

Abstract
Some rats [sign-trackers (STs)] are prone to attribute incentive salience to reward cues, which can manifest as a propensity to approach and contact pavlovian cues, and for addiction-like behavior. STs also exhibit poor attentional performance, relative to goal-trackers (GTs), which is associated with attenuated acetylcholine (ACh) levels in prefrontal cortex (Paolone et al., 2013). Here, we demonstrate a cellular mechanism, linked to ACh synthesis, that accounts for attenuated cholinergic capacity in STs. First, we found that electrical stimulation of the basal forebrain increased cortical choline transporter (CHT)-mediated choline transport in GTs, paralleled by a redistribution of CHTs to the synaptic plasma membrane. Neither increases in choline uptake nor translocation of CHTs occurred in STs. Second, and consistent with uptake/translocation alterations, STs demonstrated a reduced ability to support cortical ACh release in vivo compared with GTs after reverse-dialysis to elevate extracellular potassium levels. Third, rats were significantly more likely to develop sign-tracking behavior if treated systemically before pavlovian conditioned approach training with the CHT inhibitor VU6001221. Consistent with its proposed mechanisms, administration of VU6001221 attenuated potassium-evoked ACh levels in prefrontal cortex measured with in vivo microdialysis. We propose that loss of CHT-dependent activation of cortical cholinergic activity in STs degrades top-down executive control over behavior, producing a bias for bottom-up or stimulus-driven attention. Such an attentional bias contributes to nonadaptive reward processing and thus identifies a novel mechanism that can support psychopathology, including addiction.
SIGNIFICANCE STATEMENT The vulnerability for addiction-like behavior has been associated with psychological traits, such as the propensity to attribute incentive salience to reward cues that is modeled in rats by sign-tracking behavior. Sign-trackers tend to approach and contact cues associated with reward, whereas their counterparts, the goal-trackers, have a preference for approaching the location of the reward. Here, we show that the capacity of presynaptic cholinergic synapses to respond to stimulation by elevating presynaptic choline uptake and releasing acetylcholine is attenuated in sign-trackers. Furthermore, pharmacological inhibition of choline transport induced sign-tracking behavior. Our findings suggest that reduced levels of cholinergic neuromodulation can mediate an attentional bias toward reward-related cues, thereby allowing such cues to exert relatively greater control over behavior.
Keywords: acetylcholine, addiction, attention, choline transporter, motivation, sign-tracking
Introduction
Structural abnormalities in cortical and subcortical regions and associated impairments in attentional control, impulsivity, and compulsivity have been documented in people with addiction. These abnormalities may represent pre-existing biopsychological risk factors and may not merely be attributable to extensive drug taking per se, although drug use may further impair cortico–subcortical interactions (Volkow et al., 2005; Tomasi et al., 2007; Ersche et al., 2011, 2012; Robbins et al., 2012). Converging research in animals likewise has focused on the identification of behavioral and neurobiological indices of psychological traits that bestow vulnerability for drug taking and relapse (Piazza et al., 1989; Flagel et al., 2014, 2016).
Among such behavioral indices, sign-tracking is a powerful behavioral predictor for a propensity for drug taking and relapse (Saunders and Robinson, 2010, 2011; Meyer et al., 2012; Yager and Robinson, 2013; Robinson et al., 2014; Yager et al., 2015). Sign-tracking rats (STs) are screened from outbred populations and differentiated from goal-tracking rats (GTs) based on their behavior during a pavlovian conditioned approach (PCA) test. In a PCA trial, a lever is extended, and it predicts subsequent reward delivery. In GTs, the lever acts as a predictive conditioned stimulus (CS) that evokes a conditioned approach response (CR), which is directed toward the site of reward delivery. In STs, the predictive CS also evokes an approach CR, but it is directed toward the cue or “sign.” STs not only approach reward cues but also work more avidly for them, and such cues are more effective in instigating reward-seeking behavior in STs than GTs (Flagel et al., 2009). Approaching and contacting the CS coincides with cue-evoked dopamine signaling in the nucleus accumbens (Flagel et al., 2011; Saunders and Robinson, 2012).
Because attentional abnormalities have been considered an essential component of psychological traits associated with addiction vulnerability (Black et al., 2006; Tomasi et al., 2007; Field and Cox, 2008; Marhe et al., 2013; Kilts et al., 2014), we previously assessed the attentional capacity of STs and GTs, demonstrating that, relative to GTs, STs exhibited relatively poor and highly unstable levels of attentional performance (Paolone et al., 2013). The performance of STs suggested poor top-down attentional control, where the impact of goal-directed behavior, task maintenance, and outcome-based performance adjustment is minimized and stimulus-driven and bottom-up processes characterize performance. Furthermore, we found that cortical levels of cholinergic neuromodulation during attention performance were significantly attenuated in STs relative to GTs. These findings are consistent with the general hypothesis that levels of cholinergic neuromodulation contribute to a bias for top-down (relatively high levels of cholinergic neuromodulation) versus bottom-up (low levels) attentional control (Sarter et al., 2016a,b).
A cellular mechanism underlying the constrained capacity for cholinergic neuromodulation in STs has yet to be defined. Here, we focused on the presynaptic choline transporter (CHT; SLC5A7) because the CHT is the most critical, or rate-limiting, step for the synthesis of acetylcholine (ACh; Ferguson et al., 2004; Sarter and Parikh, 2005; Haga, 2014; Ennis and Blakely, 2016). As such, the CHT is a major determinant of the capacity of cholinergic neurons to sustain elevated levels of cholinergic neuromodulation (Sarter et al., 2016b). Additionally, genetically imposed reductions in CHT function have been found to reduce nucleus accumbens elevations in dopamine after cocaine or nicotine administration, suggesting CHT levels as a trait-level determinant of reward (Dong et al., 2013). We first examined CHT function in STs and GTs using methods adopted from our studies on genetically imposed CHT capacity variants (Parikh et al., 2013). We found that CHT-mediated choline transport and the subcellular distribution of CHTs failed to respond to stimulation in STs and that pharmacological inhibition of CHT function promotes sign-tracking behavior. These and additional results suggest that bottom-up or stimulus-driven attention, mediated via relatively low levels of cholinergic neuromodulation, is an important component of the psychological trait indexed by sign-tracking behavior.
Materials and Methods
Subjects
Adult Sprague Dawley rats (n = 261) were obtained from Harlan and Charles River and were ∼3 months of age at the beginning of the experiments. We previously reported that the expression of the ST/GT phenotype in Sprague Dawley rats and heterogeneous stock rats is not affected by sex (Pitchers et al., 2015), and thus male Sprague Dawley rats were used for the present experiments. Screening of STs and GTs in 212 rats yielded totals of 74 GTs and 68 STs (34.91 and 32.08%, respectively; see below for definition of phenotype based on PCA scores). From these rats, 34 GTs and 30 STs were randomly selected to be used for the main experiments. An additional 49 rats were used to assess the effects of VU6001221 on PCA performance. Animals were individually housed in opaque single standard cages (27.70 × 20.30 cm) in a temperature- and humidity-controlled environment (23°C, 45%) and maintained under a 12 h light/dark schedule (lights on at 8:00 AM). Food (Rodent Chow; Harlan Teklad) and water were available ad libitum. All procedures were conducted in adherence with protocols approved by the Institutional Animal Care and Use Committee of the University of Michigan and in laboratories accredited by the Association for Assessment and Accreditation of Laboratory Animal Care.
PCA screening
Apparatus and procedures.
The screening of STs and GTs using a PCA test followed established methods (Paolone et al., 2013; Robinson et al., 2014; Saunders et al., 2014; Pitchers et al., 2015; Yager et al., 2015). Briefly, rats were handled daily for at least 1 week and given ∼15 banana-flavored grain-based pellets (45 mg; BioServ) in their home cages for 2 d before the start of testing. Rats were tested in conditioning chambers (20.5 × 24.1 cm floor area, 20.2 cm high; MED Associates). A food magazine with an automatic feeder that delivered banana pellets was located in the center of one of the walls of the chamber. Infrared photobeam breaks detected magazine entries. On either the left or the right side of the magazine was a retractable lever with an LED backlight that was illuminated only when the lever extended into the chamber. Deflections of the lever were used to quantify lever contacts. The beginning of a test session was signaled by the illumination of a red house light located near the ceiling of the side of the chamber opposite to the magazine/lever. On the first day of testing (“pretraining”), rats were placed into the conditioning chambers and the house light was illuminated after a 5 min habituation period. Twenty-five pellets were then delivered on a VI-30 (0–60 s) schedule. The pretraining session lasted 12.5 min, on average, and the lever was retracted throughout the session. During this session and all subsequent PCA sessions, rats consumed all pellets. In the next five PCA sessions, the house light was turned on, and rats were then presented with 25 lever/pellet pairings delivered on a VI-90 (30–150 s) schedule. The CS for each trial was the extension of the illuminated lever into the chamber for 8 s. After retraction of the lever, a pellet was immediately delivered into the magazine. On average, the PCA test sessions lasted 37.5 min.
PCA measures and classification criteria.
Lever presses and magazine entries during the CS periods were used to quantify three measures of approach that determined the PCA index score. (1) Response bias was defined as the difference between lever presses and magazine entries, expressed as a proportion of the total responses [(lever presses − magazine entries)/(lever presses + magazine entries)]. (2) Latency score was calculated as the difference between the latency to approach the lever and the magazine after CS presentation; this difference was normalized by dividing by the maximum 8 s latency [(magazine latency − lever latency)/8]. (3) Probability difference was calculated as the difference between the probabilities of pressing the lever during the CS (i.e., the number of trials with a lever press out of 25 trials) minus the probability of entering the magazine. The PCA index score was the average of the response bias score, latency score, and probability difference. The values of this score ranged from 1.0 to −1.0, with a score of 1.0 indicating approaches and contacts of the lever on every trial and a score of −1.0 indicating approaches and contacts of the magazine entry on every trial. For the purpose of classification, rats with an averaged PCA index score from PCA sessions 4 and 5 ranging from −1.0 to −0.5 were defined as GTs (i.e., rats more likely to direct behavior toward food magazine than lever), and rats with a PCA index score between +0.5 and +1.0 were designated as STs (i.e., rats more likely to direct behavior toward the lever-CS than the food magazine). Rats that were within the range of −0.49 to +0.49, whose behavior vacillated between lever-CS and food magazine, were classified as Intermediates (INs; Meyer et al., 2012). Approach responses (response probability, number of contacts, and latency) were analyzed with repeated-measures ANOVAs with phenotype (STs, GTs) as the between-groups measure and training day as the within-subject factor.
