

Abstract
Microinfarcts occur commonly in the aging brain as a consequence of diffuse embolic events and are associated with the development of vascular dementia and Alzheimer's disease. However, the manner in which disperse microscopic lesions reduce global cognitive function and increase the risk for Alzheimer's disease is unclear. The glymphatic system, which is a brain-wide perivascular network that supports the recirculation of CSF through the brain parenchyma, facilitates the clearance of interstitial solutes including amyloid β and tau. We investigated whether glymphatic pathway function is impaired in a murine model of multiple microinfarcts induced by intraarterial injection of cholesterol crystals. The analysis showed that multiple microinfarcts markedly impaired global influx of CSF along the glymphatic pathway. Although suppression of global glymphatic function was transient, resolving within 2 weeks of injury, CSF tracers also accumulated within tissue associated with microinfarcts. The effect of diffuse microinfarcts on global glymphatic pathway function was exacerbated in the mice aged 12 months compared with the 2- to 3-month-old mice. These findings indicate that glymphatic function is focally disrupted around microinfarcts and that the aging brain is more vulnerable to this disruption than the young brain. These observations suggest that microlesions may trap proteins and other interstitial solutes within the brain parenchyma, increasing the risk of amyloid plaque formation.
SIGNIFICANCE STATEMENT Microinfarcts, small (<1 mm) ischemic lesions, are strongly associated with age-related dementia. However, how these microscopic lesions affect global cognitive function and predispose to Alzheimer's disease is unclear. The glymphatic system is a brain-wide network of channels surrounding brain blood vessels that allows CSF to exchange with interstitial fluid, clearing away cellular wastes such as amyloid β. We observed that, in mice, microinfarcts impaired global glymphatic function and solutes from the CSF became trapped in tissue associated with microinfarcts. These data suggest that small, disperse ischemic lesions can impair glymphatic function across the brain and trapping of solutes in these lesions may promote protein aggregation and neuroinflammation and eventually lead to neurodegeneration, especially in the aging brain.
Keywords: glymphatic, microinfarct, vascular dementia
Introduction
A series of publications have recently described a brain-wide perivascular network that supports rapid fluid transport in brain. Termed the glymphatic system because of its dependence upon the astroglial aquaporin-4 (AQP4) water channel, this perivascular network supports the influx of CSF through brain tissue along periarterial pathways and the clearance of interstitial fluid (ISF) and solutes including amyloid β and tau along perivascular pathways associated with large-caliber draining veins (Iliff et al., 2012; Iliff et al., 2013a; Iliff et al., 2013b; Xie et al., 2013) and meningeal lymphatic vessels (Aspelund et al., 2015; Louveau et al., 2015). Glymphatic clearance declines in aging (Kress et al., 2014), which may contribute to the vulnerability of the aging brain to neurodegenerative diseases characterized by the misaggregation of different proteins, such as amyloid β in Alzheimer's disease (Simon and Iliff, 2015).
Loss of perivascular localization of AQP4 in vascular end feet of reactive astrocytes are associated with impairment of glymphatic function in the aging and posttraumatic brain (Iliff et al., 2014; Kress et al., 2014).
Microinfarcts are a common feature of the aging brain, with 30% of the aging public exhibiting some level of microinfarct burden. Among subjects suffering from mild cognitive impairment, incidence is 55%, whereas the incidence approaches 70% among subjects with vascular dementia (Erkinjuntti et al., 1988; Esiri et al., 1997; Haglund et al., 2006; Sonnen et al., 2007). Aging is also associated with the development of small vascular lesions resulting from embolic strokes, microbleeds, or lacunar strokes in patients suffering from small vessel disease. Approximately 87% of 60- to 70-year-old individuals and nearly all 80- to 90-year-olds exhibit subcortical white matter lesions (de Leeuw et al., 2001). Because such lesions are linked with mislocalization of AQP4, a reactive gliosis (Wang et al., 2012; Ren et al., 2013), we here investigated whether multiple small ischemic lesions suppress glymphatic function. The analysis was based on a multiple microinfarct model in which cholesterol crystals are injected in the internal carotid artery of mice (Wang et al., 2012). In this embolic ischemic model, mice develop primarily ipsilateral small incomplete ischemic lesions that result in both reactive gliosis, mislocation of AQP4, and cognitive impairment. Therefore, this acute murine stroke model is ideally suited for assessing how microlesions in the absence of an underlying vascular pathology affect glymphatic function.
