular domains of myelinated axons in the peripheral nervous system. Glia 56:1532–1540. 10.1002/glia.20750 [DOI] [PubMed] [Google Scholar]
- Sherman DL, Tait S, Melrose S, Johnson R, Zonta B, Court FA, Macklin WB, Meek S, Smith AJ, Cottrell DF, Brophy PJ (2005) Neurofascins are required to establish axonal domains for saltatory conduction. Neuron 48:737–742. 10.1016/j.neuron.2005.10.019 [DOI] [PubMed] [Google Scholar]
- Susuki K, Rasband MN (2008) Spectrin and ankyrin-based cytoskeletons at polarized domains in myelinated axons. Exp Biol Med (Maywood) 233:394–400. 10.3181/0709-MR-243 [DOI] [PubMed] [Google Scholar]
- Susuki K, Yuki N, Schafer DP, Hirata K, Zhang G, Funakoshi K, Rasband MN (2012) Dysfunction of nodes of Ranvier: a mechanism for anti-ganglioside antibody-mediated neuropathies. Exp Neurol 233:534–542. 10.1016/j.expneurol.2011.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susuki K, Chang KJ, Zollinger DR, Liu Y, Ogawa Y, Eshed-Eisenbach Y, Dours-Zimmermann MT, Oses-Prieto JA, Burlingame AL, Seidenbecher CI, Zimmermann DR, Oohashi T, Peles E, Rasband MN (2013) Three mechanisms assemble central nervous system nodes of Ranvier. Neuron 78:469–482. 10.1016/j.neuron.2013.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaxton C, Pillai AM, Pribisko AL, Labasque M, Dupree JL, Faivre-Sarrailh C, Bhat MA (2010) In vivo deletion of immunoglobulin domains 5 and 6 in neurofascin (Nfasc) reveals domain-specific requirements in myelinated axons. J Neurosci 30:4868–4876. 10.1523/JNEUROSCI.5951-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaxton C, Pillai AM, Pribisko AL, Dupree JL, Bhat MA (2011) Nodes of Ranvier act as barriers to restrict invasion of flanking paranodal domains in myelinated axons. Neuron 69:244–257. 10.1016/j.neuron.2010.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh ME, Sloane LB, Fischer KE, Austad SN, Richardson A, Van Remmen H (2015) Use of nerve conduction velocity to assess peripheral nerve health in aging mice. J Gerontol A Biol Sci Med Sci 70:1312–1319. 10.1093/gerona/glu208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young P, Qiu L, Wang D, Zhao S, Gross J, Feng G (2008) Single-neuron labeling with inducible Cre-mediated knockout in transgenic mice. Nat Neurosci 11:721–728. 10.1038/nn.2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V (1998) AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol 143:1295–1304. 10.1083/jcb.143.5.1295 [DOI] [PMC free article] [PubMed] [Google Scholar]