Basal forebrain electrical stimulation
The capacity of the CHT to support CHT-mediated choline transport and associated CHT subcellular distribution was assessed in synaptosomes harvested after basal forebrain electrical stimulation (BF-ES) in vivo. BF-ES stimulation parameters were selected from prior studies showing effects on sensory encoding and facilitation of cognitive processes (Edeline et al., 1994; McLin et al., 2002; Goard and Dan, 2009). Furthermore, these parameters were previously demonstrated to upregulate CHT function, including increases in synaptic plasma membrane CHT density (Parikh et al., 2013). Rats were anesthetized with urethane (1.25–1.5 g/kg, i.p.) and placed on a stereotaxic frame. A concentric bipolar electrode (FHC) was lowered into the right BF to electrically stimulate cholinergic neurons in the region of the nucleus basalis of Meynert and the substantia innominata, by applying 1 Hz square pulse (100 μA) trains of 500 ms for 20 min using an isolated pulse stimulator (model 2100; A-M Systems). Control animals were prepared similarly, but stimulation pulses were not applied (sham). Animals were decapitated immediately after BF-ES, and the right frontal cortex and both striata were isolated. To obtain sufficient protein for the ex vivo assays, tissues from separate groups of STs and GTs were generated to determine effects of BF-ES on choline uptake capacity and subcellular CHT distribution.
Synaptosomal choline uptake and immunoblotting experiments
Tissue harvesting and generation of synaptosomal pellets.
Rats were decapitated while still under urethane anesthesia (25%; 1.25–1.5 g/kg; i.p.). The cerebellum, brain stem, and olfactory bulb were removed. The hemispheres were separated, and landmarks (hippocampus and striatum) were used to divide the right hemisphere on a rostrocaudal axis. The frontal cortex and striatum were harvested separately on an ice-cold Petri dish, and the remaining subcortical structures were discarded. Isolated tissues were homogenized in ice-cold 0.32 m sucrose. The homogenate was centrifuged for 4 min at 1000 × g. The supernatant was then centrifuged at 12,500 × g for 10 min. The liquid was removed, and the synaptosomal pellet was either used for measuring choline uptake or was frozen at −80°C and shipped to V.P. at Temple University for immunoblot analyses.
CHT-mediated choline transport.
Synaptosomes were suspended in Krebs' bicarbonate buffer containing 118 mm NaCl, 4.7 mm KCl, 1.2 mm KH2PO4, 25 mm NaHCO3, 1.7 mm CaCl2, 10 mm glucose supplemented with 100 μm ascorbic acid, and 10 μm physostigmine and was bubbled with a 95:5 mixture of O2/CO2. Choline transport assays were performed by incubating aliquots (50 μl) of the synaptosomal suspension for 5 min at 37°C in the presence and absence of 10 μm hemicholinium-3 (HC-3), a potent and selective CHT blocker (Guyenet et al., 1973). The incubation was stopped by transferring the tubes to an ice bath, followed by rapid filtration using a cell harvester (Brandel). Kinetic studies were performed using 20 nm to 6 μm choline with 10% [3H]choline. Protein concentration was measured using the Pierce BCA protein assay kit (Thermo Fisher Scientific). CHT-mediated choline uptake was determined as total choline uptake minus the uptake in the presence of HC-3 (Parikh and Sarter, 2006; Parikh et al., 2006, 2013). Maximum choline transporter velocity (Vmax) and the affinity for choline (Km) were determined using Prism 7 (GraphPad Software).
Subcellular fractionation and immunoblotting.
Subcellular fractionation was conducted to isolate the plasma membrane-enriched (LP1) and vesicular membrane-enriched (LP2) fractions as described previously (Ferguson et al., 2003; Gates et al., 2004; Apparsundaram et al., 2005; Parikh et al., 2013). Briefly, the synaptosomal pellet was lysed in 5 mm HEPES-NaOH, pH 7.4, containing 1.0 μg/ml leupeptin, 1.0 μg/ml aprotinin, 1.0 μg/ml pepstatin, and 250 μg/ml phenylmethylsulfonyl fluoride. The synaptosomal plasma membrane-enriched LP1 fraction was collected by centrifuging the lysate at 15,000 × g for 20 min. The resultant supernatant was centrifuged at 200,000 × g for 30 min to obtain the intracellular membrane-enriched LP2 fraction using a Beckman-Coulter TL100 ultracentrifuge. Proteins were extracted from each fraction with 1% SDS, 5 mm HEPES-KOH, pH 7.3, 1 mm EDTA, 1 mm EGTA, and a protease inhibitor mixture. Protein concentrations were determined using a modified Lowry Protein Assay (Pierce). Equal quantities (25 μg) of protein from each fraction were subjected to immunoblot analysis, and each sample was assayed in duplicate. Proteins were separated on 4–15% Tris-HCl polyacrylamide gels and transferred in PVDF membranes. Immunodetection of CHT bands was accomplished by incubating the membranes overnight with 1:2000 diluted rabbit anti-CHT polyclonal antibody (ABN458; Millipore). This antibody was raised against the recombinant GST-tagged full-length human CHT protein (Uniprot number Q9GZV3). The membranes were then exposed to horseradish peroxidase-conjugated anti-rabbit secondary antibody raised in donkey and ECL Advance chemiluminescent substrate (GE Healthcare). The resulting chemiluminescent signal was acquired with a Molecular Imager Chemidoc EQ system (Bio-Rad). Densitometric analysis of CHT-immunoreactive bands in the LP1 and LP2 fractions was performed by calculating the integrated pixel densities using NIH ImageJ software. The membranes were stripped for the detection of β-tubulin in all samples that served as a control to accommodate any differences in the sample loading during gel electrophoresis. CHT densities were normalized to the levels of β-tubulin-immunoreactive bands for each sample analyzed.
In addition to determining CHT densities in the LP1 and LP2 fractions, we also determined total CHT protein in cortical synaptosomes. Proteins were extracted from lysed synaptosomal pellets and subjected to the CHT immunoblotting procedure as described above.
VU6001221
VU6001221 (Vanderbilt Center of Neuroscience Discovery) is a second-generation CHT inhibitor with improved CNS penetration and in vivo pharmacokinetic characteristics (Bertron et al., 2016). VU6001221 was dissolved in 10% Tween 80 in water and administered in a volume of 1.0 ml/kg body weight. Dissolving VU6001221 required placing the drug preparation into a sonicator for ∼1 h with intermittent vortexing. The temperature of the sonicator bath was monitored and kept below 37°C.
Dose–response pilot studies indicated that VU6001221 was lethal at 6.0 and 10 mg/kg doses, whereas at 3.0 mg/kg, rats appeared sickly for >1 h, as indicated by hunched posture, lethargy, and limited consumption of food pellets during training. Thus, the effects of VU6001221 on PCA performance and stimulated extracellular ACh were tested at a dose of 0.3 mg/kg (i.p.), a dose where signs of sickness or discomfort were absent.
Effects of VU6001221 on PCA performance
PCA testing was conducted using the apparatus and procedures described above. Before the start of pavlovian training, all animals received habituation vehicle injections (intraperitoneally) for 2 consecutive days (including the pretest training day). Before training session 1, all rats were given a vehicle injection 12 min before the start of training. After training session 1, rats with a calculated PCA index value between −1.0 and 0 were selected for this experiment, for the following reason. Although the scores from session 1 are an imperfect predictor of the subsequent phenotype, rats with high PCA scores (>0.5) on session 1, indicating sign-tracking, may be more likely to emerge as STs (Flagel et al., 2007). Thus, by excluding rats with session 1 scores >0, we minimized the manifestation of sign-tracking in our experimental groups and composed experimental groups that favored the rejection of our hypothesis that administration of VU6001221 fosters sign-tracking. Selected rats were assigned to the two treatment groups (VU, n = 25; vehicle, n = 24). Rats received the appropriate injection 12 min before the start of each of the next four training sessions (sessions 2–5). All animals consumed all food pellets during each training session. At the end of each training session, the animals were returned to their home cages in the animal colony room.
Microdialysis and HPLC-mass spectrometry
Potassium-evoked cortical ACh release.
A microdialysis guide cannula and probes were custom designed. For the construction of the probe, two 75 μm (inner diameter) × 150 μm (outer diameter) fused silica capillary tubings (TSP075150; Polymicro Technologies) were glued together and inserted into a 22 gauge stainless steel tubing, which served as a shaft, with a 2 mm offset at the tip for the inlet capillary. The capillary tip was ensheathed in a 20 kDa molecular weight cutoff polyacrylonitrile membrane (AN69; Hospal). The tip and the base of the membrane were sealed with epoxy resin (Locitite; Henkel). Guide cannulae were implanted into the infralimbic region of the medial prefrontal cortex of STs and GTs (from bregma: AP, +2.9 mm; ML, −0.6 mm; DV, 2.0 mm below skull). To prevent clogging, the cannulae were equipped with stainless steel stylets. A headstage to hold the guide cannula in place was constructed using surgical screws implanted into the skull and dental cement. Rats were habituated to a test box (30.0 × 25 cm area × 23 cm height) for 30 min each day for 2 d. Furthermore, a tether was clamped to a hook adhered to the headstage to accommodate rats to the test conditions. On day 3, microdialysis sessions began with the removal of the stylet and the insertion of a probe (2.0 mm membrane tip). Animals were perfused at a rate of 1.0 μl/min with artificial CSF (aCSF; pH 7.4) containing the following (in mm): 145.00 NaCl, 2.68 KCl, 1.10 MgSO4 1.22 CaCl2, 0.50 NaH2PO4, and 1.55 NaHPO4 as well as 25 ascorbic acid. A relatively low concentration of neostigmine (10 nm) was also added to the aCSF perfusate to enhance the detection of ACh while having minimal effects on basal and activated ACh levels (Himmelheber et al., 1998). Dialysate samples were collected every 3 min, beginning 2 h after inserting probes. After the collection of five baseline samples, potassium (100 mm) was added to the perfusion medium for the collection of five additional samples. After termination of potassium perfusion, five more samples were collected.