We found that microinfarcts cause a rapid and marked decrease in brain-wide glymphatic CSF influx, an effect that was exacerbated in the aging brain compared with the young adult brain. Although this impairment was transient, resolving within 2 weeks after the injury, trapping of solutes within tissue associated with microinfarcts was observed long after global glymphatic pathway function recovered. This observation may explain why embolic strokes, microbleeds, and small-vessel disease all predispose to Alzheimer's disease and dementia. We speculate that the suppression of glymphatic clearance after multiple microinfarcts will accelerate neurodegeneration by promoting the accumulation of neurotoxic waste products, including oligomeric or fibrillary forms of amyloid β, promoting neuroinflammation or accelerating synaptic loss.
Materials and Methods
Animals.
For all experiments, 8- to 10-week-old (22–24 g) male C57BL/6j mice (Charles River Laboratories) and 12-month-old (24–30 g) male C57BL/6j mice were used. All experiments were approved by the Institutional Animal Care and Use Committee of the University of Rochester and were performed according to guidelines from the National Institutes of Health.
Induction of multiple microinfarcts.
USP-grade free cholesterol crystals (Sigma-Aldrich) sized 40–70 μm were collected as described previously (Wang et al., 2012). Briefly, cholesterol crystals were filtered through a 70 μm and then a 40 μm cell strainer (352340; BD Biosciences). Crystals 40–70 μm in size were collected and counted with a hemocytometer. Mice were randomly assigned into cholesterol crystal injection or vehicle (saline) injection groups. Animals were anesthetized with ketamine and xylazine (50 mg/kg and 10 mg/kg, i.p., respectively). The right common carotid artery (CCA), internal carotid artery (ICA), and external carotid artery (ECA) were carefully isolated under a microscope. The extracranial branch of the ICA was ligated with a 7-0 suture and the distal portion of the ECA was permanently ligated. The proximal segments of CCA, ECA, and ICA were temporarily occluded with microvascular clips and an incision was made between the ECA ligation site and the ECA clip. Polyethylene (PE10) tubing was inserted into the ECA toward the CCA and the microvascular clips were removed to restore blood flow. Then, 3000 ± 500 cholesterol crystals in 100 μl of saline or 100 μl of saline alone (for sham animals) were injected via the PE10 tubing. After injection, the tube was removed and the proximal ECA was permanently ligated and the wound was closed. The animals were closely monitored for the first 12 h after induction of injury.
Immunofluorescence.
At 3 or 14 d after injury, multiple microinfarct-treated or sham-treated animals were perfusion fixed with 4% paraformaldehyde, brains were removed and postfixed for 24 h, and 100 μm coronal vibratome sections were cut. Sections were washed and blocked with 3% normal donkey serum in PBS with 0.01% Triton X-100. Slices were incubated with the following primary antibodies: mouse anti-glial fibrillary acidic protein (GFAP, 1:1000; Millipore) and rat anti-CD68 (1:100; Serotec). Secondary detection was performed with Cy3- or Cy5-conjugated donkey anti-mouse or anti-rat antibodies (1:500; Jackson Immunoresearch). Slices were mounted with Prolong Antifade Gold with DAPI (Invitrogen). Images were acquired by laser scanning confocal microscopy (FV500; Olympus) with the same imaging parameters (laser power, photomultiplier tube voltage, and gain) were used across all sections in all groups.
In vivo two-photon imaging.
As described previously (Iliff et al., 2012; Iliff et al., 2013b; Xie et al., 2013; Kress et al., 2014), CSF tracer movement into the mouse brain was visualized in vivo through a closed cranial window by laser-scanning two-photon microscopy. Animals were anesthetized with ketamine and xylazine (50 mg/kg and 10 mg/kg, i.p., respectively), intubated, and ventilated with a small animal ventilator (CWE). Blood gasses and pH were monitored and body temperature was maintained at physiological parameters (∼100 breaths/min, tidal volume of 0.3–0.4 ml, pO2 = 80–150 mmHg, pH 7.25–7.5, body temperature 37°C). Unilateral craniotomy (1 mm lateral and 0.5 mm posterior to bregma; 3 mm in diameter) was performed to expose the cortical surface while leaving the dura intact. The dura was covered with artificial CSF (aCSF) and the window sealed by glass coverslip and Super Glue. The femoral artery was cannulated for monitoring arterial blood gas values and for intraarterial injection of 0.1 ml of blood–brain barrier-impermeable Texas Red-conjugated dextran (MW 70 kDa; 1% in saline; Invitrogen). A Mai Tai laser (SpectraPhysics) attached to a confocal scanning system (Fluoview 300; Olympus) and an upright microscope (IX51W; Olympus) was used for in vivo imaging as described previously (Iliff et al., 2012; Iliff et al., 2013b; Xie et al., 2013; Kress et al., 2014). A 20× (0.9 numerical aperture) water-immersion