

Abstract
Functioning of the glymphatic system, a network of paravascular tunnels through which cortical interstitial solutes are cleared from the brain, has recently been linked to sleep and traumatic brain injury, both of which can affect the progression of migraine. This led us to investigate the connection between migraine and the glymphatic system. Taking advantage of a novel in vivo method we developed using two-photon microscopy to visualize the paravascular space (PVS) in naive uninjected mice, we show that a single wave of cortical spreading depression (CSD), an animal model of migraine aura, induces a rapid and nearly complete closure of the PVS around surface as well as penetrating cortical arteries and veins lasting several minutes, and gradually recovering over 30 min. A temporal mismatch between the constriction or dilation of the blood vessel lumen and the closure of the PVS suggests that this closure is not likely to result from changes in vessel diameter. We also show that CSD impairs glymphatic flow, as indicated by the reduced rate at which intraparenchymally injected dye was cleared from the cortex to the PVS. This is the first observation of a PVS closure in connection with an abnormal cortical event that underlies a neurological disorder. More specifically, the findings demonstrate a link between the glymphatic system and migraine, and suggest a novel mechanism for regulation of glymphatic flow.
SIGNIFICANCE STATEMENT Impairment of brain solute clearance through the recently described glymphatic system has been linked with traumatic brain injury, prolonged wakefulness, and aging. This paper shows that cortical spreading depression, the neural correlate of migraine aura, closes the paravascular space and impairs glymphatic flow. This closure holds the potential to define a novel mechanism for regulation of glymphatic flow. It also implicates the glymphatic system in the altered cortical and endothelial functioning of the migraine brain.
Keywords: aura, glymph, interstitial fluid, lymph, neurodegeneration, trigeminovascular
Introduction
Cortical spreading depression (CSD), first described by Leao (Leao, 1944; Leão, 1986), is a slowly propagating wave of altered neural activity, consisting of a brief excitation followed by a prolonged inhibition. CSD has long been thought to be the neural event that underlies the migraine aura, the nonpainful sensory phenomena that can precede the migraine attack in 25–30% of patients, and is implicated in initiating the headache phase of migraine by activating the nociceptive pathway that provides the sensory innervation to the intracranial meninges (Bolay et al., 2002; Zhang et al., 2010, 2011).
Clinical studies showing that migraine aura patients may be prone to stroke (Scher et al., 2005; Schürks et al., 2009; Gudmundsson et al., 2010; Kurth et al., 2010), silent infarction (Kruit et al., 2004, 2006, 2010), and increased thickness of cortical areas involved in the genesis of visual (Hadjikhani et al., 2001; Granziera et al., 2006) and sensory (DaSilva et al., 2007) aura have raised the possibility that repeated occurrence of CSD may have long-term effects on the brain. In animals, CSD was shown to facilitate cortical damage in models of ischemia (Lauritzen and Strong, 2016), subarachnoid hemorrhage (Hamming et al., 2016), and traumatic brain injury (Toth et al., 2016). To date, the focus of much research has been on CSD's vascular effects, which include mild oligemia (Fabricius and Lauritzen, 1993; Unekawa et al., 2015), impairment of neurovascular coupling (Østergaard et al., 2015), and creation of an O2 supply–demand mismatch (von Bornstädt et al., 2015) that can lead to anoxic levels in metabolically compromised brain. At the cellular and molecular level, repetitive CSDs were also found to induce an inflammatory cascade resulting in the production of inducible nitric oxide synthase and COX-2 in the brain parenchyma (Karatas et al., 2013), as well activation of microglia (Gehrmann et al., 1993) and hypertrophy, proliferation, and activation of astrocytic cells (Kraig et al., 1991; Xue et al., 2009; Sukhotinsky et al., 2011). The clearance of intraparenchymal extracellular macromolecules, like those produced during the inflammatory cascade described above, is thought to be mediated mainly via the glymphatic system. The glymphatic system is a newly characterized series of extracellular compartments that clears waste from the brain parenchyma (Carare et al., 2008; Jessen et al., 2015) into paravascular spaces (PVSs) and then through the dural lymphatics to cervical lymph nodes (Aspelund et al., 2015; Louveau et al., 2015). Impairment of glymphatic pathway function in the aging brain (Kress et al., 2014), during prolonged wakefulness (Xie et al., 2013), after traumatic brain injury (Iliff et al., 2014), or in aquaporin 4 (AQP4) knock-out mice (Iliff et al., 2012) suggests that proper clearance of interstitial fluid and solutes, such as amyloid-β and hyperphosphorylated tau, through this paravascular drainage system is critical for maintaining a healthy brain.
The goal of the current study was to determine whether the glymphatic system is affected by CSD. To answer this question, we used in vivo two-photon imaging to determine whether this cortical event compromises the PVS and the clearance of interstitial solutes through the glymphatic system. As a new approach to examine the effects of CSD, we developed a novel method of visualizing fluid-filled spaces in mice that ubiquitously express GFP in all tissues. We show that CSD has a dramatic effect on the physical dimensions of the PVS, and that glymphatic flow is delayed and slowed by CSD.
Materials and Methods
Animals.
All procedures involving animals were in compliance with the experimental protocol approved by the institutional Animal Care and Use Committee of the Beth Israel Deaconess Medical Center and Harvard Medical School and adhered to the guidelines of the Committee for Research and Ethical Issues of the International Association for the Study of Pain. Mice genotypic lines include bAct-GFP [Jackson Labs (Jax) strain 6567, RRID:IMSR_JAX:006567], Ai14D × CMV-Cre (which we call Ai14Dx, Jax 7914, RRID:IMSR_JAX:007914, and 6054, RRID:IMSR_JAX:006054), and GFAP-cre × mTmG (Jax 12886, RRID:IMSR_JAX:012886, and Jax 7676, RRID:IMSR_JAX:007676). Fluorescent mice were all on C57BL/6J background. Equal numbers of male and female mice, 5–10 months old, were used throughout the experiments. The GFAP-cre-mTmG image comes from one male mouse imaged at 2 months old. Mice were kept in 12 h light/dark cycles and were housed 2–5 per cage.
Anesthesia.
Mice were deeply anesthetized using intraperitoneal injection of urethane (1.5 g/kg) and atropine (0.15 mg/kg). Using this method, we routinely kept mice at normal physiological levels (heart rate, 550–700 beats per minute; arterial O2 saturation, 90–100%; breathing rate, 120–200 breaths per minute; monitored with a MouseOx, Starr Life Science) throughout the length of the experiment. Systemic blood pressure was not monitored.
Skull thinning.
A modified thin-skull procedure was used for acute transcranial imaging based on those described previously (Grutzendler et al., 2002; Yang et al., 2010). Briefly, the scalp was shaved and sterilized, a midline scalp incision was made, and a metal plate was affixed to the skull using cyanoacrylate. An area ≤1.5 mm in diameter was thinned near the middle of the right parietal skull plate. We used a high-speed drill and a microsurgical blade to thin the skull to a thickness of 40–50 μm for the thin-skull preparation.
In vivo imaging.
Fully anesthetized mice were imaged in an Olympus FV1000MPE-E multiphoton Imaging System using a Spectra-Physics Maitai Deepsee laser (∼70 fs pulse width) and a Plan 25× 1.05 numerical aperture objective. Three color channels were collected simultaneously (420–460 nm blue in a photomultiplier tube; 495–540 green and 575–630 red in GaAsP detectors). A wavelength of 890 nm was used to excite all fluorophores, and produce second harmonic generation at ∼445 nm (blue). After skull thinning, mice were placed in the microscope with head plate still attached. Time-lapse three-dimensional image stacks were taken starting just under the skull through the meninges into the brain.
CSD initiation.