Effects of VU6001221 on potassium-evoked ACh release.
The effects of the CHT inhibitor on potassium-stimulated ACh levels were tested using the microdialysis methods described above. VU600122 was administered at the same dose used for the experiment on effects on PCA performance (0.3 mg/kg; i.p.; in 10% Tween 80 in water). Each rat was dialyzed twice, with half of the rats receiving drug during the first session. Fifteen minutes after the injection, potassium reverse-dialysis was initiated and continued for the collection of five 3 min samples. Samples were derivatized (described below) and stored at −80°C until they were analyzed by HPLC-mass spectrometry (HPLC-MS).
HPLC-MS methods, reagents, and chemicals.
All chemicals, drugs, and reagents were purchased from Sigma-Aldrich unless otherwise noted. HPLC-grade water was purchased from Thermo Fisher Scientific, and HPLC-grade acetonitrile and methanol were obtained from VWR. d4-ACh and d4-choline were obtained from C/D/N Isotopes.
Sample preparation.
Benzoyl chloride derivatization of dialysates and internal standards was performed using a slightly modified version of a previously described method (Song et al., 2012). Briefly, calibration curves were generated using standards at 0, 0.1, 0.5, 1, 5, 10, and 20 nm for ACh, serotonin (5-HT), norepinephrine (NE), and dopamine (DA) and 10- or 100-fold higher for other analytes. Internal standards were 1 mm glycine, serine and taurine, aspartate, glutamate, and adenosine; 100 μm GABA, histamine, homovanillic acid, 5-hydroxyindoleacetic acid, and 3,4-dihydroxyphenylacetic acid; and 50 μm 5-HT, NE, and DA. Internal standards were derivatized by adding 100 mm sodium carbonate buffer, followed by 2% 13C6 benzoyl chloride in acetonitrile with 0.1% formic acid. The internal standard stock was then diluted 100-fold in 50% acetonitrile in water containing 1% sulfuric acid. d4-ACh and d4-Cho were spiked into the reaction mixture to a final concentration of 50 nm and 5 μm, respectively.
HPLC.
Microdialysis samples were analyzed by a Thermo Finnigan Surveyor Plus HPLC system consisting of an Autosampler Plus and MS Pump Plus. Neurochemical separation was achieved with a Phenomenex Kinetex biphenyl LC column (50 × 2.1 mm, 1.7 μm particle size, 100 Å pore size). Mobile phase A was 10 mm ammonium formate and 0.15% (v/v) formic acid in water. Mobile phase B was acetonitrile. The mobile phase gradient for all 16 analytes was as follows: initial, 0% B; 0.1 min, 10% B; 2.3 min, 20% B; 3.7 min, 50% B; 4.0 min, 80% B; 4.5 min, 0% B; 6.5 min, 0% B. The flow rate was 200 μl/min, and the sample injection volume was 5 μl. The autosampler and column were maintained at ambient temperature throughout the analysis.
Mass spectrometry.
A Thermo Finnigan TSQ Quantum Ultra triple quadrupole mass spectrometer operating in positive mode was used for detection. Electrospray ionization (ESI) voltage was 3.5 kV, and heated ESI probe (HESI-II) was set at 300°C. Capillary temperature was 350°C, and sheath gas, aux gas, and ion sweep gas were maintained at 25, 15, and 0 arb, respectively. Intercycle delay was 200 ms. Automated peak integration was performed using Thermo XCalibur QuanBrowser version 2.1. All peaks were visually inspected to ensure proper integration. Calibration curves were constructed based on peak area ratio (Panalyte/PI.S.) versus concentrations of internal standard by linear regression.
Statistical analyses
Measures of CHT-mediated choline transport (Vmax and Km) and CHT concentrations in the subcellular fractions (LP1, LP2; above) were analyzed using two-way ANOVAs with group and stimulation condition (BF-sham or BF-ES) as factors. To verify the similarity of PCA scores among the experiments, scores were compared using a two-way ANOVA with phenotype and experiment as factors. Two-way repeated-measures ANOVAs were used to determine effects of VU600122 treatment on lever- and food cup-directed behavior during PCA training. Analyses were conducted on PCA performance measures from training sessions 2–5. PCA scores from session 1, before the onset of treatment and after the removal of rats with session 1 scores >0 (above), were compared using an unpaired t test. The distribution of STs and GTs after the final and fifth PCA session (average PCA scores from sessions 4 and 5; scores of less than −0.5 versus more than +0.5) was compared between vehicle- and VU600122-treated rats using the χ2 test. Two-way repeated-measures ANOVAs, with phenotype and potassium stimulation as factors, were used to analyze absolute neurotransmitter levels in STs and GTs. As we observed that neurotransmitter levels generally returned to baseline during the fifth collection in the presence of potassium (12–15 min of potassium reverse-dialysis), indicating depletion of synapses, this analysis was conducted over four prepotassium collections and the first four collections in the presence of potassium (12 min of reverse-dialysis). The effects of VU600122 on potassium-evoked neurotransmitter levels were conducted on the basis of percent change values and analyzed with respect to effects of stimulation and treatment. Post hoc analyses for all within-subject comparisons were performed using t test or least significant difference (LSD) test when applicable. Statistical analyses were performed using the SPSS for Windows (version 17.0; SPSS). Assumptions underlying the statistical model were assessed. In cases of violation of the sphericity assumption, Huyhn–Feldt-corrected F values, along with uncorrected degrees of freedom, are given. Alpha was set at 0.05. Exact p values are reported as recommended previously (Greenwald et al., 1996).
Results
PCA scores of STs and GTs across experiments
Sixty rats (30 GTs and 30 STs) were divided into four groups for separate analyses: (1) BF-ES-evoked choline transport; (2) BF-ES-evoked subcellular CHT distribution; (3) determination of total CHT protein density; and (4) potassium-evoked extracellular levels of ACh. As expected, PCA scores differed between STs and GTs (F(1,52) = 2120.25, p < 0.001; GTs: range, −0.90 to −0.51; mean ± SEM, −0.77 ± 0.02; STs: range, 0.40–0.93; 0.71 ± 0.02) but not between experiments (main effects of experiment and interaction with phenotype: both F(3,52) < 1.91, both p > 0.14). The PCA scores of the GTs used to measure the effects of VU6001221 on potassium-evoked ACh (−0.73 ± 0.06) did not differ from the PCA scores of GTs assigned to the other experiments (t(32) = 0.71, p = 0.48).
BF-ES does not increase CHT-mediated choline transport in STs
We previously reported that attentional performance-associated increases in cortical extracellular ACh levels were greatly attenuated in STs (Paolone et al., 2013). To identify a cellular mechanism that may account for the limited capacity of cholinergic neurons to sustained elevated levels of neurotransmission in STs, we determined the efficacy of the CHT in response to stimulation in vivo. Rats underwent 20 min of BF-ES (see Materials and Methods for justification of stimulation parameters), followed by tissue harvesting and the assessment of CHT-mediated, HC-3-dependent-choline transport. The effects of phenotype and stimulation condition (sham, BF-ES) on Vmax and Km values (see Materials and Methods) were determined.
Saturation curves and Vmax values for cortical and striatal CHT-mediated choline transport are illustrated in Figure 1 (note that the figures depict the results of multiple comparisons, where appropriate). Vmax values differed significantly between the phenotypes (F(1,19) = 8.05, p = 0.012; GT, 7.44 ± 0.55 pmol/mg protein/5 min; ST, 5.28 ± 0.66), and, importantly, the effects of BF-ES and phenotype interacted significantly (F(1,19) = 5.85, p = 0.028; main effect of stimulation, n.s.; Fig. 1). Multiple comparisons indicated that BF-ES increased Vmax values in GTs and that BF-ES-induced Vmax values were higher in GTs relative to STs (Fig. 1). Although Figure 1d suggests that striatal Vmax appeared to increase in GTs, perhaps reflecting spread of the stimulation into the adjacent striatum, striatal Vmax values were not affected by phenotype or BF-ES (main effects and interaction: all F < 2.12, all p > 0.17). Finally, there were no effects on Km values in either brain region (all F < 2.98, all p > 0.13; cortex, 2.63 ± 0.94 μm; striatum, 1.48 ± 0.17 μm).
Figure 1.
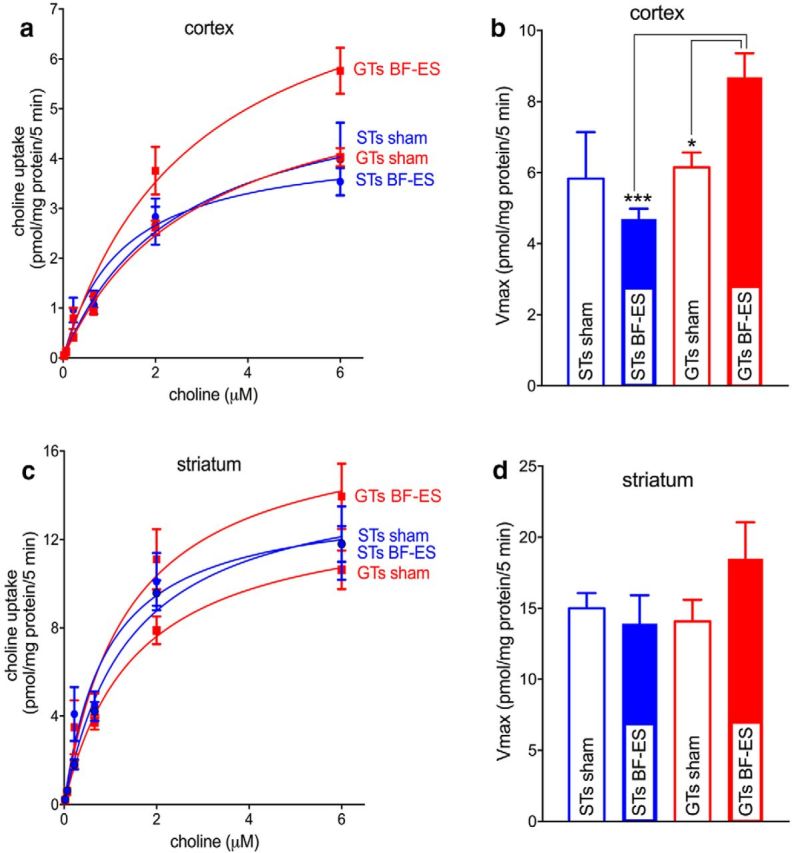
Effects of BF-ES in vivo on CHT-mediated, HC-3-dependent choline transport (n = 20; n = 5 GTs and 5 STs for BF-ES; n = 5 GTs and 5 STs for sham stimulation). After 20 min of BF-ES, cortical and striatal tissues were harvested and synaptosomes were prepared for measuring choline uptake. a, c, Saturation curves for choline uptake by synaptosomes from frontal cortical and striatal tissues. b, d, Vmax values for cortical and striatal synaptosomes. In GTs, BF-ES significantly increased the capacity of cortical CHTs to transport choline (b). No such effect was found in STs. Striatal Vmax values were unaffected by phenotype or BF-ES. BF-ES may have spread into the striatum to yield an insignificant increase in striatal synaptosomes from GTs (d). Km values were not affected by phenotype or BF-ES in either region (see Results for ANOVAs; multiple comparisons for this and subsequent figures: *p < 0.05; **p < 0.01; ***p < 0.001).
