

Abstract
The human dopamine transporter (DAT) has a tetrahedral Zn2+-binding site. Zn2+-binding sites are also recognized by other first-row transition metals. Excessive accumulation of manganese or of copper can lead to parkinsonism because of dopamine deficiency. Accordingly, we examined the effect of Mn2+, Co2+, Ni2+, and Cu2+ on transport-associated currents through DAT and DAT-H193K, a mutant with a disrupted Zn2+-binding site. All transition metals except Mn2+ modulated the transport cycle of wild-type DAT with affinities in the low micromolar range. In this concentration range, they were devoid of any action on DAT-H193K. The active transition metals reduced the affinity of DAT for dopamine. The affinity shift was most pronounced for Cu2+, followed by Ni2+ and Zn2+ (= Co2+). The extent of the affinity shift and the reciprocal effect of substrate on metal affinity accounted for the different modes of action: Ni2+ and Cu2+ uniformly stimulated and inhibited, respectively, the substrate-induced steady-state currents through DAT. In contrast, Zn2+ elicited biphasic effects on transport, i.e. stimulation at 1 μm and inhibition at 10 μm. A kinetic model that posited preferential binding of transition metal ions to the outward-facing apo state of DAT and a reciprocal interaction of dopamine and transition metals recapitulated all experimental findings. Allosteric activation of DAT via the Zn2+-binding site may be of interest to restore transport in loss-of-function mutants.
Keywords: dopamine, electrophysiology, metal, monoamine transporter, neurotransmitter transport
Introduction
The physiological role of the dopamine transporter (DAT/SLC6A3)3 is to clear the extracellular space from previously released dopamine and to replenish vesicular stores (1). Accordingly, the transport capacity of DAT shapes the synaptic response. We recently showed that the endogenous ligand Zn2+ increases the turnover rate of DAT (2). This action of Zn2+ is presumably of physiological relevance: Zn2+ is accumulated in synaptic vesicles via a dedicated transporter (ZnT3/SLC30A3) and released as a co-transmitter (3).
Numerous proteins require Zn2+ for their activity; it has been estimated that about 10% of the proteins encoded by the human genome bind Zn2+ (4). Similarly, about 30% of all enzymes are thought to require Zn2+ for catalysis (5). In addition, structural Zn2+-binding sites are important for protein stability, e.g. in transcription factors harboring the eponymous zinc fingers (6). Zn2+-binding sites rely on the permutation of specific arrangements of cysteine, histidine, aspartate, and glutamate residues and, in catalytic Zn2+ sites, of water. The resulting affinities range from picomolar to micromolar (7). However, other first-row transition metal ions can also be trapped by these binding sites (5, 8). As an approximation, the stability of their interaction increases across the period to a maximum stability for complexes containing Cu2+, resulting in the Irving-Williams series: Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+ (9). It is thought that mismetallation of proteins is prevented by controlling the concentration of metal ions in the compartment in which the target protein operates (4, 7).
Mismetallation of DAT may be of toxicological relevance. It has long been known that occupational exposure to manganese can result in a syndrome resembling idiopathic Parkinson's disease (10, 11). Studies in primates indicate that manganese poisoning impairs dopamine release in the basal ganglia (12). Loss-of-function mutations in the manganese transporter SLC39A14 result in manganese overload and cause a syndrome of childhood dystonia/parkinsonism (13). This is reminiscent of inactivating mutations of DAT/SLC6A3 (14). Inactivating mutations in SLC30A10, another manganese extruder, also gives rise to parkinsonism (15). Similarly, Wilson's disease is caused by a deficiency in the copper transporter ATP7B, which results in copper accumulation in the basal ganglia and produces, inter alia, Parkinsonian symptoms (16). Before the link between dopamine deficiency and Parkinson's disease had been established, it was noted that dopamine excretion was augmented in patients suffering from Wilson's disease (17). Accordingly, in this study, we explored the effect of transition metals on human DAT. In our analysis, we relied on electrophysiological recordings because they provided the time resolution required to dissect the actions of the transition metals on individual steps of the transport cycle. We observed that individual metals differ in their ability to affect the transport cycle of DAT because they differed in the extent to which they were subject to a reciprocal modulation by substrate.
Results
Effects of Transition Metals on the Steady-state Current through DAT
When challenged with substrate, monoamine transporters of the SLC6 family produce two types of currents that can be recorded in the whole-cell patch clamp configuration: an initial capacitive peak current that reflects the binding of substrate and co-substrate ions to the transporter and their movement in the electric field of the membrane (18, 19) and a sustained, steady-state current that reflects movement of the transporter through the transport cycle (18). These two components can be seen when dopamine is applied to a voltage-clamped HEK293 cell expressing DAT in the whole-cell patch clamp configuration (cf. Fig. 1, A–E). Zn2+ accelerates the return step, i.e. the transition of the empty inward-facing to the outward-facing state of the transporter (2). The resulting acceleration of the transport cycle resulted in an increase in the steady-state current when Zn2+ was applied while the cell was continuously superfused with dopamine (Fig. 1E). We employed Zn2+ at 10 μm because this was shown previously to be a saturating concentration (20–23). We compared the action of the transition metals Mn2+, Co2+, Ni2+, and Cu2+ (Fig. 1, A–D) with that of Zn2+ (Fig. 1E). We found Mn2+ to be ineffective at concentrations between 1 and 100 μm. In Fig. 1, A and F, we show the absence of any effect at 30 and 100 μm.
FIGURE 1.