CSD was initiated from a small craniotomy ∼3.5 mm anterior to the center of the imaging window. This site was chosen to maximize the distance from the imaging site and thus to avoid any direct effects on the imaged tissue from the CSD-inducing stimuli. To expose the dura, a small (<1 mm2) area was thinned on the frontal skull plate, ∼1 mm anterior to bregma and equidistant medial-laterally from the edges of the frontal plate, and small piece of skull was lifted with a 31 gauge needle. CSD was initiated by either pinprick or KCl crystal, which is similar to the approach in previous studies (Zhang et al., 2010; Charles and Baca, 2013). Pinprick was produced by insertion of a glass micropipette (30 μm tip) ∼500 μm into the cortex for 20 s before removal. For induction by KCl, a single crystal was placed on the cortex for 20 s and then washed off with synthetic interstitial fluid. Pinprick was used in all experiments except those in which dye was injected into the anterior craniotomy. KCl was used in these experiments to avoid a second pipette insertion into the same region of cortex. Consistent with previous studies (Zhang et al., 2010), we observed no differences in the effects of CSD induced by these two methods. Sham mice received the craniotomy but no pinprick or KCl.
Intracortical injection.
One microliter of FITC-conjugated or Texas-red-conjugated 3 kDa dextran (Thermo Fisher Scientific; 2 mg/ml) or of SR101 (Thermo Fisher Scientific; 2 mm) in synthetic interstitial fluid was slowly injected 700–1000 μm beneath the dura through a glass micropipette (tip diameter, ∼10 μm) attached to a micromanipulator. For prelabeling of PVS with Texas-red dextran (TRD) or astrocyte labeling with SR101, the dye was injected into a small craniotomy created using a 31 gauge needle on the corner of the imaging window (<1 mm from the location imaged). For determination of the rate of interstitial/glymphatic flow to the PVS, FITC-dextran was injected into the same opening used for CSD initiation (4 mm anterior to the imaging window, as described above). We found that with slow insertion of the glass electrode and injection, we could inject dye without causing CSD in most animals. Animals where CSD was initiated by the dye injection were excluded from analysis.
Field potential recording.
For verification of CSD, a small craniotomy was created at the edge of the imaging window on the side opposite the CSD initiation location, and cortical activity was recorded (electrocorticogram or local field potential) with a glass micropipette (0.9% saline, ∼1 mΩ, 7 μm tip) placed just below the surface of the cerebral cortex through the craniotomy.
Mapping of injection site.
Mice were killed after dye-flow experiments and the brain was extracted and postfixed in 4% paraformaldehyde and then 30% sucrose before parasagittal sectioning using a cryostat. Slices were mounted and images were taken using a fluorescence microscope.
Image analysis.
Images were analyzed using Fiji (http://www.fiji.sc; RRID:SCR_002285), a version of ImageJ.
Blood vessel and PVS size quantification.
Orthogonal image planes were generated in Fiji from image stacks taken of a blood vessel with surrounding PVS in bAct-GFP or Ai14Dx mice. Three to five orthogonal planes were averaged together, and each color channel was normalized across time according to the mean of the genetic fluorescent channel (green for bAct-GFP, red for Ai14Dx) at each time point to account for small changes in brightness due to objective immersion water evaporation or photobleaching. For cross-sectional area quantification, PVS and blood vessel cross sections were identified anatomically. One brightness threshold was chosen per animal such that the negative space of the lumen could be consistently identified through each time point during baseline image acquisition. The cross-sectional area of the lumen or neighboring PVS was measured at each time point using a custom Fiji macro based on the Wand tool autoselection combined with visual guidance. Areas are represented as fold change compared with their average size during baseline. For changes in vessel lumen and PVS during CSD, 320 × 320 pixel images with 20–25 slices were taken every 20 s starting ≥10 min before CSD induction and for 30 min postinduction. For linear diameter quantification (only used for penetrating-vessel PVSs and their paired surface vessels), a line between the endothelium and parenchyma was drawn on the averaged XY projections at a particular location (chosen for maximal starting PVS size) and the distance was measured for each time point. Because there was an average 15 s difference within each z stack between imaging of the surface and penetrating vessels (each image slice in the stack takes ∼2 s), penetrating-artery PVS was compared with the average of the surface-vessel PVS from the time point before and after the penetrating-vessel image. This average was weighted by proximity in time to the penetrating-vessel PVS image.
Dye-intensity quantification.
To determine the rate that the PVS fills with dye postinjection, orthogonal images of arteries with PVS were constructed as described above. The thresholded cross-sectional areas of PVS measured from the tdTomato channel in Ai14Dx mice were then applied as a selection mask for the green channel (FITC-dextran dye), and average pixel intensity in arbitrary units of fluorescence within the mask was measured. The average green channel pixel intensity due to noise within the PVS at baseline was subtracted from each subsequent time point postinjection per mouse (background noise was subtracted).
Statistics.
Two-tailed paired t test with Bonferroni–Holm correction was used to compare blood vessel lumen characteristics at baseline versus each time point post-CSD. Arterial PVS size calculations had unequal variance and were compared with the Kruskal–Wallis test. Individual time points were compared with baseline using multiple Welsh's t tests for unequal variance with Bonferroni's correction. Two-way repeated-measures ANOVA was used to compare dye intensity areas between control and CSD-induced groups (All five control mice were compared with five of six CSD mice chosen randomly as a matched requirement for repeated-measures ANOVA). One-way ANOVA and post hoc paired t tests with Bonferroni–Holm correction were used to compare time points to baseline for PVS cross-sectional area during the two CSDs induced after dye injection. The 40 min of closure was defined by time points that were both <1 SD of zero and significantly less than baseline. One control mouse was excluded because a CSD was triggered by injection. A two-tailed two-sample equal-variance t test was used to compare dye accumulation in PVS after CSD or control. Means are reported ±SEM. No sample size justification was calculated, but numbers of animals are similar to previously reported findings for arterial lumen changes (Ayata et al., 2004). No randomization was used and blind analysis was either not necessary (for lumen and PVS changes) or not possible (for dye flow within PVS it was obvious which PVS closed due to CSD).
Data availability.
All relevant data are available from the authors by request.
Results
Anatomical and functional characterization of PVS in fluorescent mice in vivo
We developed a novel in vivo method to visualize the PVS in naive uninjected mice, defining PVS here as a fluid-filled space bordered by the pia, the endothelial wall or smooth muscle of pial blood vessels, and the brain. We performed two-photon imaging through a thinned skull in mice expressing fluorescence in all tissues (here using bAct-GFP), leaving fluid-filled spaces anatomically identifiable by lack of fluorescence (Fig. 1). We visualize the PVS in superficial vessels using orthogonal reconstructions, which best show the three-dimensional relationship between vessel lumen, PVS, and overlying subarachnoid space (SAS). To verify that our identification of the PVS is consistent with previous studies (Iliff et al., 2012, 2013, 2014), we took images using GFAP-cre/mTmG mice (n = 3), where astrocytes and neurons are green, all other cellular tissue is red, and fluid-filled spaces lack fluorescence (Fig. 2a), which show that our imaged PVS is bordered by parenchyma, endothelium, and pia. We also injected 3 kDa TRD into the nearby cortex, just outside of the imaging site, of GFP mice (n = 7; Fig. 2c–f), and found that the injected dye fully filled the nonfluorescent space in the GFP mice surrounding both arteries and veins, functionally confirming its identification as PVS. Double injection of 3 kDa FITC-dextran and the astrocytic label SR101 in a wild-type C57 mouse also verified that intracortically injected dextran dye labels PVS bordered by astrocytes within the parenchyma (n = 2; Fig. 2b).
Figure 1.

Method for anatomical characterization and quantification of arterial lumen and PVS. a, A three-dimensional image stack is obtained using two-photon microscopy through the thinned skull of a bAct-GFP mouse, with representative slices shown at the level of the skull (blue is second harmonic generation created by bone), dura, pia, and brain (green). White line represents angle and position of the orthogonal reconstruction shown in b. b, Each cranial layer [including SAS (sas)], along with the arterial lumen (art) and arterial PVS (pvs). c, To quantify cross-sectional areas of PVS and lumen, the green GFP channel is thresholded and cross-sectional areas of PVS and lumen are measured from the result. PVS and lumen areas are normalized across time points to their average baseline size to compare across mice. d, Diagram of mouse skull showing the location of frontal craniotomy, where CSD was induced by pinprick (glass micropipette, black circle), and the thinned area in parietal bone (blue circle), where pial blood vessels and PVS were imaged.