lens was used to image the cortex. Excitation wavelength was 870 nm for FITC and Texas Red and emission was collected at 575–645 nm. The cerebral vasculature was imaged first with 512 × 512 pixel frames at 5 μm steps to a depth of 240 μm below cortical surface. To evaluate CSF tracer influx into the cortex, 10 μl of fluorescent CSF tracer (FITC-conjugated dextran, 40 kDa, 1% in aCSF; Invitrogen) was infused through a 30 GA needle inserted through the atlanto-occipital membrane into the cisterna magna at a rate of 2 μl/min using a syringe pump. After infusion, 240 μm depth 3D stacks were acquired at 1 min intervals for at least 45 min. As described previously (Iliff et al., 2012; Iliff et al., 2013b; Xie et al., 2013; Kress et al., 2014), analysis of CSF tracer influx into the cortex was conducted in a blinded manner using Fiji software. After imaging, 3D stacks were registered with respect to depth from the cortical surface and the imaging plane 120 μm below the cortical surface was selected for analysis. Circular ROIs 50 pixels in diameter were defined that encompassed the perivascular space immediately surrounding penetrating cerebral arterioles (which were defined morphologically based upon the pial vasculature); donut-shaped ROIs with an outer diameter of 150 pixels were defined that included brain parenchyma surrounding these perivascular ROIs, but excluded the perivascular ROIs themselves. Mean fluorescence intensity was evaluated within the circular perivascular ROI to define perivascular CSF influx and fluorescence intensity was measured within the donut-shaped ROI to define the movement of CSF tracer from the perivascular pathway through the surrounding interstitium.
Ex vivo imaging of CSF tracer influx.
The influx of fluorescent CSF tracer was evaluated throughout the brain by ex vivo imaging of brain slices fixed 30 min or 3 h after CSF tracer infusion. Fluorescent CSF tracer (ovalbumin-conjugated Alexa Fluor 647, 45 kDa; OA-45) was infused intracisternally as above. Thirty minutes or 3 h after the start of tracer infusion, animals were rapidly transcardially perfused, brains dissected and postfixed for 24 h, and then 100 μm vibratome slices were cut and mounted. For each animal, 8 slices (equivalent slices from each animal) were selected and imaged by conventional fluorescence microscopy under 4× objective power. All imaging and image analysis was conducted by a blinded observer. Whole-slice montages were generated in an automated manner using the Virtual Slice module of the Microlucida software package (MBF Bioscience). CSF tracer influx was evaluated using ImageJ software, with mean fluorescence intensity being measured for each whole slice or anatomical region of interest. Fluorescence intensity was summed and averaged across all eight brain sections to define CSF tracer influx for each individual animal. Because multiple microinfarcts within the present model are largely unilateral, the relative fluorescence intensity was measured between the hemispheres ipsilateral and contralateral to the cholesterol injection site by measuring the ratio between ipsilateral:contralateral hemisphere fluorescence intensity.
Statistical analysis.
Data are presented as mean ± SEM. All statistical analysis was performed using Prism software (GraphPad). Dynamic in vivo CSF tracer influx was evaluated with repeated-measures two-way ANOVA with Sidak's post hoc correction for multiple comparisons. The results of ex vivo imaging were evaluated at 30 min and 3 h by standard or repeated-measures 2-way ANOVA with Tukey's or Sidak's post hoc correction for multiple comparisons, respectively. p < 0.05 was considered statistically significant and adjusted p-values (padj) are provided for results of post hoc corrections.
Results
Focal microinfarcts impair global glymphatic pathway function
Three days after injection, microinfarcts were identified by immunostaining against CD68 (a marker for activated microglia and macrophages) and glial fibrillary acidic protein (GFAP, a marker of reactive astrogliosis) and were characterized by a core of CD68+ cells surrounded by large areas of astrogliosis. As described previously (Wang et al., 2012), unilateral cholesterol crystal injection into the internal carotid artery of young adult mice resulted in the formation of multiple microinfarcts distributed primarily within the deep cortex, hippocampus, and subcortical structures in the ipsilateral hemisphere (Fig. 1A,B).
Figure 1.

Focal microinfarcts are associated with widespread reactive astrogliosis. A, Multiple microinfarcts are induced in mice after intracarotid infusion of 40–70 μm cholesterol crystals. B, Three days after induction, microinfarcts (arrows) are distributed largely unilaterally with focal labeling for the reactive microglial or macrophage marker CD68. Focal microinfarcts are surrounded by regional fields of reactive astrocytes characterized by increased GFAP labeling. Inset depicts a single microinfarct. Scale bars: B, 500 μm, 100 μm.