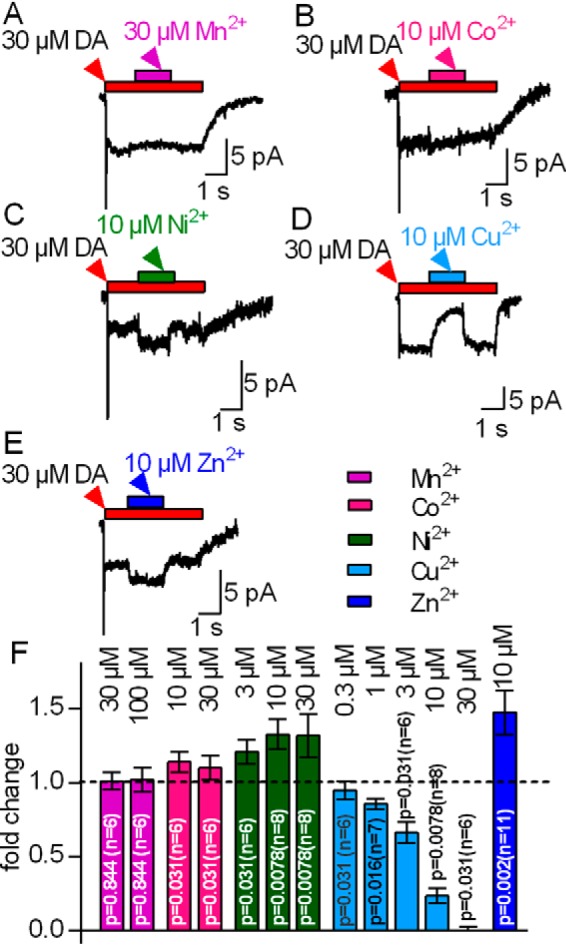
Effects of first-row transition metals on the steady-state current amplitude (Isteady-state) carried by hDAT. HEK293 cells stably expressing hDAT were voltage-clamped to −60 mV. The cells were challenged with 30 μm dopamine (DA). After 2 s, the transition metal was co-applied for 3 s as indicated. A, co-application of 30 μm Mn2+ was ineffective. B, C, and E, co-application of 10 μm Co2+, Ni2+, or Zn2+ stimulated the current. D, 10 μm Cu2+ led to inhibition. In all instances, the steady-state current recovered to its initial amplitude upon metal removal. F, the steady-state current upon co-application of the indicated transition metals was related to that seen prior to metal application (-fold change = Isteady-state after application / Isteady-state before application) to normalize for differences in current sizes in individual cells. Data represent mean ± S.D. The respective -fold changes were tested against the hypothetical value 1 (= no change, indicated as a dotted line in the bar graph) using Wilcoxon matched pairs signed-rank test.
In contrast, application of 10 μm Co2+ (Fig. 1B) led to a small but significant increase in the steady-state current amplitude by about 10%. The current did not increase further in the presence of 30 μm Co2+ (Fig. 1F). This weak stimulatory effect of Co2+ precluded further analysis. Application of 10 μm Ni2+ resulted in a robust increase in the steady-state current, which relaxed to its initial amplitude upon removal of Ni2+ (Fig. 1C). We also tested 3 and 30 μm Ni2+ (summarized in Fig. 1F). The effect elicited by 30 μm Ni2+ was essentially comparable with that caused by 10 μm Ni2+, indicating that, at saturating levels of Ni2+, the steady-state currents were about 1.3-fold larger than in the absence of any metal. Contrary to Co2+ and Ni2+, Cu2+ reversibly inhibited the steady-state current through DAT (Fig. 1D) with an IC50 of 4.4 μm (2.3–8.2 μm, 95% confidence interval). These observations suggest that all tested transition metals except Mn2+ either stimulate or inhibit the transport cycle of DAT. Two explanations can account for the inability of Mn2+ to modulate the activity of DAT: Mn2+ binds but neither stimulates nor inhibits the transport cycle, or the affinity of Mn2+ for the Zn2+-binding site of DAT is too low. We distinguished between these two possibilities by first concomitantly applying 30 μm dopamine and 100 μm Mn2+ to elicit the steady-state current through DAT. If Mn2+ had occupied the Zn2+-binding site of DAT, then it should preclude the increase in steady-state current resulting from the subsequent application of Zn2+. This, however, was not the case. As can be seen in Fig. 2A, wash-in of Zn2+ induced a robust stimulation of the current that was similar to that seen in the absence of Mn2+ (cf. Fig. 2F, first and fifth columns).
FIGURE 2.