Figure 2.

Functional definition of PVS. a, Orthogonal reconstruction from a GFAP-cre/mTmG mouse, which expresses red tdTomato fluorescence in all tissues except astrocytes and most neurons, which instead express GFP. In this mouse the parenchyma is green, including astrocytic endfeet; the rest of the tissue is red, and the skull is visible as blue second-harmonic generation. The PVS is bordered by the parenchyma (green), blood vessel endothelium, and pial membrane (red; representative of n = 3 mice). b, SR101-labeled astrocytes (red) also reveal the border of the FITC-dextran (green) filled PVS (n = 2). c–f, Labeling of PVS following local intracortical dye injection (n = 7). c, In vivo two-photon image from a bAct-GFP mouse taken near the site of intracortical injection of 3 kDa TRD (slightly above and to the left of the image, arrow), ∼5 min postinjection. Dye has diffused only a short distance through the cortex (dotted line) compared with the faster and farther-reaching bulk flow through PVS (arrowheads). Numbers in red represent dye intensity in the corresponding box. The labeling intensity is higher within PVS than within cortex, and decreases with distance from the injection site more in the cortex than the PVS. d, Dye from an intracortical injection (asterisk) fills both para-arterial space (arrow) and para-venous space (arrowhead), and these spaces can be shared and continuous with each other near large superficial blood vessels. e, Diagram of mouse skull as in Figure 1d, showing the location of the injection of 3 kDa TRD dye (red electrode) through a small craniotomy on the edge of the imaging window (blue circle) for PVS labeling. Dye was injected and allowed to label PVS before imaging. f, g, Orthogonal reconstructions of pial blood vessels after dye injection. f, Dye injected intracortically fills the PVS, but does not cross into the SAS, nor does it fill the blood vessel lumen (BV). g, Dye injected directly into the SAS, achieved by using an oblique angle when inserting the glass electrode, does not cross freely into the underlying PVS. (n = 3).
When labeling the PVS with TRD via intracortical injection, we observed that the pia was a functional barrier to 3 kDa TRD as dye did not readily enter the adjacent SAS. To further test this barrier, we also injected TRD directly into the SAS (n = 3), and found that it did not freely enter the underlying PVS (Fig. 2f).
For imaging and quantification of PVS, we targeted pial blood vessels, whose size ranged from 6.0 to 11 μm in diameter (113–388 μm2 cross-sectional area) for arteries/arterioles (average, 292 ± 52 μm2), and 9.4–21 μm in diameter (279–1370 μm2 cross-sectional area) for veins/venules (average, 675 ± 227 μm2). PVS area varied in size depending on anatomy. For example, PVS was larger if it accompanied multiple blood vessels (Fig. 2d) or blood vessel bifurcations (Fig. 2c, top right). On average, PVS cross-sectional area was 305 ± 140 μm2 near arteries, and tended to be smaller surrounding veins (90.9 ± 28 μm2), even though the veins studied were larger on average (average ratio of PVS/lumen areas was 1.26 for arteries vs 0.13 for veins). Arteries and veins could be differentiated in bAct-GFP mice by the presence of smooth muscle, seen as a thickness in the blood vessel wall (Fig. 3, compare f, g).
Figure 3.
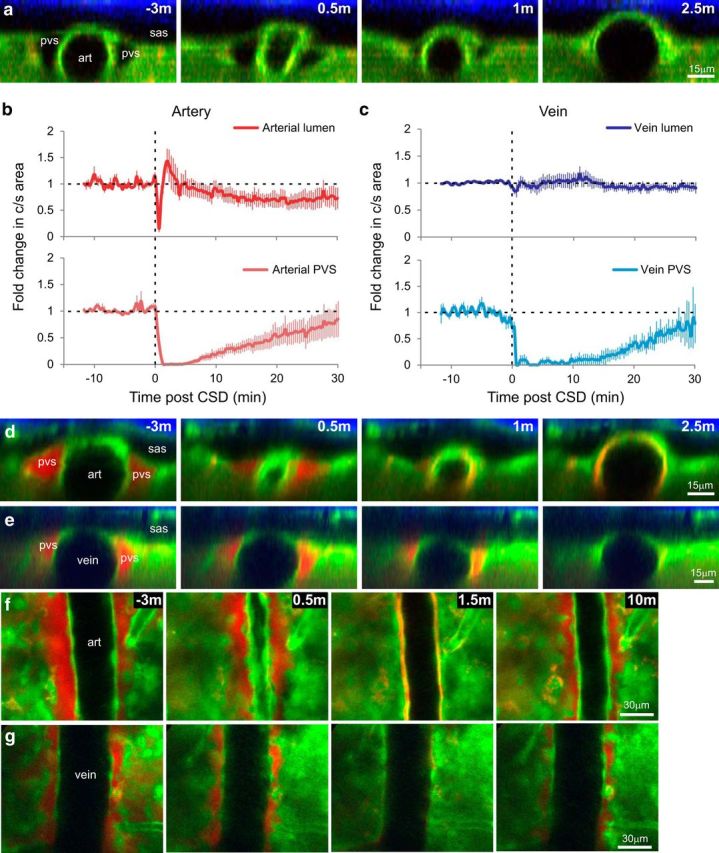
CSD causes closure of arterial and venous PVSs. a, d–g, Pre-CSD and post-CSD in vivo two-photon orthogonal (a, d, e) and XY (f, g) images of PVS near artery (art; a, d, f) and vein (e, g) in bAct-GFP mice. b, c, Quantification of CSD-induced changes in normalized cross-sectional area of lumen (top) and PVS (bottom) of arteries (b) and veins (c) pre-CSD and post-CSD measured in the uninjected mice (a). Time 0 was set at 20 s before initial arterial constriction, which lasted for 1 min and reduced cross-sectional area to 15.1 ± 6.6% of baseline. Arterial lumen then dilated for 3 min to 155 ± 18% of baseline, and constricted again, though to a lesser degree, for ≥20 min (to a minimum of 62.4 ± 12% of baseline at 22 min), while the vein lumen remained unchanged. During this period, para-arterial and paravenous spaces closed (area <1 SD of 0%) for 6 and 16 min, respectively, and partially reopened by 30 min (n = 5 arteries/PVS in 5 mice, n = 4 veins/PVS in 4 mice, error bars are ±SEM). d–g, To determine what happens to PVS solutes during closure, we also examined mice in which the PVS had been dye-filled by intracortical injection of TRD (red) before CSD induction (n = 5). When PVS closes, dye appears nearer to the arterial endothelium but not in the brain parenchyma or SAS (sas).
CSD induces closure of the PVS
To test the effects of CSD on the PVS, we induced CSD by pinprick in the frontal cortex, while imaging the superficial pial vasculature and surrounding spaces in the parieto-occipital cortex ∼3.5 mm caudally (Fig. 1d). Pinprick is a well accepted method of initiating CSD that causes negligible trauma (see Materials and Methods). Blood vessel lumen and PVS cross-sectional areas were measured in mice that were not injected with dye to avoid any brain changes due to injection (although no obvious changes were observed in dye-injected animals). The arrival of the CSD wave in the imaging window (Fig. 3a,b) was confirmed by visualizing the well reported pattern of arterial constriction and dilation (Ayata et al., 2004; Unekawa et al., 2015). CSD arrival caused a significant reduction of arterial lumen cross-sectional area to 15.1 ± 6.6% of baseline (t(4) = 12.72, p = 2.2 × 10−4, paired t test) for 1 min, followed by dilatation to 155 ± 18% of baseline at 3 min, followed by a longer phase of constriction to a minimum of 62.4 ± 12% of baseline at 22 min. That these vascular changes were indeed caused by CSD was confirmed by recording the field potential using an electrode placed within the imaging window (n = 3; Fig. 4). Apart from these vascular changes, CSD caused rapid and nearly complete closure of the PVS surrounding pial arteries, as defined by cross-sectional area reduction to <1 SD of 0% (7.75 ± 5.5 × 10−4 percentage of baseline, SD of 1.29 × 10−3, n = 5, t(4) = 1727, p = 6.7 × 10−13, Welch's t test at minute 1.66), starting within 2 min and continuing for another 6 min (H(67) = 179, p = 2.87 × 10−12 Kruskal–Wallis test, p < 7.5 × 10−4 p-critical for all closed time points, Welch's t test with Bonferroni's correction; Fig. 3a,b,d,f). The PVS then opened gradually, but remained partially constricted for ≥30 min post-CSD, when it reached 85.1 ± 34% of baseline. Induction of CSD by a single brief (20 s) application of KCl crystal to the dura instead of pinprick produced qualitatively similar changes in the dimensions of the PVS.