BF-ES does not increase CHT plasma membrane density in STs
Stimulation of cholinergic synapses, including the performance in tasks mediated via increases in cholinergic activity, increases the proportion of CHTs in the synaptic plasma membrane (Apparsundaram et al., 2000; Ferguson et al., 2003; Parikh et al., 2013). Thus, the capacity of the CHT to transport choline into the cholinergic terminal normally is mostly controlled by the net result of outward trafficking and internalization of CHTs (Ferguson and Blakely, 2004; Ribeiro et al., 2005, 2006). We investigated the effects of BF-ES on subcellular CHT distribution in STs and GTs in separate groups of rats using established centrifugation steps and immunoblotting (see Materials and Methods).
Total synaptosomal CHT levels in STs and GTs
We first determined total CHT density in synaptosomes from frontal cortex in STs and GTs (n = 5 per phenotype). Densitometric analyses of immunoblots indicated that total CHT levels did not differ between the phenotypes (t(8) = 1.17, p = 0.28; mean ± SEM, 29.23 ± 3.57; normalized integrated density).
Stimulation-induced subcellular CHT redistribution
At baseline, the majority of CHTs are located on vesicular membrane-enriched domains (LP2 fraction), and a proportion of these intracellular CHTs is available for outward mobilization and thus removal from this pool. As indicated by a main effect of BF-ES but no interaction between the effects of BF-ES and phenotype, stimulation-induced removal of a proportion of CHTs from the intracellular domain occurred in both STs and GTs [main effect of BF-ES on LP2 CHT density: F(1,15) = 7.37, p = 0.02; sham, 65.28 ± 12.82 (normalized density); BF-ES, 45.77 ± 9.86; BF-ES × phenotype: F(1,15) = 0.85, p = 0.37]. Moreover, the density of CHTs in the LP2 fraction from STs was lower than in GTs [main effect of phenotype: F(1,15) = 18.46, p = 0.001; ST, 34.28 ± 7.70; GT, 84.48 ± 8.16 (normalized density); Fig. 2a].
Figure 2.

Effects of BF-ES on subcellular CHT distribution in cortical synaptosomes (n = 19; n = 5 GTs and 5 STs for BF-ES; n = 4 GTs and 5 STs for sham stimulation; the immunoblots depict duplicates from 1 ST and 1 GT). a, Consistent with previous research, at baseline (sham stimulation), in GTs and STs, the majority of CHTs was located on intracellular domains, such as vesicular and endosomal membranes. BF-ES lowered the intracellular density of CHTs in both phenotypes [main effect, no interaction with phenotype; sham, 65.28 ± 12.82 (normalized density); BF-ES, 45.77 ± 9.86]. Moreover, STs generally exhibited lower levels of intracellular CHTs than GTs (main effect of phenotype). b, Whereas BF-ES increased the density of CHTs in the plasma membrane-enriched LP1 fraction in GTs, this was not the case in STs. Results from additional analyses are illustrated in c. In synaptosomes from sham-stimulated animals, the LP2/LP1 ratio was already near 1 (indicated by red protein symbols in membrane). Previous findings indicated that a ratio of 1 represents the limit of CHT mobilization to plasma membrane. Thus, synaptosomal CHT distribution in STs may have already reached this maximum at baseline, primarily attributable to a lower intracellular CHT density than in GTs. Second, in STs, the BF-ES-induced loss of LP2 CHTs (blue protein symbols) was not reciprocated with an increase in LP1 CHTs, suggesting stimulation-induced translocation of a portion of intracellular CHTs to domains other than synaptic plasma membrane and those captured in the present LP2 fraction.
Stimulation-induced decreases in intracellular CHT density were expected to be mirrored by increases in CHT density in the plasma membrane-enriched LP1 fraction, reflecting mobilization from intracellular domains to synaptosomal plasma membrane. This was the case for GTs, but not STs, as indicated by a significant interaction between the effects of phenotype and BF-ES (F(1,15) = 7.01, p = 0.02) and multiple comparisons (no main effects of phenotype or BF-ES: both F < 3.30, both p > 0.09; Fig. 2b).
We previously reported results, obtained in mice, suggesting that stimulation-induced CHT outward trafficking does not go beyond an LP2/LP1 ratio of 1 (Parikh et al., 2013). The present results in GTs are consistent with this view. In sham-stimulated GTs, LP2/LP1 ratios reflected the greater concentration of CHT in the vesicular membrane-enriched LP2 fraction [LP2/LP1 ratios (mean ± SEM), 3.47 ± 0.43]. After BF-ES in GTs, this ratio approached 1 (0.97 ± 0.26) and was significantly lower than in sham-stimulated GTs (Mann–Whitney U test, p = 0.01), indicating that as a result of BF-ES, outward mobilization of CHTs in GTs reached this upper limit.
However, in STs, LP2/LP1 ratios, after sham stimulation and BF-ES, did not follow the findings originally observed in mice. First, in sham-stimulated STs, the LP2/LP1 ratio was already near 1 (1.09 ± 0.21), suggesting that, at baseline and without stimulation, the CHT distribution ratio in STs already reached the level previously seen after BF-ES in mice (Parikh et al., 2013). Consistent with the view that equal densities of CHTs in the two subcellular fractions mirror an upper limit of synaptic plasma membrane CHT density in STs BF-ES did not significantly alter the LP2/LP1 CHT distribution ratio (0.82 ± 0.32; p = 0.92).
Because BF-ES in STs lowered intracellular CHT density without increasing CHT density in the plasma membrane-enriched LP1 fraction, a portion of CHTs in STs, after BF-ES, may have been translocated to subcellular domains not captured by our LP1 and LP2 fractions. Across individual values and separate groups of stimulated and sham-stimulated animals, BF-ES in GTs yielded a 39% lower CHT LP2 concentration and a 99% increase in CHT LP1 density. (This disproportional increase in LP1 CHT density primarily may have been attributable to errors resulting from a comparison between separate groups of sham-stimulated and BF-ES-stimulated animals.) In STs, however, BF-ES resulted in a 45% loss in LP2 CHTs and also in a loss in LP1 CHTs (−11%). The effects of BF-ES in GTs and STs are summarized and illustrated in Figure 2c. The failure of cholinergic synapses of STs to respond to stimulation by increasing plasma membrane CHT density is consistent with the finding, from separate rats, that BF-ES in STs fails to increase CHT-mediated choline transport (Fig. 1).
Sustaining increases in cholinergic neurotransmission in vivo in GTs but not STs
We previously reported that levels of cortical extracellular ACh in attention task-performing STs exhibited relatively small increases (∼70%) compared with the ∼200% increases in GTs and that the STs' low ACh levels were associated with their relatively poor attentional performance (Paolone et al., 2013). Given the seemingly fundamental CHT capacity limit in STs described above, we reasoned that the relatively low capacity of STs' cholinergic neurons may be demonstrated merely by testing the effects of depolarization of cholinergic neurons on extracellular ACh levels. To this end, we reverse-dialyzed potassium (100 mm) for 15 min in awake STs and GTs undergoing microdialysis in the prelimbic cortex. Furthermore, we analyzed the effects of eight additional neurotransmitters and metabolites to potentially reject the presence of global phenotype-associated differences in neurotransmitter levels. The analysis of the effects of potassium stimulation and phenotype was conducted using absolute neurotransmitter levels (Table 1).
Table 1.
Effects of potassium stimulation on extracellular levels of neurotransmitters and metabolites
| Analyte | Basal levels (nm) | Phenotype effect on basal | Levels during stimulation (nm) | Main effect of stimulation | Phenotype × stimulation |
|---|---|---|---|---|---|
| Acetylcholine | |||||
| GTs | 312.93 ± 46.25 | n.s. | 992.89 ± 161.90 | F(1,9) = 47.66, p < 0.001 | F(1,9) = 5.92, p = 0.038 |
| STs | 328.59 ± 93.17 | 654.19 ± 116.68 | |||
| Choline | |||||
| GTs | 3620.77 ± 349.77 | n.s. | 4271.18 ± 527.00 | F(1,9) = 3.49, p = 0.10 | F(1,9) = 0.01, p = 0.98 |
| STs | 4540.33 ± 1608.44 | 5173.66 ± 1788.80 | |||
| Adenosine | |||||
| GTs | 88.96 ± 19.56 | n.s. | 433.95 ± 87.78 | F(1,9) = 25.91, p = 0.001 | F(1,9) = 8.29, p = 0.018 |
| STs | 58.16 ± 17.72 | 153.85 ± 45.03 | |||
| GABA | |||||
| GTs | 190.23 ± 61.07 | n.s. | 4649.34 ± 747.17 | F(1,9) = 27.36, p = 0.001 | F(1,9) = 0.26, p = 0.62 |
| STs | 534.50 ± 247.99 | 4198.82 ± 1659.85 | |||
| Glutamate | |||||
| GTs | 9918.11 ± 3578.66 | n.s. | 26346.11 ± 6299.28 | F(1,9) = 25.14, p = 0.001 | F(1,9) = 0.25, p = 0.63 |
| STs | 17595.26 ± 5371.42 | 37649.19 ± 9947.55 | |||
| Norepinephrine | |||||
| GTs | 5.87 ± 2.87 | n.s. | 27.90 ± 6.01 | F(1,9) = 21.47, p = 0.001 | F(1,9) = 0.54, p = 0.48 |
| STs | 13.69 ± 6.55 | 30.34 ± 7.33 | |||
| Serotonin | |||||
| GTs | 8.98 ± 0.46 | n.s. | 36.18 ± 9.91 | F(1,9) = 14.48, p = 0.004 | F(1,9) = 1.00, p = 0.34 |
| STs | 8.12 ± 2.46 | 24.00 ± 6.59 | |||
| Dopamine | |||||
| GTs | 5.41 ± 2.09 | n.s. | 18.25 ± 5.96 | F(1,9) = 23.26, p = 0.001 | F(1,9) = 0.13, p = 0.73 |
| STs | 8.63 ± 2.83 | 23.52 ± 5.46 |
Values were corrected for in vitro probe recovery rates.