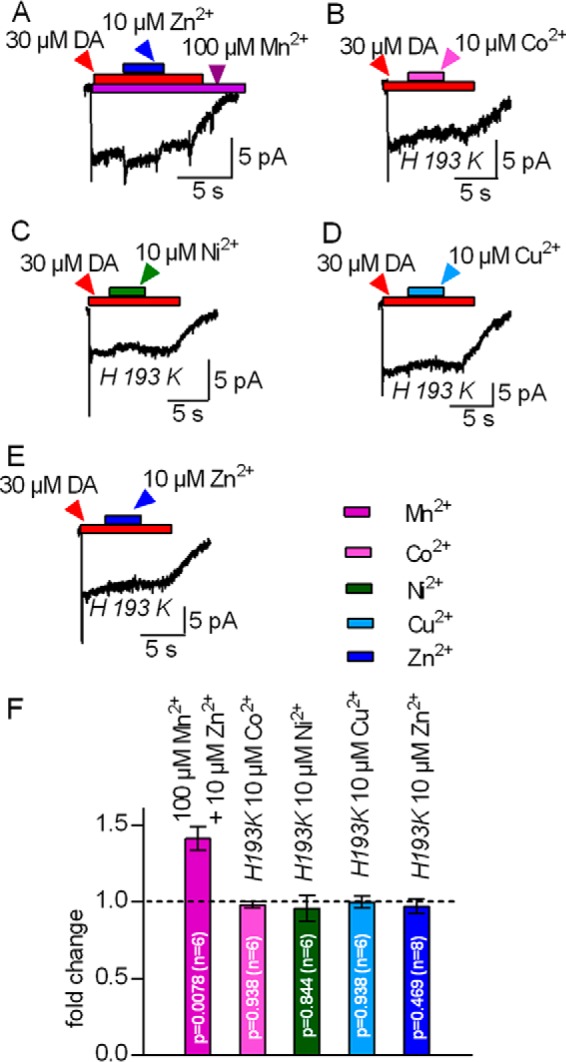
Interaction of Co2+, Ni2+, and Cu2+ but not Mn2+ with the endogenous Zn2+-binding site of DAT. A–E, HEK293 cells stably expressing hDAT (A) or transiently transfected with the mutant hDAT-H193K (B–E) were clamped to −60 mV. Currents were evoked by 30 μm dopamine. A, representative trace of a current recorded in the continuous presence of 100 μm Mn2+. Co-application of 10 μm Zn2+ increased the current amplitude. B–E, representative traces of currents carried by DAT-H193K. Co-application of 10 μm Co2+, Ni2+, Cu2+, or Zn2+ did not affect the current. F, summary of A–E. The steady-state current upon co-application of the indicated transition metals was related to that seen prior to metal application (-fold change); Zn2+ increased the current carried by hDAT in the continuous presence of 100 μm Mn2+. Currents by hDAT-H193K were unchanged upon co-application of 10 μm Co2+, Ni2+, Cu2+, or Zn2+, respectively. Data represent mean ± S.D. The respective -fold changes were tested against the hypothetical value 1 (= no change, indicated as a dotted line in the bar graph) using Wilcoxon matched pairs signed-rank test.
We also verified that both the stimulatory action of Co2+ and Ni2+ and the inhibitory action of Cu2+ required the Zn2+-binding site of DAT by examining their action on steady-state currents through DAT-H193K. In this mutant, the Zn2+-coordinating histidine is replaced by lysine, the residue found at the equivalent position of the norepinephrine transporter. The norepinephrine transporter is the closest relative of DAT but is insensitive to Zn2+. Accordingly, the mutation of His193 to Lys eliminates high-affinity Zn2+ binding to DAT (20, 21). It is evident from the current traces shown in Fig. 2, B–E, and the summary shown in Fig. 1F that both the stimulation of the steady-state current by Co2+, Ni2+, and Zn2+ and its inhibition by Cu2+ were abrogated in DAT-H193K. This comparison is based on recordings done in a stable cell line and transiently transfected cells for wild-type DAT (Fig. 1) and the DAT-H193K mutant (Fig. 2), respectively. We also recorded currents through wild-type DAT in transiently transfected HEK293 cells. These experiments recapitulated the data shown in Fig. 1, i.e. stimulation of substrate-induced currents by Co2+, Ni2+, and Zn2+, their inhibition by Cu2+, and the absence of any effect in the presence of Mn2+ (data not shown). Hence, we rule out that differences between transiently and stably transfected cells can account for the distinct effects of transition metals on wild-type and DAT-H913K.
Transition Metal-induced Shifts of EC50 for Substrate-induced Steady-state Currents through hDAT
The substrate-induced steady-state current reflects cycling of DAT through the forward transport mode (18). Dopamine increased the steady-state current through DAT in a concentration-dependent manner (Fig. 3A). This concentration-response curve was shifted by all transition metals; representative original traces are shown in Fig. 3, B–E, for dopamine-induced steady-state currents in the presence of 10 μm Zn2+, Co2+, Ni2+, and Cu2+, respectively. The pertinent analysis for all transition metals is summarized in Fig. 3E; the concentration-response curve was adequately described by a saturation hyperbola. More importantly, all transition metals reduced the apparent affinity of dopamine, but the magnitude of the shift differed. In the absence of any metal, the EC50 for the currents induced by dopamine was 0.8 μm (0.6–1 μm, 95% confidence interval) and increased to EC50 Zn2+ = 1.6 μm (1.2–2.2 μm), EC50 Co2+ = 2.4 μm (2.1–2.8 μm), EC50 Ni2+ = 4.3 μm (3.5–4.7 μm), and EC50 Cu2+ = 36.5 μm (17.1–72.2 μm) in the presence of Zn2+, Co2+, Ni2+, and Cu2+, respectively.
FIGURE 3.