Figure 4.

Simultaneous field potential recording of CSD and imaging of pial blood vessels. a, Field potential recording of a CSD-induced mouse with representative images of pial vascular changes marked according to the timing of their occurrence. Scale bar, 20 μm (results representative of n = 3 mice). b, Experimental setup. Note imaged area is ∼0.5 mm closer to the CSD initiation site than the recording electrode.
When we induced CSD by pinprick in mice whose PVS was prelabeled with local TRD injection (n = 5), the resulting closure appeared to force the dye between the smooth muscle and the endothelium of the arterial wall (Fig. 3d,f) and between individual smooth muscle cells (Fig. 5). Judging by its brightness, it appears as if the concentration of the dye increased drastically in and near the endothelial cell layer. This pattern of labeling is reminiscent of the periarterial labeling described histologically following intraparenchymal tracer injection and attributed to uptake along basement membrane (Carare et al., 2008). The absence of dye from the SAS or surrounding cortex during each phase of CSD further suggests that there are potential barriers to efflux from the PVS, even during CSD (Fig. 3d).
Figure 5.

Dye in the PVS during CSD. a, XY images of a pial artery during CSD postintracortical injection of 3 kDa FITC-dextran into a wild-type nonfluorescent mouse (images representative of n = 2 mice). Dye in the PVS near a blood vessel lumen (BV) appears to concentrate within the PVS ∼30 s to 1 min, and is then brightest between smooth muscle and near the endothelium when the PVS closes completely at ∼2 min. b, Detail of box indicated in a at −1 min. Smooth muscle (SM) can be seen at the border of the BV and the PVS. Note that the SM cells enlarge as the blood vessel constricts (as expected; 30 s), and that the dye is forced between and around SM cells as the PVS closes (2 min).
Because interstitial fluid has been reported to flow out of the brain through the paravenous (as opposed to para-arterial) space (Iliff et al., 2012), we also measured CSD's effects on pial veins and venous PVS. CSD had minimal effect on vein luminal cross-sectional area (Fig. 3c,e,g), but nonetheless caused paravenous space to close completely within 1 min (n = 4, t(3) = 160.9, p = 3.86 × 10−5, paired t test), remaining <1 SD of zero for 16 min but statistically significantly different from baseline for time points in the range of minute 1.3 to 10 (p < 2.2 × 10−5 p-critical), and then gradually opening to 79.3 ± 37% of its original size over 30 min.
Since previous studies (Iliff et al., 2012, 2013, 2014) have largely dealt with penetrating vasculature, as opposed to surface vasculature, we also imaged the PVS in penetrating arteries. Closure of the PVS was also apparent around penetrating arteries (Fig. 6), although the changes could not be as precisely quantified compared with orthogonal reconstructions because of the smaller baseline size of the PVS, as well as the greater movement of the parenchyma. To compare PVS closure in penetrating and surface arteries, we measured the diameter of the PVS, or distance between the endothelium and the parenchyma, in a consistent location for both surface and penetrating arteries (16–35 μm below) in animals not injected with dye. Penetrating-artery PVS closed during CSD with a similar time frame as surface-vessel PVS (Fig. 6d); a repeated-measures ANOVA showed no significant effect of vessel location, penetrating or surface (F(1,176) = 0.01, p = 0.95), and there was no significant interaction of the groups with time (F(44,176) = 1.1, p = 0.39). The only difference was a tendency for the penetrating-vessel PVS to begin closing slightly before the surface-vessel PVS. At time point 0, here defined as when the surface artery is constricted maximally, the penetrating PVS has closed ∼60% more than the surface-vessel PVS (t(2) = 21.9, p = 0.002 paired t test).
Figure 6.

Penetrating-vessel PVS closes with dynamics similar to those of surface vessels. a, b, Two optical planes from a three-dimensional image stack from a bAct-GFP mouse whose PVS was labeled with a nearby intracortical injection of TRD that includes a surface artery with a penetrating branch. a, A surface pial artery that branches (arrow) and penetrates into the cortex. b, Artery in a as seen 30 μm below surface. c, A Y-Z orthogonal reconstruction along the dotted line in a and b shows the path of the descending artery, and the shared PVS between the surface (arrow) and penetrating (arrowhead) vessel. d, Change in PVS of surface (black) and penetrating (gray) vessel, which was quantified as linear distance between the endothelium and parenchyma, and represented as fold change from average baseline size, in nondye-injected animals. PVS changes of surface and penetrating vessels occur with a similar time course, except that closure tends to occur earlier in penetrating vessels. At the zero time point (time of maximal surface artery constriction) penetrating PVS shows a significantly greater reduction than the pial surface PVS (*p = 0.002, paired t test, n = 3). e–g, CSD causes PVS closure for surface (e) and penetrating (f) artery (16 μm deeper; images of penetrating artery taken 7 s after images of surface artery were taken) in a dye-injected animal. Dotted circle in f is likely a macrophage that has taken up dye. Note that the 20 s time point shows nearly complete closure of penetrating artery PVS while surface PVS is still open. g, Color intensity (arbitrary units) profiles over the dotted line in f show how the Texas-red dextran within PVS of penetrating vessel becomes brighter as it is forced into a smaller volume. Representative of n = 3 dye-injected mice.
CSD impairs glymphatic flow
Because the PVS is considered essential for clearance of brain solutes (Abbott, 2004; Carare et al., 2008; Iliff et al., 2012, 2014; Jessen et al., 2015), we sought to determine the effects of CSD on interstitial/glymphatic flow. Accordingly, we injected dye (3 kDa FITC-dextran) into the frontal cortex (3.5 mm anterior to the imaging window) of ubiquitously labeled td-Tomato red fluorescent mice, and observed how quickly the dye filled the posterior PVS in CSD-induced and non-CSD-induced mice (Fig. 7a,b). The switch from GFP to tdTomato mice and consequently from red to green dextran injection was done to show that the technique was not dependent on the strain of mouse or the wavelength of the dye. To evaluate more accurately the effect of CSD on solute clearance, we attempted to maximize the period the PVS was closed by inducing two rather than one wave of CSD. We used topical application of a crystal of KCl for 20 s instead of pinprick to induce CSD, to avoid repeated needle insertion in a restricted region of cortex (see Materials and Methods). In control animals that received no CSD induction, the dye injection alone produced no significant change in PVS (Fig. 7c, control). Induction of two CSDs, 3 min before and 20 min after dye injection, which caused significant reduction of PVS for 40 min (F(17,90) = 1.39, p < 3.8 × 10−4 p-critical, compared with baseline, Bonferroni–Holm, as well as t(9) = 5.13, p ≤ 0.037 compared with control, Welch's t test; Fig. 7c,f), significantly delayed and reduced accumulation of dye in PVS under the imaging window (13.6 ± 8.8 a.u. in CSD vs 52.3 ± 18 a.u. in control, F(5,40) = 3.61, p = 0.046 at 150 min, p = 0.0086, two-way repeated-measures ANOVA, n = 5 control; n = 6 CSD; see Materials and Methods; Fig. 7d–f).
Figure 7.