Three days after unilateral intracarotid cholesterol crystal injection, we evaluated glymphatic pathway function by injecting a fluorescent tracer (Texas Red-conjugated dextran, 70 kDa) into the subarachnoid CSF at the cisterna magna. Perivascular CSF tracer influx was evaluated in vivo in the cerebral cortex via a closed cranial window by laser scanning two-photon microscopy as described previously (Iliff et al., 2012; Iliff et al., 2013b; Xie et al., 2013; Kress et al., 2014) (Fig. 2A,B). Within the cortex of sham-treated animals, CSF tracer moved into the cortex first along penetrating cerebral arterioles (Fig. 2B1,C; perivascular ROIs), spreading through the surrounding interstitium (Fig. 2B1,D; parenchymal ROIs). Compared with sham-treated animals, 3 d after induction of multiple microinfarcts, CSF tracer influx along periarterial spaces into the ipsilateral cortex was virtually abolished (Fig. 2B2,C,D; p < 0.0001 sham vs ipsilateral; 2-way repeated-measures ANOVA). Similarly, the perivascular movement of CSF tracer through the contralateral hemisphere was markedly slowed compared with sham-treated animals (Fig. 2B3,C,D; p < 0.01 sham vs contralateral, 2-way repeated-measures ANOVA).
Figure 2.
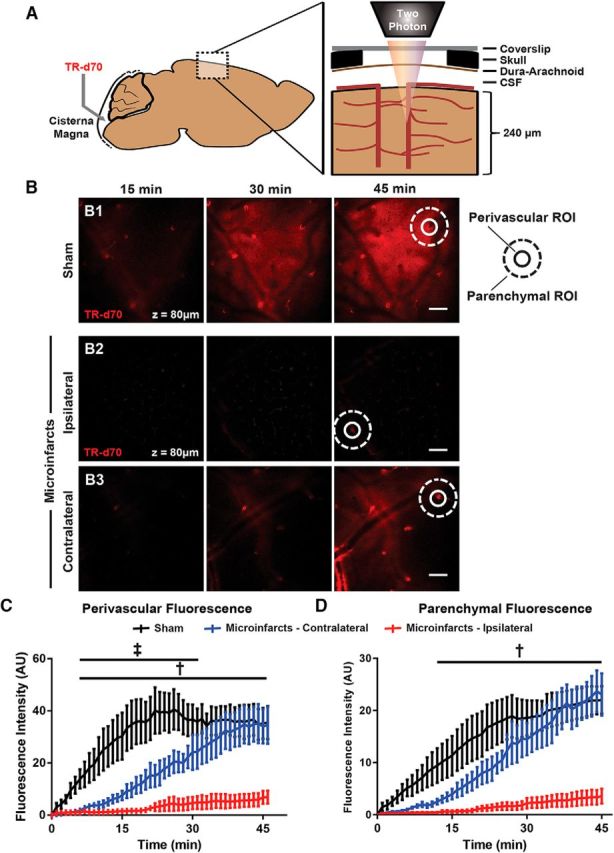
CSF tracer influx along the glymphatic pathway is reduced after multiple microinfarcts. A, Cortical influx of CSF tracer after intracisternal infusion was evaluated in vivo through a closed cranial window by two-photon microscopy. B, Representative imaging planes 120 μm below cortical surface 15, 30, and 45 min after intracisternal tracer infusion of sham-treated mice (B1) or those subjected to multiple microinfarcts and imaged over the ipsilateral (B2) or contralateral (B3) hemispheres 3 d after injury. Representative perivascular (circular) and parenchymal (donut-shaped) ROIs are shown. C, CSF tracer influx is dramatically slowed within both the ipsilateral and contralateral hemispheres (‡padj < 0.05 ipsilateral vs sham; †padj < 0.05 contralateral vs sham; n = 4 per group). D, CSF tracer movement through the surrounding parenchyma was slowed markedly after microinfarcts (†padj < 0.05 ipsliateral vs sham). Scale bars in B, 100 μm.
To evaluate global glymphatic function, 3 d after induction of multiple microinfarcts, fluorescent CSF tracer (OA-45) was injected intracisternally and animals were perfusion fixed 30 min later. Brains were removed, sectioned, and imaged by whole-slice fluorescence imaging to define CSF tracer distribution throughout the brain. At 30 min after injection, CSF tracer distributed extensively and bilaterally through the brain volume (Fig. 3A), with the most intense labeling associated with perivascular spaces surrounding penetrating arteries. In animals subjected to multiple microinfarcts, CSF tracer influx into the brain was significantly reduced at 30 min after injection (Fig. 3B,C; p < 0.01, sham vs microinfarcts, t test). Despite a largely unilateral lesion burden (Wang et al., 2012), CSF tracer influx was globally depressed after multiple microinfarcts. When glymphatic function was evaluated 14 d after injury, CSF tracer influx 30 min after injection was not impaired significantly compared with sham-treated animals (Fig. 3D), suggesting that impairment of global glymphatic function after diffuse microinfarcts is transient.