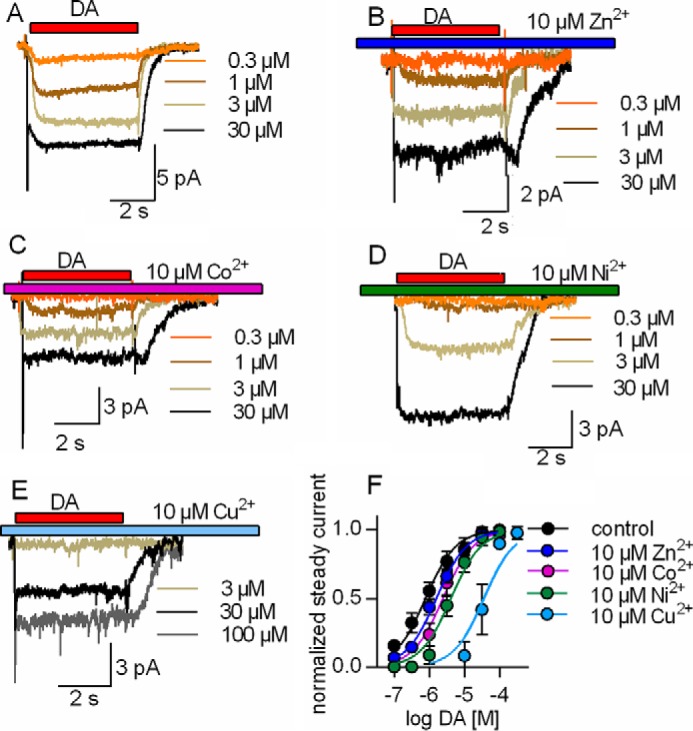
Rightward shift by Zn2+, Co2+, Ni2+, and Cu2+ in the EC50 of dopamine in eliciting the steady-state current through DAT. HEK293 cells stably expressing hDAT were clamped to −60 mV. Currents through DAT were evoked by 5-s applications of increasing dopamine concentrations (0.3–100 μm) in the absence and presence of 10 μm Zn2+, Co2+, Ni2+, and Cu2+. A–E, representative traces of currents through hDAT evoked by increasing dopamine concentrations in the absence (A) or presence of 10 μm Zn2+, Co2+, Ni2+, and Cu2+, respectively (B–E). F, the concentration-response curve of the steady-state current was shifted to higher dopamine concentrations in the presence of the metals. The amplitudes of the steady-state current were normalized to the current at saturation. The EC50 values were estimated by a fit to a binding hyperbola. EC50 control = 0.8 μm (0.6–1, n = 9), EC50 Zn2+ = 1.6 μm (1.2–2.2, n = 8), EC50 Co2+ = 2.4 μm (2.1–2.8, n = 8), EC50 Ni2+ = 4.3 μm (3.5–4.7, n = 6), and EC50 Cu2+ = 36.5 μm (17.1–72.2, n = 6). Data represent mean and 95% confidence interval (in parentheses).
Sole Activation of DAT by Ni2+
Taken together, the data summarized in Figs. 1–3 indicate that transition metals induced distinct effects on the transport cycle of DAT despite their binding to the same site: Cu2+ was uniformly inhibitory. The action of Zn2+ depends on the substrate concentration and the internal Na+ concentration (see below and Refs. 2, 22–24). Accordingly, we selected Ni2+ as an additional representative of the transition metals to understand their action on DAT because, contrary to Co2+, Ni2+ elicited a robust enhancement of the steady-state current. We recently demonstrated that current stimulation by Zn2+ is caused by an enhancement of the turnover rate of DAT (2). We verified that this was also true for Ni2+ by examining the turnover rate; the pertinent protocol and representative current traces are shown in Fig. 4. The approach relies on the paired application of substrate (i.e. dopamine) pulses. The first application elicits a robust peak current that corresponds to the conformational transition associated with substrate binding and drives DAT into the transport cycle, which gives rise to the sustained steady-state current. This steady-state current decays when substrate is withheld. During this decay, only a fraction of the transporters are available for binding of dopamine because they have not yet reached the outward-facing conformation. Full recovery of the peak current is achieved only after all transporters have completed the transport cycle. Hence, the rate of peak current recovery is a measure of the turnover rate. The time-dependent recovery of the peak current is evident from the original traces shown in Fig. 4A. The shorter the interval between the first and the second substrate pulse, the smaller a peak current was elicited by the second dopamine pulse. Full recovery was accomplished within 2 s with a time course that was adequately described by a monoexponential rise (Fig. 4B, open symbols. In the presence of Ni2+, the peak current recovered at a faster rate (Fig. 4B, closed symbols). Thus, similar to Zn2+, Ni2+ significantly accelerated the transport cycle of hDAT (p < 0.0001, F test).
FIGURE 4.
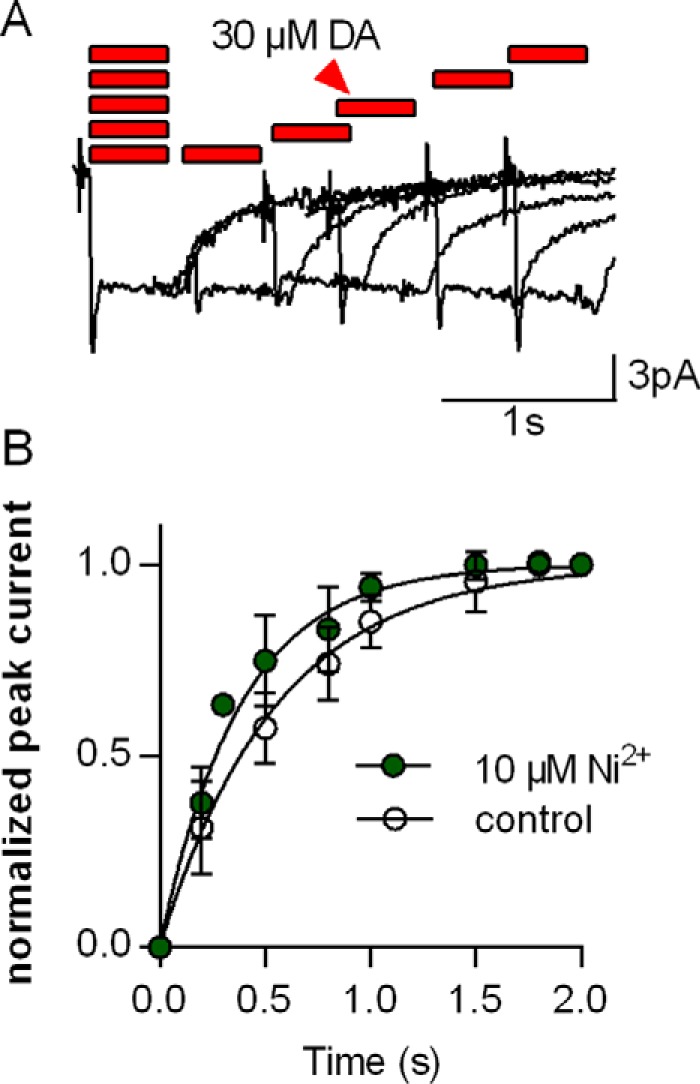
Ni2+-induced increase in the turnover rate of DAT. HEK293 cells stably expressing hDAT were voltage-clamped to −60 mV. A, protocol to measure the turnover rate of DAT. 30 μm dopamine was applied to the cell for 0.5 s. This was followed by a second pulse (30 μm dopamine for 0.5 s) applied after different wash intervals (0.1, 0.2, 0.5, 0.8, 1, 1.5, and 2 s). The peak current recovered over time and reached its full amplitude after prolonged dopamine-free intervals. B, the peak current recovers faster in the presence of 10 μm Ni2+ (green circle) in comparison with the control. The peak currents were normalized to the respective largest peak current from the same cell. The time course of the peak current recovery was fit to a monoexponential function (black lines, R2 = 0.96 and 0.96). The time constants estimated by the fits were as follows: control, 0.55 s (0.51–0.60, n = 6); Ni2+, 0.38 s (0.35–0.42, n = 6). Data represent mean and 95% confidence interval (in parentheses).