CSD impairs interstitial/glymphatic flow. Interstitial/glymphatic flow was measured by the rate at which green 3 kDa FITC-dextran (FD) dye appeared in the parieto-occipital PVS after being injected into the frontal cortex of ubiquitously fluorescent red mice, here using the tdTomato-expressing Ai14Dx strain, exemplifying fluorophore versatility in this technique. a, Diagram of mouse skull showing where CSD was induced with a crystal of KCl, and 3 kDa FITC-dextran was injected (green electrode), along with the imaging window (blue circle). b, Parasagittal view showing location and spread of FD injection in relation to imaging site. c, d, Induction of two CSDs, one before injection and one 20 min after, caused PVS to close significantly for 40 min (c, red time points), and extended the time it took for FD to reach the imaging window by ∼40 min (d), and reduced total accumulation nearly fourfold (b; * denotes p < 0.05, n = 5 control; n = 6 CSD, see Materials and Methods). e, f, Orthogonal (xz) views of pial artery and its PVS taken before and after FD injection (green) in control (e) and CSD (f) mice. Note correlation between CSD-induced PVS closure and paucity of green dye in the PVS (f).
Discussion
Using in vivo two-photon imaging, we show that CSD, a known instigator of migraine, produced a dramatic alteration in both the structure and function of the glymphatic system—a recently described paravascular pathway for waste clearance from brain interstitial fluid. Our key findings are that (1) CSD produces a rapid closure of the PVS around both arteries and veins on the pial surface of the cerebral cortex and (2) this closure is accompanied by a reduction in the outflow of interstitial fluid from the parenchyma into the PVS. While previous studies of the glymphatic system have reported alterations in the outflow of interstitial fluid through the paravascular clearance system, this is the first observation of physical PVS closure in connection with an abnormal cortical event that underlies a neurological disorder. These findings raise the possibility that such alterations may constitute a novel mechanism for regulation of glymphatic flow. In the context of migraine, transient impairment of glymphatic flow may compromise the clearance of a variety of extracellular excitatory and inflammatory chemicals known to increase post-CSD, and consequently lead to the development of localized cortical hyperexcitability (Aurora et al., 1998; Coppola et al., 2007) and structural thickening (Maleki et al., 2011, 2012, 2013).
Mechanism of PVS closure
While we show that CSD causes a physical closure of the PVS, we cannot yet fully explain the mechanism. This closure is not likely to be simply a result of the CSD-induced changes in vessel diameter, because it was maximal throughout both the initial dilation and constriction exhibited by the arteries, and because the closure of the paravenous space (i.e., PVS near pial veins) occurred in the absence of any detectable change in the diameter of the pial vein. Based on reports of neuronal (Takano et al., 2007) and astrocytic endfeet (Tomita et al., 2011) swelling after CSD, it may be more likely that the closure of the PVS is secondary to such swelling—especially considering that astrocytic endfeet form the outer wall of the PVS (Jessen et al., 2015). Also, we observed that the PVS around penetrating arteries may close seconds before it closes around the surface arteries, which raises the possibility that the CSD-induced parenchymal swelling spreads from deep layer I up to the pia.
Mechanism of slowed dye clearance
Our finding that closure of the PVS by CSD was accompanied by a reduction in dye clearance from the injection site suggests that this closure may impede the outflow of interstitial fluid from the brain into the PVS, and slow fluid movement within the PVS. This impairment of flow is consistent with the prior body of evidence on the critical role played by the paravascular pathway in clearance of solutes from the interstitial fluid (Jessen et al., 2015; Bakker et al., 2016). Current knowledge identifies AQP4, a channel that regulates water transport through cell membranes, as a key player in the dynamics of glymphatic flow (Iliff et al., 2012; Nagelhus and Ottersen, 2013). In fact, the presence of AQP4 in astrocytic endfeet makes it an ideal candidate to mediate (1) CSD-induced astrocytic endfeet swelling (Tomita et al., 2011); (2) CSD-induced accumulation of solutes, such as potassium, ATP, and glutamate in the interstitial fluid (Schock et al., 2007; Charles and Brennan, 2009; Ayata and Lauritzen, 2015), potentially due to reduced interstitial flow; and (3) the closure of the PVS. If AQP4 modulation could prevent PVS closure and subsequent flow impairment, then it could be a potential target for migraine treatment.
The glymphatic system and headache
As recognized by the International Headache Classification Committee [Headache Classification Committee of the International Headache Society (IHS), 2013], the aura and the headache phase of migraine sufferers may be separated by 20–40 min (Liveing, 1873; Blau, 1992) or appear simultaneously (Hansen et al., 2012). Attempting to understand these relationships, we showed recently that activation of meningeal nociceptors (Zhang et al., 2010) and central trigeminovascular neurons (Zhang et al., 2011) may coincide with the occurrence of CSD or begin 15–35 min after. CSD is accompanied by a transient increase in the extracellular concentration of a number of substances that have the ability to activate nociceptors. These include potassium (Vyskocil et al., 1972; Ayata and Lauritzen, 2015), glutamate (Molchanova et al., 2004), and ATP (Schock et al., 2007), as well as an inflammatory cascade that results in COX-2 production and meningeal mast cell degranulation (Karatas et al., 2013). Each of these substances has been shown to produce strong activation of nociceptors [glutamate (MacIver and Tanelian, 1993; Du et al., 2001; Cairns et al., 2002); ATP (Cook and McCleskey, 2002; Molliver et al., 2002; Dussor et al., 2008; Zhao and Levy, 2015); potassium (Strassman et al., 1996)]. Immediate onset of headache after aura could be triggered by such chemicals released into the cortical interstitial fluid during CSD, which then enter the PVS around surface pial vessels and activate nociceptors that innervate those vessels (O'Connor and van der Kooy, 1986; Suzuki et al., 1989) or the pial surface itself (Kosaras et al., 2009) in certain areas of the brain. Delayed onset of headache could be caused by the temporary blockade of outflow of excitatory or inflammatory chemicals from the cortex following CSD, which might prevent these chemicals from reaching dural nociceptors until the PVS reopens. This is supported by evidence that glymphatic flow is eventually drained through dural lymphatic vessels that pass through the walls of densely innervated dural sinuses and large veins (Edvinsson and Uddman, 1981; O'Connor and van der Kooy, 1986; Edvinsson et al., 1987). Our results argue against the possibility that the excitatory chemicals released in the cortex during CSD pass directly through the pia and diffuse through the SAS to the overlying dura, because we found that cortical solutes do not diffuse into the SAS, and instead accumulate in the PVS, where they remain segregated from the SAS.
The potential relationship between headache and the glymphatic system is also supported by evidence for inhibition of glymphatic clearance of solutes and metabolites from the brain parenchyma during prolonged wakefulness (Xie et al., 2013) and after traumatic brain injury (Plog et al., 2015), conditions associated with migraine headache. Sleep disturbances, especially insomnia, reliably precipitate headache (Kelman and Rains, 2005) and show a strong association with migraine chronification (Jennum and Jensen, 2002; Dodick et al., 2003; Rains and Poceta, 2006; Rains et al., 2008; Lateef et al., 2011), whereas post-traumatic headache fulfilling migraine criteria is the most frequent symptom after traumatic brain injury (Theeler et al., 2013; Lucas et al., 2014; Anderson et al., 2015). The common finding of a reduction in glymphatic flow now shown for three disparate headache-associated phenomena raises the possibility that alterations in the glymphatic flow might be a general mechanism linking disruption of brain homeostasis to headache. Based on our findings, it may be worthwhile to determine whether partial closure of the PVS plays any role in the inhibition of glymphatic flow during prolonged wakefulness or after traumatic brain injury. In this case, accumulation of interstitial waste, which may occur during sleep deprivation (Xie et al., 2013), may lower the threshold for migraine onset, whereas enhanced glymphatic clearance of solutes from the brain during sleep (Xie et al., 2013) may partially explain the mechanisms by which sleep terminates migraine.
Although there is no experimental evidence on which one can speculate on CSD's impact on glymphatic flow during wake versus sleep states, our data raise the possibility that CSD can suppress glymphatic flow during wakefulness beyond what is observed with wakefulness alone. Along this line, it may also be reasonable to propose that the occurrence of aura during the sleep state may have a more robust effect as it can suppress glymphatic flow at a time it is at maximal value.