Basal extracellular levels of all nine analytes did not differ between the phenotypes (Table 1). With respect to ACh, this finding is consistent with the absence of phenotype effects on basal CHT-mediated uptake and comparable LP1 CHT densities in unstimulated STs and GTs (above). As expected, potassium stimulation significantly increased levels of all analytes except choline, including ACh (main effect of stimulation: F(1,9) = 47.66, p < 0.001; Fig. 3a), adenosine, GABA, glutamate, norepinephrine, serotonin, and dopamine (Table 1). Choline levels were not expected to increase in response to potassium stimulation, in part because microdialysis measures primarily reflect the relatively stable 3–4 μm extracellular concentration of choline while not being capable of detecting highly local changes that may reflect CHT-mediated clearance or hydrolysis of ACh (Klein et al., 1998; Sarter and Kim, 2015).
Figure 3.
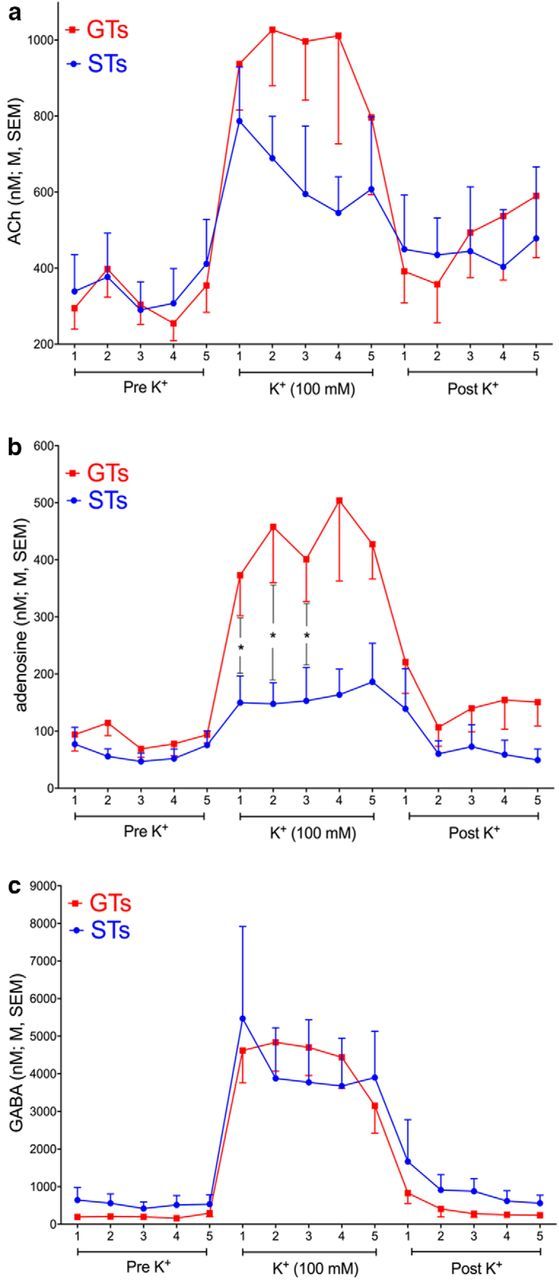
Effects of reverse-dialysis of potassium (100 mm) on cortical levels of ACh, adenosine, and GABA (see Table 1 for a summary of effects on all neurotransmitters and metabolites that were determined in this experiment; n = 11; n = 6 GTs and 5 STs). K+ was perfused for 15 min (five 3 min collections). a, b, Significant interactions between the effects of phenotype and stimulation were found only for ACh (a) and adenosine (b; see Table 1 for ANOVAs). Because of the variability of absolute ACh levels, post hoc comparisons did not locate the interaction (see Results for additional analyses of data expressed as percent change from baseline). Adenosine levels were less variable and were significantly higher in GTs for the first three collections during stimulation. c, GABA levels increased similarly in GTs and STs (see Table 1 for other analytes that reached comparable levels during stimulation in the two phenotypes).
As indicated by a significant interaction between the effects of phenotype and stimulation, stimulation-induced increases in ACh levels were higher in GTs than in STs (F(1,9) = 5.92, p = 0.038; Fig. 3a). Post hoc multiple comparisons did not locate this interaction, reflecting the variability of absolute nanomolar ACh levels. When expressed as percent change from baseline, stimulation-induced ACh levels in GTs reached 201.81 ± 27.28% over baseline compared with 109.07 ± 20.83% in STs (across collections), and levels differed significantly between the phenotypes for collections 2 and 3 (both p < 0.03).
Unexpectedly, K+-evoked increases in the extracellular level of a second analyte, adenosine, was also greater in GTs than STs (phenotype × stimulation: F(1,9) = 5.85, p = 0.039; Table 1; Fig. 3b). When expressed as percent change from baseline, adenosine levels in GTs reached 410.56 ± 80.26% over baseline when compared with 180.67 ± 37.82% in STs.
Among the analytes exhibiting similar K+-evoked increases in GTs and STs, Figure 3c depicts GABA, as a portion of GABA may have originated from cholinergic terminals (Saunders et al., 2015). As summarized in Table 1, the effects of phenotype and stimulation did not interact significantly with respect to GABA, glutamate, serotonin, noradrenaline, and dopamine.
CHT inhibition favors sign-tracking behavior
Sign-tracking behavior in the PCA test has been interpreted in terms of the attribution of incentive properties to the CS and is associated with enhanced mesolimbic dopamine signaling (Flagel et al., 2009; Flagel et al., 2011). Given the attenuated CHT capacity and cholinergic neurotransmission in STs, we hypothesized that sign-tracking behavior may also be mediated by relatively low levels of cholinergic activity. Such a finding would support the possibility that a stimulus-driven attentional bias may contribute to the emergence of sign-tracking behavior. We tested this hypothesis by assessing the effects of a CHT inhibitor, VU600122, on PCA performance (see Materials and Methods for selection and justification of dose). The first PCA session was conducted in the absence of drug to exclude all rats with PCA index scores >0 and thus rats that were more likely to emerge as STs. Drug was then given before sessions 2–5 to determine whether, compared with the effects of vehicle, inhibition of the CHT yields a greater proportion of rats classified eventually as STs.
PCA scores, obtained from session 1, of rats to be treated with vehicle (n = 24) or VU600122 (n = 25) did not differ significantly (t(47) = 0.46, p = 0.65; vehicle, −0.30 ± 0.04; VU, −0.33 ± 0.04; see Fig. 5). As expected, lever- and food cup-directed behavior (learning the conditioned response) evolved across sessions for all rats, regardless of treatment (main effect of session on all parameters: all F(3,141) > 5.53, all p < 0.002; Fig. 4). Furthermore, rats treated with vehicle or VU600122 equally learned to discriminate between CS and non-CS periods as indicated by a decrease in non-CS food cup contacts across days of training (effect of treatment: F(1,141) = 2.159, p = 0.148; effect of session: F(3,141) = 52.41, p < 0.001; Fig. 4d). Administration of VU600122 facilitated all aspects of lever-directed behavior [main effects of treatment on lever contact probability, number of lever contacts (Fig. 4c), and lever approach latency (Fig. 4e); all F(1,141) > 4.09, all p < 0.05]. Conversely, administration of VU600122 facilitated the decrease of food cup-directed behavior (all F(1,141) > 4.07, p < 0.05; Fig. 4b,f).
Figure 5.

Individual PCA scores for rats treated with vehicle (a) or VU600122 (b) (final numbers per phenotype and treatment are indicated on the right of the two graphs). Note that session 1 scores were obtained in the absence of treatment and that rats with PCA scores >0, and thus showing an early trend for sign-tracking, were excluded from this experiment to bias it against the expected effect of the drug. VU600122 or vehicle was given before sessions 2–5. Individual rats' PCA scores from sessions 4 and 5 were averaged to assign the final phenotype. Compared with vehicle, treatment with VU600122 significantly increased the proportion of STs after the final session 5.
Figure 4.
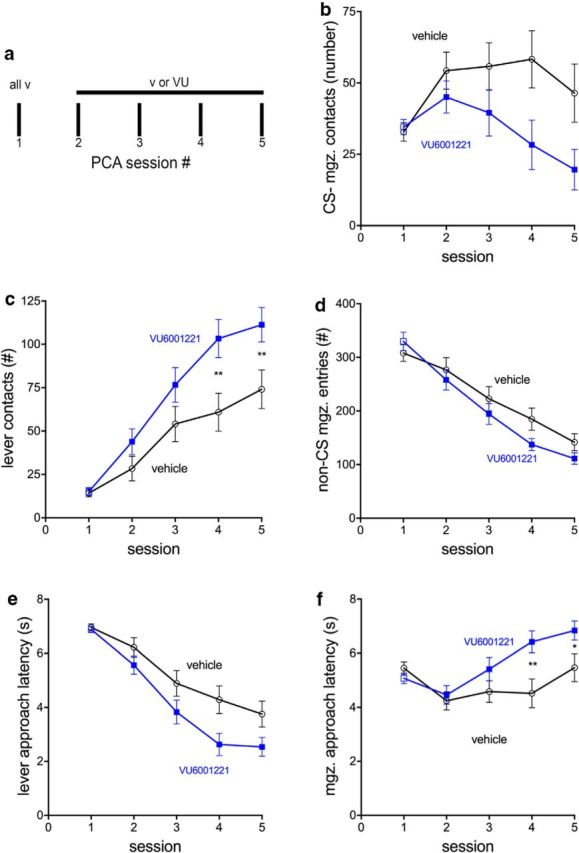
Effects of the CHT inhibitor VU600122 on pavlovian conditioned approach behavior toward a lever (CS) versus the location of food delivery (food cup; n = 49; n = 25 treated with VU600122; n = 24 treated with vehicle). Behavior was measured over five training sessions to determine lever- and food cup-directed behavior after administration. a, Drug or vehicle was given before sessions 2–5. Data from session 1, and after the administration of vehicle, were used to exclude rats that exhibited an early trend for sign-tracking behavior, to allow for a more “aggressive” test of the hypothesis that VU600122 fosters sign-tracking. b–f, The plots depict mean ± SEM for food cup or magazine (mgz.) contacts evoked by the CS (b), number of lever contacts (c), number of non-CS food magazine entries (d), latency to first lever contact after CS presentation (e), and latency to the first food magazine entry after CS presentation (f). Compared with the effects of vehicle, administration of VU600122 facilitated lever-oriented behavior and reduced CS-evoked magazine-oriented behavior (for main effects of treatment and interactions, see Results; multiple comparisons indicated in c and f were based on significant interactions between the effects of VU600122 and session; see Results).