This analysis of currents through DAT is consistent with the interpretation that Ni2+ acts as a stimulator of substrate uptake. We verified this conjecture by comparing cellular uptake of dopamine by hDAT in the absence and presence of 1 and 10 μm Zn2+ (Fig. 5A) and Ni2+ (Fig. 5C). Uptake of [3H]dopamine uptake was stimulated at both Ni2+ concentrations. In contrast, [3H]dopamine uptake was stimulated by 1 μm Zn2+ but inhibited by 10 μm Zn2+. This biphasic action of Zn2+ on substrate uptake in HEK293 cells has not been reported previously (20, 21). However, in earlier studies, the action of Zn2+ on substrate uptake by hDAT was explored at substrate concentrations below the Km of the transporter. Thus, we surmise that the stimulation at low Zn2+ concentrations escaped detection in studies (20, 21) that examined the action of Zn2+ at non-saturating substrate concentrations. We explored the biphasic effect of Zn2+ in the presence of saturating substrate concentrations (30 μm dopamine) in the absence and presence of 0.1, 1, 3, 10, and 30 μm Zn2+ (Fig. 5B). Under these conditions, the stimulation of maximal transport velocity peaks at about 1 μm Zn2+. We also explored the effect of Ni2+ at saturating substrate concentrations (30 μm dopamine) in the absence and presence of 1, 10, and 30 μm Ni2+. We found substrate uptake stimulated for Ni2+ concentrations up to 30 μm (Fig. 5D). At concentrations exceeding 30 μm, Ni2+ causes a nonspecific inhibition of transport, i.e. this inhibition is also seen in DAT-H193K (data not shown). Accordingly, it was not possible to examine how higher concentrations of Ni2+ affected substrate transport via the transition metal ion-binding site.
FIGURE 5.
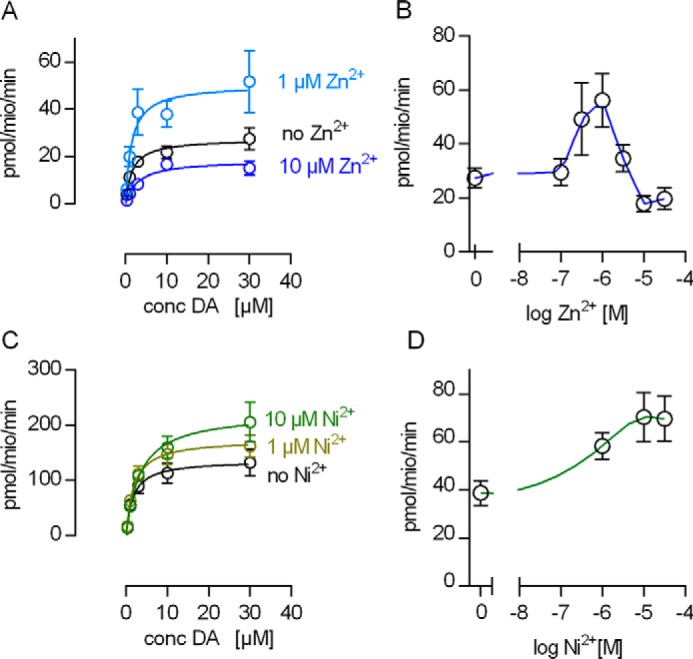
Ni2+-mediated stimulation of substrate uptake by hDAT. A and C, [3H]dopamine (DA) uptake in the absence and in the presence of 1 and 10 μm Zn2+ (A) or Ni2+ (C), respectively. Each data point in A and C is the mean of five independent experiments. The data were fit to the Michaelis-Menten equation, and the estimated parameters were as follows (Km in micromolar, Vmax in picomoles per million per minute). A, control, Km = 1.6 (0.6–2.7, Vmax = 27.3 (22.6–31.9); 1 μm Zn2+, Km = 1.5 (0.5–3.0), Vmax = 50.3 (35.8–64.0); 10 μm Zn2+, Km = 2.9 (0.9–4.9), Vmax = 18.3 (14.0–21.0). C, control, Km = 1.6 (0.3–3.0), Vmax = 136 (107.5–164.7); 1 μm Ni2+, Km = 1.8 (0.7–2.9), Vmax = 174.3 (144.0–204.0); 10 μm Ni2+, Km = 3.4 (1.4–5.3), Vmax = 223.2 (180.9–265.0). Data represent mean and 95% confidence interval (in parentheses). The estimated Vmax values were significantly different between 0, 1, and 10 μm Zn2+ (p < 0.0008, F-test) and between 0, 1, and 10 μm Ni2+ (p < 0.0024, F-test). B and D, Vmax (= [3H]dopamine uptake at a final concentration of 30 μm dopamine) as a function of the applied Zn2+ or Ni2+ concentration. The blue line in B and the green line in D show spline fits through the data points. The error bars indicate standard deviation (n = 6).