Migraine aura, impaired glymphatic system, and brain health
Our finding of decreased glymphatic flow is potentially of significance for understanding the long-term effects of migraine aura on brain health. Impairment of glymphatic flow and clearance of interstitial fluid in the aging brain (Kress et al., 2014), after traumatic brain injury (Iliff et al., 2014; Plog et al., 2015), and during prolonged wakefulness (Xie et al., 2013) increase accumulation of amyloid-β and/or tau proteins (both responsible for neurodegenerative diseases) by reducing the clearance from the brain parenchyma through the PVSs. Each of these conditions (aging, traumatic brain injury, sleep disruption) are also major risk factors for the development of neurodegenerative diseases, such as Alzheimer's disease (Lindsay et al., 2002; Ju et al., 2014), chronic traumatic encephalopathy (Goldstein et al., 2012), and dementia (Montagna et al., 2003; Saper et al., 2010; Moretti et al., 2012; Smith et al., 2013). Clinically, indications for impaired glymphatic functions after occurrence of CSD raise the possibility that migraine aura can facilitate localized degenerative processes. Scientifically, it can provide a new framework for understanding the variety of structural and functional alterations seen in the migraine brain.
Another potential risk to the migraine brain arises from our observation that CSD causes a transient spike in the concentration of interstitial solutes in the space between the smooth muscle and endothelial wall. Given that some of these solutes may include inflammatory molecules, the repeated occurrence of such events could contribute to endothelial dysfunction in pial and cortical arteries. Migraine aura patients exhibiting elevated levels of biomarkers of coagulation activity, fibrinolysis, inflammation, and oxidative stress (Tietjen et al., 2001, 2009), as well as enhanced arterial stiffness and vascular tone (Vanmolkot et al., 2007; Liman et al., 2012), call for further attempts to delineate CSD's impact on the endothelium.
Caveats
One technical caveat that should be noted in studies of the glymphatic system, as discussed previously by Nedergaard (Iliff et al., 2012), is that the dural puncture required for the injection of tracer might itself alter glymphatic flow, and our measurements were done in anesthetized animals, which, in common with sleeping animals, exhibit a higher baseline level of glymphatic flow than animals that are awake.
Footnotes
This work was supported by National Institutes of Health Grants R37 NS079678 and RO1 NS069847 (R.B.), and by a grant from R. Chemers Neustein (A.J.S.). We thank Dr. Rony Nir for assistance with statistical analyses.
The authors declare no competing financial interests.
References
- Abbott NJ. (2004) Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int 45:545–552. 10.1016/j.neuint.2003.11.006 [DOI] [PubMed] [Google Scholar]
- Anderson K, Tinawi S, Lamoureux J, Feyz M, de Guise E (2015) Detecting migraine in patients with mild traumatic brain injury using three different headache measures. Behav Neurol 2015:693925. 10.1155/2015/693925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, Wiig H, Alitalo K (2015) A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 212:991–999. 10.1084/jem.20142290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurora SK, Ahmad BK, Welch KM, Bhardhwaj P, Ramadan NM (1998) Transcranial magnetic stimulation confirms hyperexcitability of occipital cortex in migraine. Neurology 50:1111–1114. 10.1212/WNL.50.4.1111 [DOI] [PubMed] [Google Scholar]
- Ayata C, Lauritzen M (2015) Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 95:953–993. 10.1152/physrev.00027.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata C, Shin HK, Salomone S, Ozdemir-Gursoy Y, Boas DA, Dunn AK, Moskowitz MA (2004) Pronounced hypoperfusion during spreading depression in mouse cortex. J Cereb Blood Flow Metab 24:1172–1182. [DOI] [PubMed] [Google Scholar]
- Bakker EN, Bacskai BJ, Arbel-Ornath M, Aldea R, Bedussi B, Morris AW, Weller RO, Carare RO (2016) Lymphatic clearance of the brain: perivascular, paravascular and significance for neurodegenerative diseases. Cell Mol Neurobiol 36:181–194. 10.1007/s10571-015-0273-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blau JN. (1992) Classical migraine: symptoms between visual aura and headache onset. Lancet 340:355–356. 10.1016/0140-6736(92)91415-5 [DOI] [PubMed] [Google Scholar]
- Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA (2002) Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med 8:136–142. 10.1038/nm0202-136 [DOI] [PubMed] [Google Scholar]
- Cairns BE, Gambarota G, Svensson P, Arendt-Nielsen L, Berde CB (2002) Glutamate-induced sensitization of rat masseter muscle fibers. Neuroscience 109:389–399. 10.1016/S0306-4522(01)00489-4 [DOI] [PubMed] [Google Scholar]
- Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH, Weller RO (2008) Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol 34:131–144. 10.1111/j.1365-2990.2007.00926.x [DOI] [PubMed] [Google Scholar]
- Charles AC, Baca SM (2013) Cortical spreading depression and migraine. Nat Rev Neurol 9:637–644. 10.1038/nrneurol.2013.192 [DOI] [PubMed] [Google Scholar]
- Charles A, Brennan K (2009) Cortical spreading depression—new insights and persistent questions. Cephalalgia 29:1115–1124. 10.1111/j.1468-2982.2009.01983.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook SP, McCleskey EW (2002) Cell damage excites nociceptors through release of cytosolic ATP. Pain 95:41–47. 10.1016/S0304-3959(01)00372-4 [DOI] [PubMed] [Google Scholar]
- Coppola G, Pierelli F, Schoenen J (2007) Is the cerebral cortex hyperexcitable or hyperresponsive in migraine? Cephalalgia 27:1427–1439. 10.1111/j.1468-2982.2007.01500.x [DOI] [PubMed] [Google Scholar]
- DaSilva AF, Granziera C, Snyder J, Hadjikhani N (2007) Thickening in the somatosensory cortex of patients with migraine. Neurology 69:1990–1995. 10.1212/01.wnl.0000291618.32247.2d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodick DW, Eross EJ, Parish JM, Silber M (2003) Clinical, anatomical, and physiologic relationship between sleep and headache. Headache 43:282–292. 10.1046/j.1526-4610.2003.03055.x [DOI] [PubMed] [Google Scholar]
- Du J, Koltzenburg M, Carlton SM (2001) Glutamate-induced excitation and sensitization of nociceptors in rat glabrous skin. Pain 89:187–198. 10.1016/S0304-3959(00)00362-6 [DOI] [PubMed] [Google Scholar]
- Dussor G, Zylka MJ, Anderson DJ, McCleskey EW (2008) Cutaneous sensory neurons expressing the Mrgprd receptor sense extracellular ATP and are putative nociceptors. J Neurophysiol 99:1581–1589. 10.1152/jn.01396.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson L, Uddman R (1981) Adrenergic, cholinergic and peptidergic nerve fibres in dura mater—involvement in headache? Cephalalgia 1:175–179. 10.1046/j.1468-2982.1981.0104175.x [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Ekman R, Jansen I, McCulloch J, Uddman R (1987) Calcitonin gene-related peptide and cerebral blood vessels: distribution and vasomotor effects. J Cereb Blood Flow Metab 7:720–728. 10.1038/jcbfm.1987.126 [DOI] [PubMed] [Google Scholar]
- Fabricius M, Lauritzen M (1993) Transient hyperemia succeeds oligemia in the wake of cortical spreading depression. Brain Res 602:350–353. 10.1016/0006-8993(93)90701-N [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Mies G, Bonnekoh P, Banati R, Iijima T, Kreutzberg GW, Hossmann KA (1993) Microglial reaction in the rat cerebral cortex induced by cortical spreading depression. Brain Pathol 3:11–17. 10.1111/j.1750-3639.1993.tb00720.x [DOI] [PubMed] [Google Scholar]