Figure 3.

CSF influx is globally impaired after multiple microinfarcts. CSF tracer influx was by ex vivo whole-slice imaging 30 min after intracisternal infusion of fluorescent tracer. A–C, Representative images showing that, 3 d after multiple microinfarcts (B), CSF tracer influx is globally impaired compared with sham-treated controls (A). C, CSF tracer influx is dramatically impaired 30 min after tracer infusion, whereas within 3 h of infusion, CSF tracer has entered into the injured brain (**p < 0.01, t test; n = 4–5 per group). D, At 14 d after injury, no significant differences in CSF tracer intensity are observed between animals subjected to multiple microinfarcts or sham operation. Scale bars in A and B, 500 μm.
We next evaluated regional differences in the impairment of glymphatic pathway function with experimental microinfarcts 3 d after injury. CSF tracer influx was evaluated in brain slices fixed 3 h after intracisternal infusion of OA-45 and fluorescence intensity was quantified within ROIs corresponding to the cerebral cortex, corpus callosum, subcortical structures (including the striatum, thalamus, and hypothalamus), and hippocampus. As observed 30 min after tracer infusion, global fluorescence intensity was significantly reduced 3 h after tracer infusion in animals subjected to microinfarcts (Fig. 4A,B; padj = 0.029, 2-way ANOVA with Tukey's post hoc correction). The effect of microinfarcts on glymphatic function in the young adult brain were most pronounced in the subcortical white matter of the corpus callosum and the hippocampus (Fig. 4C; padj = 0.026, 0.025, respectively; 2-way ANOVA with Tukey's post hoc correction).
Figure 4.
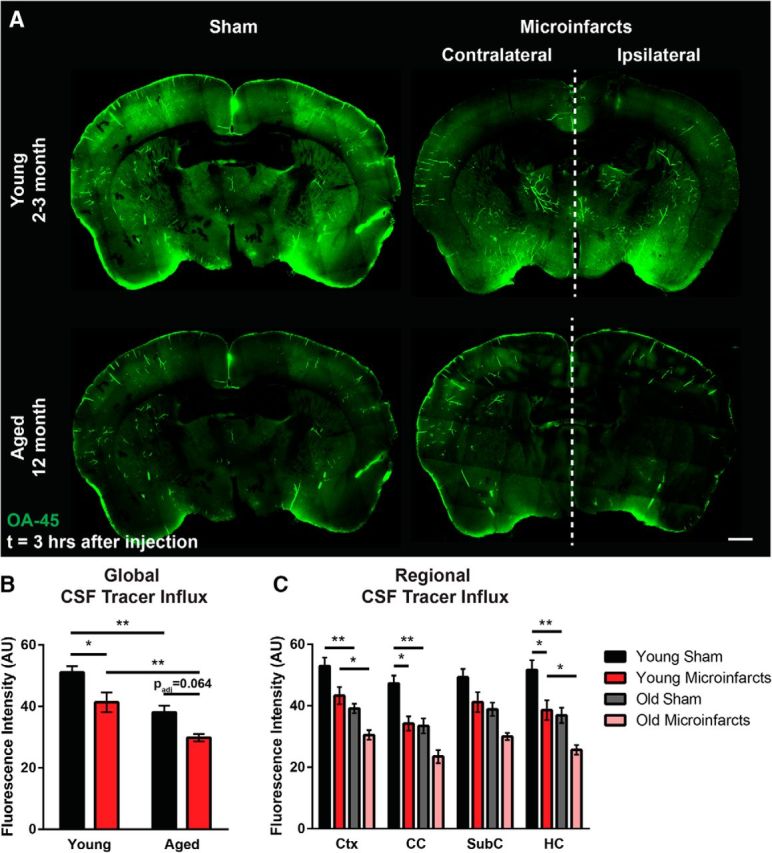
Impairment of glymphatic function by multiple microinfarcts is exacerbated in the aging brain. Regional CSF tracer influx was evaluated in brain tissue fixed 3 h after intracisternal OA-45 infusion. A, Representative whole-slice montages showing CSF tracer distribution in young adult (2- to 3-month-old) and aged (12-month-old) brain 3 d after experimental microinfarcts or sham operation. B, Quantification of global CSF tracer fluorescence showing that CSF tracer influx is reduced in the aged brain, whereas multiple microinfarcts reduce influx into both the young and the aged brain (*padj < 0.05, **padj < 0.01; 2-way ANOVA with Tukey's post hoc correction; n = 7–8 per group). C, Regional CSF tracer influx was evaluated in ROIs including the cortex (Ctx); the corpus callosum (CC); subcortical structures including the striatum, thalamus, and hypothalamus (SubC); and the hippocampus (HC). Slowing of CSF tracer influx into the young brain after multiple microinfacts was most pronounced in the CC and HC; loss of glymphatic function in the aging versus young brain was greatest in the Ctx and HC (*padj < 0.05, **padj < 0.01; 2-way ANOVA with Tukey's post hoc correction; n = 7–8 per group). Scale bars in A, 500 μm.