Stimulation of Currents through DAT by Ni2+ at High [Na+]i
The action of Zn2+ depends on the intracellular sodium concentration. At low [Na+]i, Zn2+ acts as an activator of dopamine transport, but it is inhibitory when [Na+]i is high. This is the case in HEK293 cells overexpressing DAT (2, 22–24). Accordingly, the finding that Ni2+ increased substrate uptake into HEK293 cells suggested that Ni2+ can also stimulate DAT at high [Na+]i. We tested this conjecture by comparing the action of 10 μm Zn2+ and 10 μm Ni2+ on currents by DAT in the presence of 25 mm Na+i. Fig. 6A shows representative currents through hDAT that were induced by the rapid application of 30 μm dopamine followed by the concomitant superfusion with 10 μm Ni2+ (Fig. 6A, left panel) or 10 μm Ni2+ (Fig. 6A, right panel) for 5 s. It is evident from these recordings that, in the presence of 25 mm [Na+]i, Zn2+ inhibits the steady-state current whereas Ni2+ does not. Although we failed to detect current inhibition in the presence of 10 μm Ni2+, it is worth pointing out that the stimulatory effect of Ni2+ was lower with 25 mm [Na+]i than the one observed with 6 mm [Na+]i (Fig. 1C). This suggests that, although the inhibitory action of [Na+]i is smaller with Ni2+ than with Zn2+, it is not fully eliminated.
FIGURE 6.
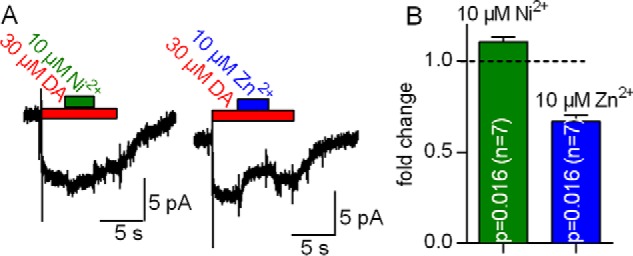
Inhibition by Zn2+ but not by Ni2+ of steady-state currents through DAT in the presence of 25 mm [Na+]i. A, a HEK293 cell stably expressing hDAT was clamped to −60 mV with [Na+]i set to 25 mm. The cell was challenged with 30 μm dopamine to elicit a current. Although co-application of 10 μm Ni2+ led to a small increase in the current amplitude, co-application of 10 μm Zn2+ decreased it. B, -fold change of the steady-state current at 25 mm [Na+]i in the presence of 10 μm Ni2+ (green column) and 10 μm Zn2+ (blue column). Data represent mean ± S.D. The respective -fold changes were tested against the hypothetical value 1 (= no change, indicated as a dotted line in the bar graph) using Wilcoxon matched pairs signed-rank test.
Differential Modulation of the Peak Current Amplitude by Ni2+ and Zn2+
The substrate-induced peak current reflects the initiation of the transport cycle, where substrate and co-substrate ions are bound by the transporter and moved through the electric field of the membrane (18, 19). Zn2+ decreases the amplitude of this substrate-induced peak current (2), presumably because Zn2+ does not only favor the return of the empty transporter (forward transport mode) but also the return of the substrate-bound transporter to the outward-facing state (substrate exchange mode) (2). Accordingly, the charge that gives rise to the inwardly directed peak current is moved into the opposite direction, and, hence, the peak current amplitude is decreased. Ni2+ and Zn2+ differed in their ability to stimulate steady-state transport. In the micromolar range, Ni2+ uniformly stimulated substrate uptake, but Zn2+ gave rise to a stimulation at 1 μm and inhibition at 10 μm (cf. Fig. 5A). This suggests that Zn2+ and Ni2+ do not affect the conformational transitions hDAT must undergo during the transport cycle in the same way. Hence, we compared the effect of Ni2+ on the dopamine-induced peak current by employing the protocol shown in Fig. 7A. We applied 10 μm Ni2+ or Zn2+ for 5 s to a HEK293 cell expressing hDAT. Subsequently, test pulses of 30 μm dopamine were applied immediately or 0.2, 0.5, 1, and 2 s after removal of the metal ion. As can be seen in Fig. 7B, the effect of Ni2+ on the peak current was much smaller than that of Zn2+. Although Zn2+ decreased the peak current amplitude by about 30%, Ni2+ only affected it to a very minor extent.
FIGURE 7.