- Goldstein LE, Fisher AM. Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, et al. (2012) Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 4:134ra160. 10.1126/scitranslmed.3003716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granziera C, DaSilva AF, Snyder J, Tuch DS, Hadjikhani N (2006) Anatomical alterations of the visual motion processing network in migraine with and without aura. PLoS Med 3:e402. 10.1371/journal.pmed.0030402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grutzendler J, Kasthuri N, Gan WB (2002) Long-term dendritic spine stability in the adult cortex. Nature 420:812–816. 10.1038/nature01276 [DOI] [PubMed] [Google Scholar]
- Gudmundsson LS, Scher AI, Aspelund T, Eliasson JH, Johannsson M, Thorgeirsson G, Launer L, Gudnason V (2010) Migraine with aura and risk of cardiovascular and all cause mortality in men and women: prospective cohort study. BMJ 341:c3966. 10.1136/bmj.c3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RB, Sorensen AG, Moskowitz MA (2001) Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A 98:4687–4692. 10.1073/pnas.071582498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamming AM, Wermer MJ, Umesh Rudrapatna S, Lanier C, van Os HJ, van den Bergh WM, Ferrari MD, van der Toorn A, van den Maagdenberg AM, Stowe AM, Dijkhuizen RM (2016) Spreading depolarizations increase delayed brain injury in a rat model of subarachnoid hemorrhage. J Cereb Blood Flow Metab 36:1224–1231. 10.1177/0271678X15619189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JM, Lipton RB, Dodick DW, Silberstein SD, Saper JR, Aurora SK, Goadsby PJ, Charles A (2012) Migraine headache is present in the aura phase: a prospective study. Neurology 79:2044–2049. 10.1212/WNL.0b013e3182749eed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headache Classification Committee of the International Headache Society (IHS) (2013) The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia 33:629–808. 10.1177/0333102413485658 [DOI] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M (2012) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4:147ra111. 10.1126/scitranslmed.3003748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, Deane R, Nedergaard M (2013) Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci 33:18190–18199. 10.1523/JNEUROSCI.1592-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, Singh I, Deane R, Nedergaard M (2014) Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci 34:16180–16193. 10.1523/JNEUROSCI.3020-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennum P, Jensen R (2002) Sleep and headache. Sleep Med Rev 6:471–479. 10.1053/smrv.2001.0223 [DOI] [PubMed] [Google Scholar]
- Jessen NA, Munk AS, Lundgaard I, Nedergaard M (2015) The glymphatic system: a beginner's guide. Neurochem Res 40:2583–2599. 10.1007/s11064-015-1581-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YE, Lucey BP, Holtzman DM (2014) Sleep and Alzheimer disease pathology—a bidirectional relationship. Nat Rev Neurol 10:115–119. 10.1038/nrneurol.2013.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatas H, Erdener SE, Gursoy-Ozdemir Y, Lule S, Eren-Koçak E, Sen ZD, Dalkara T (2013) Spreading depression triggers headache by activating neuronal Panx1 channels. Science 339:1092–1095. 10.1126/science.1231897 [DOI] [PubMed] [Google Scholar]
- Kelman L, Rains JC (2005) Headache and sleep: examination of sleep patterns and complaints in a large clinical sample of migraineurs. Headache 45:904–910. 10.1111/j.1526-4610.2005.05159.x [DOI] [PubMed] [Google Scholar]
- Kosaras B, Jakubowski M, Kainz V, Burstein R (2009) Sensory innervation of the calvarial bones of the mouse. J Comp Neurol 515:331–348. 10.1002/cne.22049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraig RP, Dong LM, Thisted R, Jaeger CB (1991) Spreading depression increases immunohistochemical staining of glial fibrillary acidic protein. J Neurosci 11:2187–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, Xie L, Kang H, Xu Q, Liew JA, Plog BA, Ding F, Deane R, Nedergaard M (2014) Impairment of paravascular clearance pathways in the aging brain. Ann Neurol 76:845–861. 10.1002/ana.24271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruit MC, van Buchem MA, Hofman PA, Bakkers JT, Terwindt GM, Ferrari MD, Launer LJ (2004) Migraine as a risk factor for subclinical brain lesions. JAMA 291:427–434. 10.1001/jama.291.4.427 [DOI] [PubMed] [Google Scholar]
- Kruit MC, Launer LJ, Ferrari MD, van Buchem MA (2006) Brain stem and cerebellar hyperintense lesions in migraine. Stroke 37:1109–1112. 10.1161/01.STR.0000206446.26702.e9 [DOI] [PubMed] [Google Scholar]
- Kruit MC, van Buchem MA, Launer LJ, Terwindt GM, Ferrari MD (2010) Migraine is associated with an increased risk of deep white matter lesions, subclinical posterior circulation infarcts and brain iron accumulation: the population-based MRI CAMERA study. Cephalalgia 30:129–136. 10.1111/j.1468-2982.2009.01904.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurth T, Kase CS, Schürks M, Tzourio C, Buring JE (2010) Migraine and risk of haemorrhagic stroke in women: prospective cohort study. BMJ 341:c3659. 10.1136/bmj.c3659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lateef T, Swanson S, Cui L, Nelson K, Nakamura E, Merikangas K (2011) Headaches and sleep problems among adults in the United States: findings from the National Comorbidity Survey-Replication study. Cephalalgia 31:648–653. 10.1177/0333102410390395 [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Strong AJ (2016) ‘Spreading depression of Leäo’ and its emerging relevance to acute brain injury in humans. J Cereb Blood Flow Metab pii:0271678X16657092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leao A. (1944) Spreading depression of activity in cerebral cortex. J Neurophysiol 7:359–390. [DOI] [PubMed] [Google Scholar]
- Leão AA. (1986) Spreading depression. Funct Neurol 1:363–366. [PubMed] [Google Scholar]
- Liman TG, Neeb L, Rosinski J, Wellwood I, Reuter U, Doehner W, Heuschmann PU, Endres M (2012) Peripheral endothelial function and arterial stiffness in women with migraine with aura: a case-control study. Cephalalgia 32:459–466. 10.1177/0333102412444014 [DOI] [PubMed] [Google Scholar]
- Lindsay J, Laurin D, Verreault R, Hébert R, Helliwell B, Hill GB, McDowell I (2002) Risk factors for Alzheimer's disease: a prospective analysis from the Canadian Study of Health and Aging. Am J epidemiol 156:445–453. 10.1093/aje/kwf074 [DOI] [PubMed] [Google Scholar]
- Liveing E. (1873) On megrim, sick headache. Nijmegen: Arts and Boeve. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, Harris TH, Kipnis J (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523:337–341. 10.1038/nature14432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas S, Hoffman JM, Bell KR, Dikmen S (2014) A prospective study of prevalence and characterization of headache following mild traumatic brain injury. Cephalalgia 34:93–102. 10.1177/0333102413499645 [DOI] [PubMed] [Google Scholar]
- MacIver MB, Tanelian DL (1993) Structural and functional specialization of A delta and C fiber free nerve endings innervating rabbit corneal epithelium. J Neurosci 13:4511–4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleki N, Becerra L, Nutile L, Pendse G, Brawn J, Bigal M, Burstein R, Borsook D (2011) Migraine attacks the basal ganglia. Mol Pain 7:71. 10.1186/1744-8069-7-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleki N, Becerra L, Brawn J, Bigal M, Burstein R, Borsook D (2012) Concurrent functional and structural cortical alterations in migraine. Cephalalgia 32:607–620. 10.1177/0333102412445622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleki N, Becerra L, Brawn J, McEwen B, Burstein R, Borsook D (2013) Common hippocampal structural and functional changes in migraine. Brain Struct Funct 218:903–912. 10.1007/s00429-012-0437-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molchanova S, Kööbi P, Oja SS, Saransaari P (2004) Interstitial concentrations of amino acids in the rat striatum during global forebrain ischemia and potassium-evoked spreading depression. Neurochem Res 29:1519–1527. 10.1023/B:NERE.0000029564.98905.5c [DOI] [PubMed] [Google Scholar]