The effects of the treatment and session interacted significantly with respect to the number of lever contacts (Fig. 4c) and magazine approach latency (Fig. 4f; both F < 3.24, both p < 0.02), reflecting relatively more lever contacts and slower magazine approaches, respectively, by rats treated with VU600122 (all other interactions between treatment and session: all F(3,141) < 2.60, all p > 0.06).
As an alternative analysis of the effects of VU600122, we determined the distribution of STs and GTs (as defined in Materials and Methods) after the final PCA session 5. Recall that after session 1 and before drug administration, PCA scores for all animals selected for this experiment were <0 and thus suggested an early propensity for goal-tracking (Fig. 5). As the drug treatment was expected to favor sign-tracking, only animals with such session 1 scores were selected to bias the experiment against the expected effect. With vehicle treatment, the final distribution was 10 STs, 6 GTs, and 8 rats with intermediate scores (INs; Fig. 5a). In contrast, after the treatment with VU600122, we obtained 19 STs, 2 GTs, and 4 INs (Fig. 5b), indicating a significantly greater proportion of STs in the drug-treated group (χ2 = 6.11, p = 0.047). Thus, treatment with VU600122 fostered the manifestation of sign-tracking behavior.
Attenuation of elevated ACh levels by VU600122
This experiment tested the hypothesis that CHT inhibitor VU600122 will attenuate the capacity of cholinergic neurons to sustain elevated levels of neurotransmission. We again used potassium as a means to upregulate cholinergic activity over a 15 min period. We conducted this experiment in GTs that exhibit robust K+-evoked increases in extracellular ACh levels (Fig. 3a). VU600122 was administered intraperitoneally 15 min before the onset of potassium reverse-dialysis. As before (above), reverse-dialysis of K+ reliably increased cortical ACh levels by over 200% (rats treated with vehicle or VU600122; averaged over five collections, pre- vs post-K+; from 76.54 ± 23.22 nm to 232.29 ± 68.55 nm; t(3) = 10.98, p = 0.045). Administration of VU600122 significantly attenuated the increases in extracellular ACh levels (main effect of treatment: F(1,12) = 16.34, p = 0.027; Fig. 6a; treatment × collection period: F(4,12) = 1.28, p = 0.18). This evidence supports the notion that VU600122 inhibits CHT function.
Figure 6.

Attenuation of potassium-evoked increases in cortical extracellular ACh levels by VU600122 (n = 4 GTs). VU600122 was administered at the same dose (0.3 mg/kg) used to demonstrate the manifestation of sign-tracking behavior. VU600122 significantly attenuated potassium-induced increases in ACh levels (main effect of drug; see Results).
Discussion
Summary of major findings
Compared with GTs, the capacity of cholinergic neurons of STs to sustain elevated levels of neurotransmission is diminished. To uncover the cellular mechanisms accounting for the attenuated cholinergic capacity in STs, we focused on the rate-limiting step for ACh synthesis, the CHT. In STs, CHT-mediated choline transport was unresponsive to stimulation. Consistent with this finding, stimulation also failed to increase the density of CHT in the synaptic plasma membrane. Given that these results indicate an essential capacity limit of cholinergic synapses of STs to adapt to increases in activity, we then demonstrated that a straightforward method of stimulating cholinergic neurons to release ACh, based on reverse-dialysis of potassium, was sufficient to reveal the attenuated cholinergic capacity in STs. Finally, we demonstrated that pharmacological inhibition of CHT capacity fosters the development of sign-tracking behavior.
Subcellular CHT distribution in STs
Total CHT protein in cortical synaptosomes from STs and GTs did not differ. In contrast, baseline CHT levels in the vesicular membrane-enriched LP2 fraction from sham-stimulated rats were lower in STs relative to GTs (Fig. 2c). In GTs, CHTs primarily reside in vesicular membrane (Ferguson et al., 2003; Parikh et al., 2013) whereas in STs, a significant proportion of intracellular CHTs may be located in subcellular compartments, such as early endosomes and mitochondria, which may not have been captured by the subcellular fractionation method used to generate the LP2 fraction. Alternatively, the epitope for our antibody may have been masked by post-translational modifications associated with a shift in the distribution of CHT and/or the capacity for transporter mobilization with BF-ES.
As a result of BF-ES in STs, a proportion of CHTs was removed from the vesicular membrane-enriched LP2 fraction, but this was not mirrored by an increase in LP1 CHT density, suggesting that CHTs were mobilized to intracellular compartments not associated with CHT mobilization to plasma membrane. After stimulation in STs, intracellular CHTs may be modified (e.g., phosphorylation, ubiquitylation, or nitrosylation) so that they are rendered unavailable for synaptic plasma membrane relocation (Gates et al., 2004; Ribeiro et al., 2006; Cuddy et al., 2012; Yamada et al., 2012; Hartnett et al., 2014). As total synaptosomal CHT densities did not differ between the phenotypes, we assume that subcellular CHT distribution anomalies observed in STs, and the associated failure of CHTs to increase choline transport in response to stimulation, reflect an alteration in distribution or post-translational modifications, rather than degradation. Ongoing genome-wide association studies for sign- and goal-tracking (Parker et al., 2014) may reveal molecular “access points” for exploring the signaling pathways involved in CHT dysfunction in STs.
Localization of dysregulated CHTs
It is not clear whether the disruption of CHT function in STs is present in the entire nervous system or only in localized brain regions. Additional considerations suggest that unresponsive CHTs in STs are widely distributed. Low or unresponsive cortical levels of adenosine, observed in STs in the current experiments, may promote widespread neuroinflammatory mechanisms (Haskó et al., 2005), which in turn have the potential to reduce the capacity of the CHT (Koshy Cherian et al., 2015). Consistent with the speculation that STs may exhibit elevated levels of neuroinflammatory signaling, STs were previously found to show elevated plasma corticosterone levels during PCA training (Flagel et al., 2009). Future research will need to determine whether unresponsive CHTs are present throughout the nervous system and whether neuroinflammatory signaling contributes widely to CHT dysfunction and thus this phenotype. As unresponsive CHTs may be widely distributed in the nervous system of STs, the sign-tracking-fostering effects of the CHT inhibitor may have been mediated via effects on cholinergic modulation of telencephalic, striatal, and mesolimbic regions.
Stimulation parameters
BF-ES stimulation parameters were adopted from studies on cholinergic functions (see Materials and Methods), and the effects of reverse-dialysis of potassium confirmed that cholinergic neurons of STs are relatively unresponsive to stimulation. Because we observed that performance of an attention-demanding motor task likewise revealed unresponsive CHT-mediated choline transport in STs (Kucinski et al., 2015), it seems likely that the effects of BF-ES mobilized vesicular populations that are comparable, or at least partly overlap, with releasable pools of cholinergic vesicles mobilized by behavior.
CHT inhibition-induced sign-tracking: cognitive mechanisms
To determine whether unresponsive CHTs contribute to the emergence of sign-tracking behavior per se, we administered the CHT inhibitor VU600122 to rats with initial PCA scores indicative of a propensity for goal-tracking. After the administration of this drug before PCA sessions 2–5, a significantly greater proportion of rats evolved as STs than in rats treated with vehicle. This finding suggests that sign-tracking per se, not just the attentional impairments that have been viewed as a component of the trait indexed by sign-tracking behavior (Paolone et al., 2013), is fostered by the cholinergic hypofunction that results from unresponsive CHTs.
Based on the relatively poor attentional performance of animals heterozygous for the CHT and humans expressing a CHT subcapacity variant, we previously concluded that relatively low levels of cholinergic neuromodulation mediate a bias for bottom-up, or stimulus-driven, attention while minimizing the role of top-down, or goal-driven, control (Sarter et al., 2016b). This hypothesis may also apply to the manifestation of sign-tracking behavior. The extension of the lever during PCA training represents a highly salient event (Singer et al., 2016). Owing to their bottom-up bias, the lever may capture the attention of STs more effectively than is the case in GTs, fostering approach to the lever by the former. In contrast, GTs may perform in accordance with a bias for goal-driven or top-down attention, deploying attention toward the location of the reward (see also Haight et al., 2016). In accordance with this hypothesis, VU600122 shifted the bias of prospective GTs toward a more bottom-up-driven behavior and thus toward the pavlovian cue. After the initial attention to the cue, mediated via relatively low levels of cholinergic neuromodulation, motivational processes that increasingly assign incentive value to the cue and that are mediated via mesolimbic dopamine signaling (Flagel et al., 2011) likely augmented and stabilized cue-oriented behavior and thus the manifestation of the ST phenotype.
Relationships between stimulus-driven attention capture and reward processing
It remains unclear whether there are more direct relationships between, on the one hand, the attentional bias of STs and associated CHT dysfunction and, on the other hand, the motivational processes and the underlying dopaminergic responses in the nucleus accumbens that sustain the power of the reward cue to control behavior. The attentional bias of STs may be a critical mechanism toward the initial development of this phenotype. Thereafter, the cue may rapidly acquire incentive significance in STs and thereby sustain cue-oriented behavior. However, perceptual and motivational aspects of cue saliency may be extremely difficult to dissociate (Maunsell, 2004; Anderson et al., 2013; Shomstein and Johnson, 2013; van Hemel-Ruiter et al., 2015); rather, interwoven attentional–motivational processes likely contribute essentially to the sign-tracking behavioral trait.