Aging exacerbates effect of microinfarcts on glymphatic function
Because microinfarcts are a common feature of the aged (Vinters et al., 2000; Sonnen et al., 2011; Kövari et al., 2013), rather than the healthy young brain, we compared the effect of multiple microinfarcts on glymphatic function in 12-month-old animals versus the 2- to 3-month-old young adult animals evaluated above. Confirming our prior findings in aged animals (Kress et al., 2014), we observed that global CSF tracer influx was significantly reduced in sham-treated 12-month-old compared with sham-treated 2- to 3-month-old animals (Fig. 4A,B; padj = 0.002, 2-way ANOVA with Tukey's post hoc correction). In general, we observed that multiple microinfarcts in 2- to 3-month-old young animals reduced glymphatic function to levels observed in intact aged animals, whereas experimental microinfarcts in aged animals exerted an additive impairment of glymphatic pathway function. In aged animals subjected to multiple microinfarcts, global CSF tracer influx was further and significantly reduced compared young animals after injury (Fig. 4A,B; padj = 0.008, 2-way ANOVA with Tukey's post hoc correction). When the effect upon glymphatic function was evaluated in different brain regions, impairment of CSF influx into cortex and hippocampus in the presence of multiple microinfarcts was exacerbated in aged compared with young adult animals (Fig. 4C; padj = 0.033, 0.030, 2-way ANOVA with Tukey's post hoc correction).
Trapping of CSF proteins in focal microinfarcts
When evaluating the influx of the CSF tracer OA-45 (a 45 kDa protein) into brain tissue 3 d after induction of multiple microinfarcts, we observed that, at 3 h after injection, OA-45 appeared to be accumulating in discrete loci within brain tissue (Fig. 5A,B). To evaluate this, we immunolabeled slices from animals injected intracisternally with OA-45 and fixed 3 h after injection. Immunofluorescence for CD68 was used to define ROIs encompassing the core of each microinfarct (Fig. 5A,B, solid line ROIs). These ROIs were projected to the mirror-image locations in the contralateral hemisphere (dashed line ROIs) to allow comparison between microinfarct cores and corresponding intact tissue. To evaluate tracer retention associated with microinfarcts versus that in surrounding tissue, ROIs were also defined encompassing the tissue surrounding each microinfact core (Fig. 5A, inset). Analysis of fluorescence intensity revealed that, at 3 d after injury, CSF tracer intensity was increased markedly in the microinfarct cores compared with surrounding tissue or contralateral ROIs (Fig. 5C; padj < 0.05; repeated measures 2-way ANOVA with Tukey's post hoc correction). At 14 d after injury, similar CSF tracer trapping within microinfarct cores was observed (Fig. 5D; padj < 0.05, repeated-measures 2-way ANOVA with Tukey's post hoc correction). Therefore, whereas global glymphatic pathway function largely recovered within 14 d after induction of multiple microinfarcts, trapping of CSF solutes within these focal lesions persisted.
Figure 5.

CSF solutes are trapped in multiple microinfarcts. A, B, When evaluating CSF tracer influx 3 h after infusion, fluorescent tracer accumulated within tissue associated with microinfarct cores. Tracer intensity was evaluated in mircoinfarct cores defined by positive CD68 labeling (inset, white circle ROIs), in parenchymal regions surrounding microinfarct cores (inset, donut-shaped shaded ROIs), and in mirror-image ROIs in the contralateral hemisphere (dashed ROIs). C, Three days after the induction of injury, CSF tracer intensity was markedly higher in microinfarct cores compared with surrounding tissue (*padj < 0.05, **padj = 0.01; repeated measures 2-way ANOVA with Tukey's post hoc correction; n = 6 per group). D, Trapping of CSF within microinfarct cores continued 14 d after induction of injury (*padj < 0.05, **padj = 0.01; repeated measures 2-way ANOVA with Tukey's post hoc correction; n = 6 per group). Scale bars in A and B, 500 μm.
Discussion
This study showed that induction of multiple microinfarcts suppressed brain-wide perivascular CSF–ISF exchange acutely. Surprisingly, CSF influx was reduced globally, including in the hemisphere contralateral to the microinfarcts, suggesting that even small focal microvascular lesions can trigger a global changes in glymphatic pathway function. Global impairment of perivascular CSF influx was accompanied by a marked trapping of CSF tracers within the cores of microinfarcts. Although global glymphatic function normalized within 14 d of injury, CSF tracer trapping persisted, suggesting that a more subtle and focal form of glymphatic impairment can continue long after the restoration of global perivascular fluid exchange.