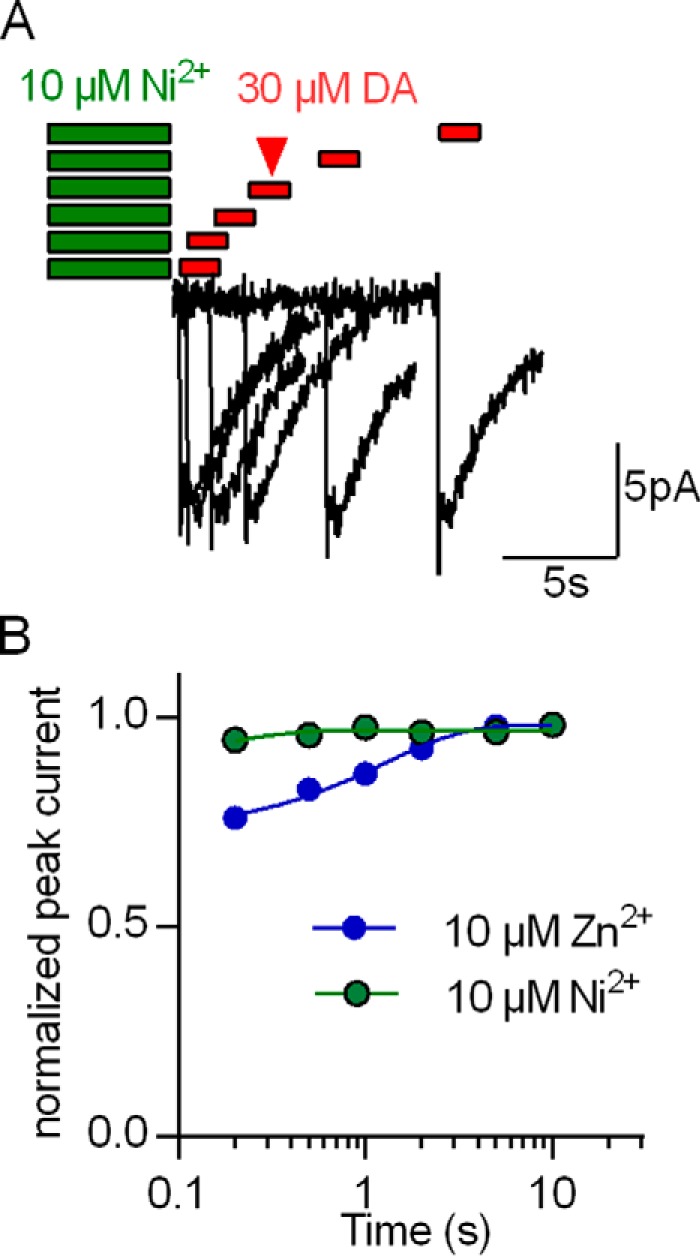
Comparison of Ni2+- and Zn2+-induced changes in the peak current. A, protocol for determining the effect of Ni2+ (or Zn2+) on the peak current by hDAT. A HEK293 cell stably expressing hDAT was clamped to −60 mV and exposed to 10 μm Ni2+ for 5 s. After wash periods of 0.2, 0.5, 1, 2, and 5 s, a peak current was triggered by rapid application of 30 μm dopamine (0.5 s). The original traces are from a representative experiment that was reproduced in seven independent experiments with 10 μm Ni2+ and Zn2+, respectively. B, recovery of the peak current following application of 10 μm Ni2+ (green circles) and 10 μm Zn2+ (blue circles), respectively. The peak current amplitudes were normalized to the respective largest current and plotted as a function of the wash time interval. The two sets of data points were each fit to a monoexponential function (green and blue lines). The fits yielded the following parameters: Ni2+, Trecovery = 0.15 s (0.07–5.66), blocked fraction 13% (10–17); Zn2+, Trecovery = 1.25 s (0.82–2.5), blocked fraction 28% (22–33). Data represent mean and 95% confidence interval (in parentheses). The estimated blocked fractions were significantly different (p < 0.0001, F-test).
Amphetamine-induced Substrate Release in the Presence of Zn2+ and Ni2+
During amphetamine-induced substrate release, DAT operates in the substrate exchange mode rather than the forward transport mode, i.e. amphetamine is carried inward and exchanged for dopamine (25, 26, 27). As outlined above, Zn2+ inhibits the peak current because it favors substrate exchange. Accordingly, Zn2+ increases amphetamine-induced substrate release mediated by DAT (21). In contrast, Ni2+ reduced the peak current amplitude only to a very modest extent (cf. Fig. 7) and did not block the steady-state current (i.e. the forward transport mode) in the presence of elevated [Na+]i. Based on our model, these findings predict that, contrary to Zn2+, Ni2+ does not enhance amphetamine-induced substrate efflux. This conjecture was verified (Fig. 8). In hDAT-expressing HEK293 cells that had been preloaded with [3H]MPP+, superfusion with 3 μm d-amphetamine (AMPH) caused a robust release of radioactive substrate that was further augmented by the concomitant application of 10 μm Zn2+. In contrast, Ni2+ did not enhance amphetamine-induced substrate release.
FIGURE 8.
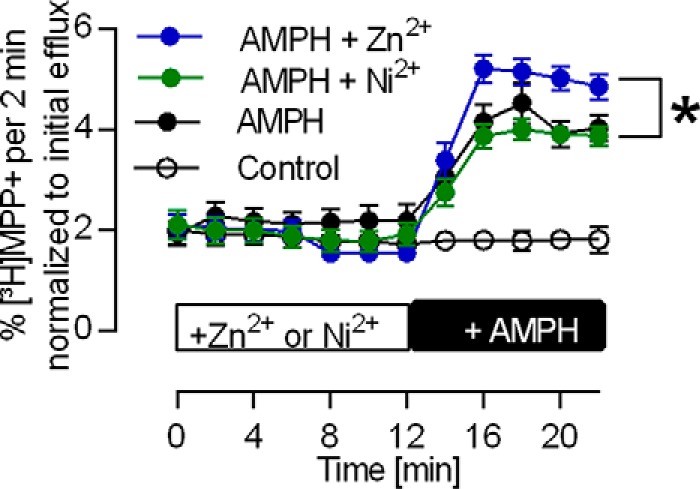
Stimulation of amphetamine-induced [3H]MPP+ release by Zn2+ but not Ni2+. HEK293 cells transfected with hDAT loaded with [3H]MPP+ were superfused with KHP buffer for 45 min (see “Experimental Procedures”). Thereafter (time point 0), 2-min fractions were collected. The prior interval of 45 min allowed for establishing a stable baseline as verified by the first seven fractions. After 12 min, 3 μm AMPH was added, which resulted in [3H]MPP+ release (black circles). This increase was larger in the presence of 10 μm Zn2+ (blue circles) but unchanged in the presence of 10 μm Ni2+ (green circles). The data are averages of 12 experiments. The data points at 20 and 22 min were used to conduct a one-way analysis of variance (Kruskal-Wallis test) followed by Dunn's multiple comparisons test (post hoc test). The difference between AMPH and AMPH + 10 μm Zn2+ is significant (p = 0.026).