- Molliver DC, Cook SP, Carlsten JA, Wright DE, McCleskey EW (2002) ATP and UTP excite sensory neurons and induce CREB phosphorylation through the metabotropic receptor, P2Y2. Eur J Neurosci 16:1850–1860. 10.1046/j.1460-9568.2002.02253.x [DOI] [PubMed] [Google Scholar]
- Montagna P, Gambetti P, Cortelli P, Lugaresi E (2003) Familial and sporadic fatal insomnia. Lancet Neurol 2:167–176. 10.1016/S1474-4422(03)00323-5 [DOI] [PubMed] [Google Scholar]
- Moretti L, Cristofori I, Weaver SM, Chau A, Portelli JN, Grafman J (2012) Cognitive decline in older adults with a history of traumatic brain injury. Lancet Neurol 11:1103–1112. 10.1016/S1474-4422(12)70226-0 [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Ottersen OP (2013) Physiological roles of aquaporin-4 in brain. Physiol Rev 93:1543–1562. 10.1152/physrev.00011.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor TP, van der Kooy D (1986) Pattern of intracranial and extracranial projections of trigeminal ganglion cells. J Neurosci 6:2200–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Østergaard L, Dreier JP, Hadjikhani N, Jespersen SN, Dirnagl U, Dalkara T (2015) Neurovascular coupling during cortical spreading depolarization and -depression. Stroke 46:1392–1401. 10.1161/STROKEAHA.114.008077 [DOI] [PubMed] [Google Scholar]
- Plog BA, Dashnaw ML, Hitomi E, Peng W, Liao Y, Lou N, Deane R, Nedergaard M (2015) Biomarkers of traumatic injury are transported from brain to blood via the glymphatic system. J Neurosci 35:518–526. 10.1523/JNEUROSCI.3742-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rains JC, Poceta JS (2006) Headache and sleep disorders: review and clinical implications for headache management. Headache 46:1344–1363. 10.1111/j.1526-4610.2006.00578.x [DOI] [PubMed] [Google Scholar]
- Rains JC, Poceta JS, Penzien DB (2008) Sleep and headaches. Curr Neurol Neurosci Rep 8:167–175. 10.1007/s11910-008-0027-9 [DOI] [PubMed] [Google Scholar]
- Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE (2010) Sleep state switching. Neuron 68:1023–1042. 10.1016/j.neuron.2010.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher AI, Terwindt GM, Picavet HS, Verschuren WM, Ferrari MD, Launer LJ (2005) Cardiovascular risk factors and migraine: the GEM population-based study. Neurology 64:614–620. 10.1212/01.WNL.0000151857.43225.49 [DOI] [PubMed] [Google Scholar]
- Schock SC, Munyao N, Yakubchyk Y, Sabourin LA, Hakim AM, Ventureyra EC, Thompson CS (2007) Cortical spreading depression releases ATP into the extracellular space and purinergic receptor activation contributes to the induction of ischemic tolerance. Brain Res 1168:129–138. 10.1016/j.brainres.2007.06.070 [DOI] [PubMed] [Google Scholar]
- Schürks M, Rist PM, Bigal ME, Buring JE, Lipton RB, Kurth T (2009) Migraine and cardiovascular disease: systematic review and meta-analysis. BMJ 339:b3914. 10.1136/bmj.b3914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Johnson VE, Stewart W (2013) Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol 9:211–221. 10.1038/nrneurol.2013.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassman AM, Raymond SA, Burstein R (1996) Sensitization of meningeal sensory neurons and the origin of headaches. Nature 384:560–564. 10.1038/384560a0 [DOI] [PubMed] [Google Scholar]
- Sukhotinsky I, Dilekoz E, Wang Y, Qin T, Eikermann-Haerter K, Waeber C, Ayata C (2011) Chronic daily cortical spreading depressions suppress spreading depression susceptibility. Cephalalgia 31:1601–1608. 10.1177/0333102411425865 [DOI] [PubMed] [Google Scholar]
- Suzuki N, Hardebo JE, Owman C (1989) Origins and pathways of cerebrovascular nerves storing substance P and calcitonin gene-related peptide in rat. Neuroscience 31:427–438. 10.1016/0306-4522(89)90385-0 [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, Nedergaard M (2007) Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci 10:754–762. 10.1038/nn1902 [DOI] [PubMed] [Google Scholar]
- Theeler B, Lucas S, Riechers RG 2nd, Ruff RL (2013) Post-traumatic headaches in civilians and military personnel: a comparative, clinical review. Headache 53:881–900. 10.1111/head.12123 [DOI] [PubMed] [Google Scholar]
- Tietjen GE, Al-Qasmi MM, Athanas K, Dafer RM, Khuder SA (2001) Increased von Willebrand factor in migraine. Neurology 57:334–336. 10.1212/WNL.57.2.334 [DOI] [PubMed] [Google Scholar]
- Tietjen GE, Herial NA, White L, Utley C, Kosmyna JM, Khuder SA (2009) Migraine and biomarkers of endothelial activation in young women. Stroke 40:2977–2982. 10.1161/STROKEAHA.109.547901 [DOI] [PubMed] [Google Scholar]
- Tomita M, Tomita Y, Unekawa M, Toriumi H, Suzuki N (2011) Oscillating neuro-capillary coupling during cortical spreading depression as observed by tracking of FITC-labeled RBCs in single capillaries. Neuroimage 56:1001–1010. 10.1016/j.neuroimage.2011.02.078 [DOI] [PubMed] [Google Scholar]
- Toth P, Szarka N, Farkas E, Ezer E, Czeiter E, Amrein K, Ungvari Z, Hartings JA, Buki A, Koller A (2016) Traumatic brain injury-induced autoregulatory dysfunction and spreading depression-related neurovascular uncoupling: pathomechanism, perspectives, and therapeutic implications. Am J Physiol Heart Circ Physiol 311:H1118–H1131. 10.1152/ajpheart.00267.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unekawa M, Tomita Y, Toriumi H, Osada T, Masamoto K, Kawaguchi H, Itoh Y, Kanno I, Suzuki N (2015) Hyperperfusion counteracted by transient rapid vasoconstriction followed by long-lasting oligemia induced by cortical spreading depression in anesthetized mice. J Cereb Blood Flow Metab 35:689–698. 10.1038/jcbfm.2014.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanmolkot FH, Van Bortel LM, de Hoon JN (2007) Altered arterial function in migraine of recent onset. Neurology 68:1563–1570. 10.1212/01.wnl.0000260964.28393.ed [DOI] [PubMed] [Google Scholar]
- von Bornstädt D, Houben T, Seidel JL, Zheng Y, Dilekoz E, Qin T, Sandow N, Kura S, Eikermann-Haerter K, Endres M, Boas DA, Moskowitz MA, Lo EH, Dreier JP, Woitzik J, Sakadžić S, Ayata C (2015) Supply-demand mismatch transients in susceptible peri-infarct hot zones explain the origins of spreading injury depolarizations. Neuron 85:1117–1131. 10.1016/j.neuron.2015.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyskocil F, Kritz N, Bures J (1972) Potassium-selective microelectrodes used for measuring the extracellular brain potassium during spreading depression and anoxic depolarization in rats. Brain Res 39:255–259. 10.1016/0006-8993(72)90802-5 [DOI] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O'Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M (2013) Sleep drives metabolite clearance from the adult brain. Science 342:373–377. 10.1126/science.1241224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue JH, Yanamoto H, Nakajo Y, Tohnai N, Nakano Y, Hori T, Iihara K, Miyamoto S (2009) Induced spreading depression evokes cell division of astrocytes in the subpial zone, generating neural precursor-like cells and new immature neurons in the adult cerebral cortex. Stroke 40:e606–613. 10.1161/STROKEAHA.109.560334 [DOI] [PubMed] [Google Scholar]
- Yang G, Pan F, Parkhurst CN, Grutzendler J, Gan WB (2010) Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nat Protoc 5:201–208. 10.1038/nprot.2009.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R (2010) Activation of meningeal nociceptors by cortical spreading depression: implications for migraine with aura. J Neurosci 30:8807–8814. 10.1523/JNEUROSCI.0511-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Levy D, Kainz V, Noseda R, Jakubowski M, Burstein R (2011) Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol 69:855–865. 10.1002/ana.22329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Levy D (2015) Modulation of intracranial meningeal nociceptor activity by cortical spreading depression: a reassessment. J Neurophysiol 113:2778–2785. 10.1152/jn.00991.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are available from the authors by request.