The glymphatic system, a brain-wide perivascular network, was first described in 2012. Perivascular CSF influx facilitates the exchange of CSF and ISF through brain tissue, supporting the clearance of interstitial wastes such as amyloid β (Iliff et al., 2012; Iliff et al., 2013a; Iliff et al., 2013b; Xie et al., 2013; Simon and Iliff, 2016). In models of regional ischemic (Gaberel et al., 2014) or traumatic brain injury (Iliff et al., 2014), as well as in the aging brain (Kress et al., 2014), global glymphatic pathway function is slowed, suggesting that this impairment could contribute to both neuroinflammatory and neurodegenerative processes.
Because CSF and ISF move into and out of brain tissue along defined anatomical routes centered around macroscopic cerebral arterial and venous pathways and white matter tracks, we surmised that focal microvascular lesions such as microinfarcts located in strategic anatomical loci may impair glymphatic function over large areas. In the present mouse model of multiple microinfacts, which features microscopic (200–400 μm), primarily unilateral lesions localized to the deep cortical layers, subcortical gray matter, and hippocampus (Wang et al., 2012), glymphatic function was impaired globally, including in the contralateral hemisphere. The mechanism underlying the global impairment of glymphatic pathway function after focal microvascular lesion remains unknown. One possibility is that focal ischemic injury evokes widespread changes in glial water transport. Glymphatic function is dependent upon the expression of the astroglial water channel AQP4, which is localized specifically to perivascular astrocytic end feet that ensheath the cerebral vasculature. Deletion of the Aqp4 gene impairs perivascular CSF influx and interstitial solute clearance (Iliff et al., 2012), whereas loss of perivascular localization of AQP4 in the aging (Kress et al., 2014) and posttraumatic (Ren et al., 2013; Iliff et al., 2014) brain is associated with loss of glymphatic function. In the present model of multiple microinfarcts, wide swaths of gray matter surrounding focal microvascular lesions exhibit pronounced astrogliosis and the loss of perivascular AQP4 localization that resolve only between 14 and 28 d after injury (Wang et al., 2012). Therefore, widespread regions of perivascular AQP4 mislocalization associated with focal microinfarcts may underlie the transient global impairment of glymphatic transport observed in the present study.
The observation that fluorescent CSF tracers infused into the CSF at the cisterna magna enter and remain trapped within microinfarcts even 14 d after injury after the restoration of global glymphatic pathway function, was unexpected and yet may have broad implications. A persistent question in neurology centers involves the basis of the strong association between the presence of a relatively small number of microscopic ischemic lesions and declines in global cognitive function in vascular and mixed dementia. The trapping of solutes (including OA-45) from the CSF compartment within diffuse microinfarcts suggests that a range of solutes from widely dispersed anatomical sources may accumulate within these focal lesions. These solutes might include inflammatory cytokines, cytotoxic wastes such as soluble amyloid β oligomers, or other drivers of neuronal pathology. The present phenomena appear to result from impairment of interstitial solute clearance and glymphatic dysfunction and may be related to focal blood–brain barrier breakdown. Such trapping of compounds within the lesions may serve as persistent foci for chronic neuroinflammation.
Advancing age, ischemic injury, microvascular lesion burden, and traumatic brain injury are all established risk factors for the development of dementia. However, the cellular or molecular mechanisms linking these features and events remain unclear. In prior studies, we and others have demonstrated that glymphatic pathway function is globally impaired in the aging brain (Kress et al., 2014) and after ischemic (Gaberel et al., 2014) or traumatic (Iliff et al., 2014) brain injury. In the present study, we observe that multiple microinfarcts can similarly impair global glymphatic pathway function. Further, our results demonstrate that focal lesions are also associated with more subtle and local glymphatic pathway impairment. Just as impairment of global glymphatic pathway function is proposed to impair widespread neural and network function, the disruption of solute transport surrounding these lesions may similarly drive outsized changes in neuronal network function because of the close association of these deep lesions with the underlying white matter. Therefore, the cellular and molecular mechanisms underlying global impairment of glymphatic pathway function and focal solute trapping within discrete microinfarcts, as well as their clinical significance in the setting of vascular dementia, are clearly an important subject for future study.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke–National Institutes of Health, the American Heart Association, the Leducq Foundation, and the European Union's Horizon 2020 Research and Innovation Programme (Grant 666881, SVDs@target). We thank Yonghong Liao for expert technical support.