In contrast to these conceptual challenges, it will be important and feasible to address the question of whether CHT subcapacity also contributes to the cue-evoked dopamine signal in the nucleus accumbens that mediates the transformation of the CS into a motivational magnet for STs (Flagel et al., 2011). The interactions between the activity of accumbens cholinergic interneurons and dopamine signaling are not well understood (Crespo et al., 2006; Collins et al., 2016), and thus the impact of potentially unresponsive CHTs on these interactions are difficult to predict (but see Dong et al., 2013). It will be important to investigate the possibility that accumbens CHT subcapacity favors pavlovian cue-evoked dopamine signaling in STs.
Relevance for addiction vulnerability and therapy
Sign-tracking is one behavioral index of a complex psychological trait associated with a propensity for certain addiction-like behaviors. Maladaptive motivational processes that render previously neutral cues to be attractive and wanted were postulated as the core characteristics of this trait (Robinson and Berridge, 2008; Robinson and Flagel, 2009; Robinson et al., 2014; Yager et al., 2015). The present results suggest that such a conceptualization of this trait is incomplete. A bias for bottom-up, or stimulus-driven, attention is either a major additional component or even a causal factor for sign-tracking behavior, and the associated CHT subcapacity appears to be a causal neuronal mechanism for sign-tracking in general. Given that poor attentional control of drug cues has been considered an essential psychological risk factor for addiction (Field and Cox, 2008), rodent as well as human studies (Garofalo and di Pellegrino, 2015) that capture the sign-tracking trait may be highly useful in the development of therapies that restore the capacity for sustained cholinergic activity and thereby the ability of drug cues to control behavior. Moreover, genotyping for the CHT I89V subcapacity variant in humans may provide a cohort enriched for addiction therapies targeting the cholinergic restoration of the normal attentional control mechanisms that assist in suppressing the power of drug cues.
Footnotes
This work was supported by United States Public Health Service Grant DA031656 (M.S.) and the Dystonia Medical Research Foundation (C.W.L. and R.D.B.). We thank Drs. Shelly Flagel and Terry Robinson (University of Michigan) for comments on this manuscript.
The authors declare no competing financial interests.
References
- Anderson BA, Laurent PA, Yantis S (2013) Reward predictions bias attentional selection. Front Hum Neurosci 7:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apparsundaram S, Ferguson SM, George AL Jr, Blakely RD (2000) Molecular cloning of a human, hemicholinium-3-sensitive choline transporter. Biochem Biophys Res Commun 276:862–867. 10.1006/bbrc.2000.3561 [DOI] [PubMed] [Google Scholar]
- Apparsundaram S, Martinez V, Parikh V, Kozak R, Sarter M (2005) Increased capacity and density of choline transporters situated in synaptic membranes of the right medial prefrontal cortex of attentional task-performing rats. J Neurosci 25:3851–3856. 10.1523/JNEUROSCI.0205-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertron JL, Ennis EA, Tarr CJ, Wright J, Dickerson JW, Locuson CW, Blobaum AL, Rook JM, Blakely RD, Lindsley CW (2016) Optimization of the choline transporter (CHT) inhibitor ML352: development of VU6001221, an improved in vivo tool compound. Bioorg Med Chem Lett 26:4637–4640. 10.1016/j.bmcl.2016.08.062 [DOI] [PubMed] [Google Scholar]
- Black YD, Maclaren FR, Naydenov AV, Carlezon WA Jr, Baxter MG, Konradi C (2006) Altered attention and prefrontal cortex gene expression in rats after binge-like exposure to cocaine during adolescence. J Neurosci 26:9656–9665. 10.1523/JNEUROSCI.2391-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AL, Aitken TJ, Greenfield VY, Ostlund SB, Wassum KM (2016) Nucleus accumbens acetylcholine receptors modulate dopamine and motivation. Neuropsychopharmacology 41:2830–2838. 10.1038/npp.2016.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo JA, Sturm K, Saria A, Zernig G (2006) Activation of muscarinic and nicotinic acetylcholine receptors in the nucleus accumbens core is necessary for the acquisition of drug reinforcement. J Neurosci 26:6004–6010. 10.1523/JNEUROSCI.4494-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddy LK, Gordon AC, Black SA, Jaworski E, Ferguson SS, Rylett RJ (2012) Peroxynitrite donor SIN-1 alters high-affinity choline transporter activity by modifying its intracellular trafficking. J Neurosci 32:5573–5584. 10.1523/JNEUROSCI.5235-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Dani JA, Blakely RD (2013) Choline transporter hemizygosity results in diminished basal extracellular dopamine levels in nucleus accumbens and blunts dopamine elevations following cocaine or nicotine. Biochem Pharmacol 86:1084–1088. 10.1016/j.bcp.2013.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edeline JM, Maho C, Hars B, Hennevin E (1994) Non-awaking basal forebrain stimulation enhances auditory cortex responsiveness during slow-wave sleep. Brain Res 636:333–337. 10.1016/0006-8993(94)91033-2 [DOI] [PubMed] [Google Scholar]
- Ennis EA, Blakely RD (2016) Choline on the move: perspectives on the molecular physiology and pharmacology of the presynaptic choline transporter. Adv Pharmacol 76:175–213. 10.1016/bs.apha.2016.03.001 [DOI] [PubMed] [Google Scholar]
- Ersche KD, Barnes A, Jones PS, Morein-Zamir S, Robbins TW, Bullmore ET (2011) Abnormal structure of frontostriatal brain systems is associated with aspects of impulsivity and compulsivity in cocaine dependence. Brain 134:2013–2024. 10.1093/brain/awr138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersche KD, Jones PS, Williams GB, Turton AJ, Robbins TW, Bullmore ET (2012) Abnormal brain structure implicated in stimulant drug addiction. Science 335:601–604. 10.1126/science.1214463 [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Blakely RD (2004) The choline transporter resurfaces: new roles for synaptic vesicles? Mol Interv 4:22–37. 10.1124/mi.4.1.22 [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Savchenko V, Apparsundaram S, Zwick M, Wright J, Heilman CJ, Yi H, Levey AI, Blakely RD (2003) Vesicular localization and activity-dependent trafficking of presynaptic choline transporters. J Neurosci 23:9697–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Bazalakova M, Savchenko V, Tapia JC, Wright J, Blakely RD (2004) Lethal impairment of cholinergic neurotransmission in hemicholinium-3-sensitive choline transporter knockout mice. Proc Natl Acad Sci U S A 101:8762–8767. 10.1073/pnas.0401667101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field M, Cox WM (2008) Attentional bias in addictive behaviors: a review of its development, causes, and consequences. Drug Alcohol Depend 97:1–20. 10.1016/j.drugalcdep.2008.03.030 [DOI] [PubMed] [Google Scholar]
- Flagel SB, Watson SJ, Robinson TE, Akil H (2007) Individual differences in the propensity to approach signals vs goals promote different adaptations in the dopamine system of rats. Psychopharmacology (Berl) 191:599–607. 10.1007/s00213-006-0535-8 [DOI] [PubMed] [Google Scholar]
- Flagel SB, Akil H, Robinson TE (2009) Individual differences in the attribution of incentive salience to reward-related cues: implications for addiction. Neuropharmacology 56 [Suppl 1]:139–148. 10.1016/j.neuropharm.2008.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagel SB, Clark JJ, Robinson TE, Mayo L, Czuj A, Willuhn I, Akers CA, Clinton SM, Phillips PE, Akil H (2011) A selective role for dopamine in stimulus-reward learning. Nature 469:53–57. 10.1038/nature09588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagel SB, Waselus M, Clinton SM, Watson SJ, Akil H (2014) Antecedents and consequences of drug abuse in rats selectively bred for high and low response to novelty. Neuropharmacology 76:425–436. 10.1016/j.neuropharm.2013.04.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagel SB, Chaudhury S, Waselus M, Kelly R, Sewani S, Clinton SM, Thompson RC, Watson SJ Jr, Akil H (2016) Genetic background and epigenetic modifications in the core of the nucleus accumbens predict addiction-like behavior in a rat model. Proc Natl Acad Sci U S A 113:E2861–E2870. 10.1073/pnas.1520491113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofalo S, di Pellegrino G (2015) Individual differences in the influence of task-irrelevant Pavlovian cues on human behavior. Front Behav Neurosci 9:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates J Jr, Ferguson SM, Blakely RD, Apparsundaram S (2004) Regulation of choline transporter surface expression and phosphorylation by protein kinase C and protein phosphatase 1/2A. J Pharmacol Exp Ther 310:536–545. 10.1124/jpet.104.066795 [DOI] [PubMed] [Google Scholar]
- Goard M, Dan Y (2009) Basal forebrain activation enhances cortical coding of natural scenes. Nat Neurosci 12:1444–1449. 10.1038/nn.2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald AG, Gonzalez R, Harris RJ, Guthrie D (1996) Effect sizes and p values: what should be reported and what should be replicated? Psychophysiology 33:175–183. 10.1111/j.1469-8986.1996.tb02121.x [DOI] [PubMed] [Google Scholar]
- Guyenet P, Lefresne P, Rossier J, Beaujouan JC, Glowinski J (1973) Inhibition by hemicholinium-3 of [14C] acetylcholine synthesis and [3H] choline high-affinity uptake in rat striatal synaptosomes. Mol Pharmacol 9:630–639. [PubMed] [Google Scholar]
- Haga T. (2014) Molecular properties of the high-affinity choline transporter CHT1. J Biochem 156:181–194. 10.1093/jb/mvu047 [DOI] [PubMed] [Google Scholar]
- Haight JL, Fuller ZL, Fraser KM, Flagel SB (2016) A food-predictive cue attributed with incentive salience engages subcortical afferents and efferents of the paraventricular nucleus of the thalamus. Neuroscience 340:135–152. 10.1016/j.neuroscience.2016.10.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartnett S, Zhang F, Abitz A, Li Y (2014) Ubiquitin C-terminal hydrolase L1 interacts with choline transporter in cholinergic cells. Neurosci Lett 564:115–119. 10.1016/j.neulet.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskó G, Pacher P, Vizi ES, Illes P (2005) Adenosine receptor signaling in the brain immune system. Trends Pharmacol Sci 26:511–516. 10.1016/j.tips.2005.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmelheber AM, Fadel J, Sarter M, Bruno JP (1998) Effects of local cholinesterase inhibition on acetylcholine release assessed simultaneously in prefrontal and frontoparietal cortex. Neuroscience 86:949–957. 10.1016/S0306-4522(98)00097-9 [DOI] [PubMed] [Google Scholar]
- Kilts CD, Kennedy A, Elton AL, Tripathi SP, Young J, Cisler JM, James GA (2014) Individual differences in attentional bias associated with cocaine dependence are related to varying engagement of neural processing networks. Neuropsychopharmacology 39:1135–1147. 10.1038/npp.2013.314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J, Köppen A, Löffelholz K (1998) Regulation of free choline in rat brain: dietary and pharmacological manipulations. Neurochem Int 32:479–485. 10.1016/S0197-0186(97)00127-7 [DOI] [PubMed] [Google Scholar]
- Koshy Cherian A, Tronson NC, Parikh V, Blakely RD, Sarter M (2015) Elevated brain cytokine levels associated with cognitive vulnerability of CHT+/- mice following repeated mild traumatic brain injury. Soc Neurosci Abstr 41:625.06/Z11. [Google Scholar]
- Kucinski A, Koshy Cherian A, Valuskova P, Yegla B, Parikh V, Robinson T, Sarter M (2015) Prone to addiction as well as to falls: poor attention in sign-tracking rats extends to complex movement control and is associated with regression of choline transporter capacity. Soc Neurosci Abstr 41:253.19/V37. [Google Scholar]
- Marhe R, Luijten M, van de Wetering BJ, Smits M, Franken IH (2013) Individual differences in anterior cingulate activation associated with attentional bias predict cocaine use after treatment. Neuropsychopharmacology 38:1085–1093. 10.1038/npp.2013.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunsell JH. (2004) Neuronal representations of cognitive state: reward or attention? Trends Cogn Sci 8:261–265. 10.1016/j.tics.2004.04.003 [DOI] [PubMed] [Google Scholar]
- McLin DE 3rd, Miasnikov AA, Weinberger NM (2002) Induction of behavioral associative memory by stimulation of the nucleus basalis. Proc Natl Acad Sci U S A 99:4002–4007. 10.1073/pnas.062057099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer PJ, Lovic V, Saunders BT, Yager LM, Flagel SB, Morrow JD, Robinson TE (2012) Quantifying individual variation in the propensity to attribute incentive salience to reward cues. PLoS One 7:e38987. 10.1371/journal.pone.0038987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolone G, Angelakos CC, Meyer PJ, Robinson TE, Sarter M (2013) Cholinergic control over attention in rats prone to attribute incentive salience to reward cues. J Neurosci 33:8321–8335. 10.1523/JNEUROSCI.0709-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Sarter M (2006) Cortical choline transporter function measured in vivo using choline-sensitive microelectrodes: clearance of endogenous and exogenous choline and effects of removal of cholinergic terminals. J Neurochem 97:488–503. 10.1111/j.1471-4159.2006.03766.x [DOI] [PubMed] [Google Scholar]
- Parikh V, Apparsundaram S, Kozak R, Richards JB, Sarter M (2006) Reduced expression and capacity of the striatal high-affinity choline transporter in hyperdopaminergic mice. Neuroscience 141:379–389. 10.1016/j.neuroscience.2006.03.055 [DOI] [PubMed] [Google Scholar]
- Parikh V, St Peters M, Blakely RD, Sarter M (2013) The presynaptic choline transporter imposes limits on sustained cortical acetylcholine release and attention. J Neurosci 33:2326–2337. 10.1523/JNEUROSCI.4993-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CC, Chen H, Flagel SB, Geurts AM, Richards JB, Robinson TE, Solberg Woods LC, Palmer AA (2014) Rats are the smart choice: rationale for a renewed focus on rats in behavioral genetics. Neuropharmacology 76:250–258. 10.1016/j.neuropharm.2013.05.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza PV, Deminiere JM, Le Moal M, Simon H (1989) Factors that predict individual vulnerability to amphetamine self-administration. Science 245:1511–1513. 10.1126/science.2781295 [DOI] [PubMed] [Google Scholar]
- Pitchers KK, Flagel SB, O'Donnell EG, Woods LC, Sarter M, Robinson TE (2015) Individual variation in the propensity to attribute incentive salience to a food cue: influence of sex. Behav Brain Res 278:462–469. 10.1016/j.bbr.2014.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro FM, Black SA, Cregan SP, Prado VF, Prado MA, Rylett RJ, Ferguson SS (2005) Constitutive high-affinity choline transporter endocytosis is determined by a carboxyl-terminal tail dileucine motif. J Neurochem 94:86–96. 10.1111/j.1471-4159.2005.03171.x [DOI] [PubMed] [Google Scholar]
- Ribeiro FM, Black SA, Prado VF, Rylett RJ, Ferguson SS, Prado MA (2006) The “ins” and “outs” of the high-affinity choline transporter CHT1. J Neurochem 97:1–12. 10.1111/j.1471-4159.2006.03695.x [DOI] [PubMed] [Google Scholar]
- Robbins TW, Gillan CM, Smith DG, de Wit S, Ersche KD (2012) Neurocognitive endophenotypes of impulsivity and compulsivity: towards dimensional psychiatry. Trends Cogn Sci 16:81–91. 10.1016/j.tics.2011.11.009 [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC (2008) Review. The incentive sensitization theory of addiction: some current issues. Philos Trans R Soc Lond B Biol Sci 363:3137–3146. 10.1098/rstb.2008.0093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Flagel SB (2009) Dissociating the predictive and incentive motivational properties of reward-related cues through the study of individual differences. Biol Psychiatry 65:869–873. 10.1016/j.biopsych.2008.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Yager LM, Cogan ES, Saunders BT (2014) On the motivational properties of reward cues: individual differences. Neuropharmacology 76:450–459. 10.1016/j.neuropharm.2013.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Kim Y (2015) Interpreting chemical neurotransmission in vivo: techniques, time scales, and theories. ACS Chem Neurosci 6:8–10. 10.1021/cn500319m [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Parikh V (2005) Choline transporters, cholinergic transmission and cognition. Nat Rev Neurosci 6:48–56. 10.1038/nrn1588 [DOI] [PubMed] [Google Scholar]
- Sarter M, Lustig C, Berry AS, Gritton H, Howe WM, Parikh V (2016a) What do phasic cholinergic signals do? Neurobiol Learn Memory 130:135–141. 10.1016/j.nlm.2016.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Lustig C, Blakely RD, Koshy Cherian A (2016b) Cholinergic genetics of visual attention: Human and mouse choline transporter capacity variants influence distractibility. J Physiol Paris 110:10–18. 10.1016/j.jphysparis.2016.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Granger AJ, Sabatini BL (2015) Corelease of acetylcholine and GABA from cholinergic forebrain neurons. eLife 4:e06412 10.7554/eLife.06412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders BT, Robinson TE (2010) A cocaine cue acts as an incentive stimulus in some but not others: implications for addiction. Biol Psychiatry 67:730–736. 10.1016/j.biopsych.2009.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders BT, Robinson TE (2011) Individual variation in the motivational properties of cocaine. Neuropsychopharmacology 36:1668–1676. 10.1038/npp.2011.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders BT, Robinson TE (2012) The role of dopamine in the accumbens core in the expression of Pavlovian-conditioned responses. Eur J Neurosci 36:2521–2532. 10.1111/j.1460-9568.2012.08217.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders BT, O'Donnell EG, Aurbach EL, Robinson TE (2014) A cocaine context renews drug seeking preferentially in a subset of individuals. Neuropsychopharmacology 39:2816–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shomstein S, Johnson J (2013) Shaping attention with reward: effects of reward on space- and object-based selection. Psychol Sci 24:2369–2378. 10.1177/0956797613490743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer BF, Bryan MA, Popov P, Scarff R, Carter C, Wright E, Aragona BJ, Robinson TE (2016) The sensory features of a food cue influence its ability to act as an incentive stimulus and evoke dopamine release in the nucleus accumbens core. Lear Mem 23:595–606. 10.1101/lm.043026.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P, Mabrouk OS, Hershey ND, Kennedy RT (2012) In vivo neurochemical monitoring using benzoyl chloride derivatization and liquid chromatography–mass spectrometry. Anal Chem 84:412–419. 10.1021/ac202794q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi D, Goldstein RZ, Telang F, Maloney T, Alia-Klein N, Caparelli EC, Volkow ND (2007) Thalamo-cortical dysfunction in cocaine abusers: implications in attention and perception. Psychiatry Res 155:189–201. 10.1016/j.pscychresns.2007.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hemel-Ruiter ME, de Jong PJ, Ostafin BD, Wiers RW (2015) Reward sensitivity, attentional bias, and executive control in early adolescent alcohol use. Addict Behav 40:84–90. 10.1016/j.addbeh.2014.09.004 [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Ma Y, Fowler JS, Wong C, Ding YS, Hitzemann R, Swanson JM, Kalivas P (2005) Activation of orbital and medial prefrontal cortex by methylphenidate in cocaine-addicted subjects but not in controls: relevance to addiction. J Neurosci 25:3932–3939. 10.1523/JNEUROSCI.0433-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yager LM, Robinson TE (2013) A classically conditioned cocaine cue acquires greater control over motivated behavior in rats prone to attribute incentive salience to a food cue. Psychopharmacology (Berl) 226:217–228. 10.1007/s00213-012-2890-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yager LM, Pitchers KK, Flagel SB, Robinson TE (2015) Individual variation in the motivational and neurobiological effects of an opioid cue. Neuropsychopharmacology 40:1269–1277. 10.1038/npp.2014.314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Imajoh-Ohmi S, Haga T (2012) The high-affinity choline transporter CHT1 is regulated by the ubiquitin ligase Nedd4-2. Biomed Res 33:1–8. 10.2220/biomedres.33.1 [DOI] [PubMed] [Google Scholar]