The authors declare no competing financial interests.
References
- Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, Wiig H, Alitalo K (2015) A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 212:991–999. 10.1084/jem.20142290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw FE, de Groot JC, Achten E, Oudkerk M, Ramos LM, Heijboer R, Hofman A, Jolles J, van Gijn J, Breteler MM (2001) Prevalence of cerebral white matter lesions in elderly people: a population based magnetic resonance imaging study: the Rotterdam Scan Study. J Neurol Neurosurg Psychiatry 70:9–14. 10.1136/jnnp.70.1.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkinjuntti T, Haltia M, Palo J, Sulkava R, Paetau A (1988) Accuracy of the clinical diagnosis of vascular dementia: a prospective clinical and post-mortem neuropathological study. J Neurol Neurosurg Psychiatry 51:1037–1044. 10.1136/jnnp.51.8.1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esiri MM, Wilcock GK, Morris JH (1997) Neuropathological assessment of the lesions of significance in vascular dementia. J Neurol Neurosurg Psychiatry 63:749–753. 10.1136/jnnp.63.6.749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaberel T, Gakuba C, Goulay R, Martinez De Lizarrondo S, Hanouz JL, Emery E, Touze E, Vivien D, Gauberti M (2014) Impaired glymphatic perfusion after strokes revealed by contrast-enhanced MRI: a new target for fibrinolysis? Stroke 45:3092–3096. 10.1161/STROKEAHA.114.006617 [DOI] [PubMed] [Google Scholar]
- Haglund M, Passant U, Sjöbeck M, Ghebremedhin E, Englund E (2006) Cerebral amyloid angiopathy and cortical microinfarcts as putative substrates of vascular dementia. Int J Geriatr Psychiatry 21:681–687. 10.1002/gps.1550 [DOI] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4:147ra111. 10.1126/scitranslmed.3003748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Lee H, Yu M, Feng T, Logan J, Nedergaard M, Benveniste H (2013a) Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest 123:1299–1309. 10.1172/JCI67677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, Deane R, Nedergaard M (2013b) Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci 33:18190–18199. 10.1523/JNEUROSCI.1592-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, Singh I, Deane R, Nedergaard M (2014) Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci 34:16180–16193. 10.1523/JNEUROSCI.3020-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- K̈ovari E, Herrmann FR, Hof PR, Bouras C (2013) The relationship between cerebral amyloid angiopathy and cortical microinfarcts in brain ageing and Alzheimer's disease. Neuropathol Appl Neurobiol 39:498–509. 10.1111/nan.12003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, Xie L, Kang H, Xu Q, Liew JA, Plog BA, Ding F, Deane R, Nedergaard M (2014) Impairment of paravascular clearance pathways in the aging brain. Ann Neurol 76:845–861. 10.1002/ana.24271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, Harris TH, Kipnis J (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523:337–341. 10.1038/nature14432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Iliff JJ, Yang L, Yang J, Chen X, Chen MJ, Giese RN, Wang B, Shi X, Nedergaard M (2013) ‘Hit and Run’ model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J Cereb Blood Flow Metab 33:834–845. 10.1038/jcbfm.2013.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MJ, Iliff JJ (2016) Regulation of cerebrospinal fluid (CSF) flow in neurodegenerative, neurovascular and neuroinflammatory disease. Biochim Biophys Acta 1862:442–451. 10.1016/j.bbadis.2015.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD, Craft S, Leverenz JB, Montine TJ (2007) Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol 62:406–413. 10.1002/ana.21208 [DOI] [PubMed] [Google Scholar]
- Sonnen JA, Santa Cruz K, Hemmy LS, Woltjer R, Leverenz JB, Montine KS, Jack CR, Kaye J, Lim K, Larson EB, White L, Montine TJ (2011) Ecology of the aging human brain. Arch Neurol 68:1049–1056. 10.1001/archneurol.2011.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinters HV, Ellis WG, Zarow C, Zaias BW, Jagust WJ, Mack WJ, Chui HC (2000) Neuropathologic substrates of ischemic vascular dementia. J Neuropathol Exp Neurol 59:931–945. 10.1093/jnen/59.11.931 [DOI] [PubMed] [Google Scholar]
- Wang M, Iliff JJ, Liao Y, Chen MJ, Shinseki MS, Venkataraman A, Cheung J, Wang W, Nedergaard M (2012) Cognitive deficits and delayed neuronal loss in a mouse model of multiple microinfarcts. J Neurosci 32:17948–17960. 10.1523/JNEUROSCI.1860-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O'Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M (2013) Sleep drives metabolite clearance from the adult brain. Science 342:373–377. 10.1126/science.1241224 [DOI] [PMC free article] [PubMed] [Google Scholar]