Discussion
DAT has a tetrahedral Zn2+-binding site. The coordination sphere is comprised of H193 in extracellular loop 2 (EL2), His375 and Glu396, which reside on top of transmembrane helix 7 in the first helical part of extracellular loop 4 (EL4A) and on top of transmembrane helix 8 in the second helix of extracellular loop 4 (EL4B), respectively (28), and Asp206 in EL2 (29). The extracellular loops are thought to be flexible, in particular the extended EL2 (29). Accordingly, it is not surprising that this binding site can accommodate transition metals other than Zn2+ even when their optimum coordination geometries differ (5). In fact, our observations are in line with the general prediction of the Irving-William series. It was, however, surprising that the effects elicited by individual metal ions, in particular Zn2+, Ni2+, and Cu2+, differed substantially. Ni2+ and Cu2+ uniformly stimulated and inhibited, respectively, the forward transport mode of DAT. In contrast, Zn2+ elicited biphasic effects. In addition, and most importantly, Zn2+ was the only transition metal ion that also promoted the substrate exchange mode. The different modes of action of the metals must be accounted for by a plausible model of the transport cycle. It is a priori reasonable to posit that occupancy of the Zn2+-binding site produces the same primary effect. In fact, this was the case. All effective metals shifted the EC50 of dopamine for eliciting the substrate-induced steady-state current, albeit to a different extent. The shift was most pronounced for Cu2+, followed by Ni2+ and Zn2+ (and Co2+). These shifts indicate that Zn2+, and, to a larger extent, Ni2+ and Cu2+, destabilized the substrate-bound conformation(s). Destabilization is most parsimoniously explained by preferred binding of these metals to the apo conformation(s) of DAT. Thermodynamics dictate that the effect on affinity must be reciprocal. If the transition metal lowers the affinity of DAT for substrate, then raising the substrate concentration must lower the affinity for metals. In addition, because individual metals differed in their ability to shift the EC50 of dopamine, they must discriminate between apo and substrate-bound transporters to different extents, with Zn2+ and Cu2+ having the least and most pronounced affinity differences, respectively.
We implemented this information into a kinetic model (18) that was extended to account for metal-bound states (Fig. 9A). This allowed for recapitulating all experimental findings. 1) It is, for instance, possible to account for the observation that low Zn2+ concentrations (1 μm) increased substrate uptake in the presence of high [Na+]i and for the biphasic effect of Zn2+ on substrate uptake. The conformation that displays the highest affinity for Zn2+ is the outward-facing apo state of DAT; the Zn2+-binding site is fully occupied at 1 μm. Upon substrate binding, the affinity at the binding site decreases, and Zn2+ dissociates. Thus, at low concentrations (1 μm), Zn2+ interacts almost exclusively with the apo conformations. This leads to an acceleration of the rate-limiting step in the transport cycle and stimulation of substrate uptake. At elevated concentrations (e.g. 10 μm), Zn2+ also binds to the substrate-loaded conformations. This gives rise to Zn2+-induced inhibition when [Na+]i is high (Fig. 9, E and G). 2) The key actions of Ni2+ were reproduced when the observation that Ni2+ induced a larger shift in the EC50 of dopamine than Zn2+ was incorporated into the model. These actions are stimulation of the dopamine-induced steady-state current by Ni2+ (Fig. 9B), the modest effect of Ni2+ on the peak current (Fig. 9H), and the stimulatory action of Ni2+ despite elevated [Na+]i (Fig. 9F). 3) Finally, the model also accounts for current inhibition by Cu2+ (Fig. 9C). In the presence of Cu2+, the shift in EC50 was most pronounced, i.e. larger than with any other transition metal (cf. Figs. 3C and 9D). This observation indicated that Cu2+ had the highest preference for the apo conformations. Hence, its uniform inhibitory action can be rationalized as follows. In the absence of Cu2+, 30 μm dopamine is a saturating concentration that induces maximal currents and allows the transporter to cycle at maximum velocity in the forward transport mode. In contrast, Cu2+ lowers the affinity of dopamine to an extent that a dopamine concentration of 30 μm does not suffice for saturation (Fig. 9D). This leads to a decrease in current amplitude, which reflects inhibition of dopamine transport by Cu2+. Thus, the parsimonious explanation for the action of transition metals on DAT is the reciprocal effect of substrate and metal: binding of one lowers the affinity for the respective other. Given the location of the coordinating residues, it is attractive to assume that occupancy of the Zn2+-binding site by metals precludes the transition of DAT from the outward open to the occluded state. By definition, this reduces the apparent substrate affinity because the substrate can more readily dissociate from the outward open than from the occluded state. Conversely, in the occluded state, the relative position of the residues that coordinate the transition metal is likely to change. This is predicted to translate into a decline in affinity for the metal.
FIGURE 9.

A kinetic model of the action of first-row transition metals on the transport cycle of DAT. A, the model scheme, which is the same as in Li et al. (2). X indicates transition metals. For each metal there is a separate set of parameters (see supplemental Table 1). Parameters are shared between these sets when they describe conformational rearrangements and binding reactions within DAT that are unaffected by the metals. Red and orange arrows indicate metal binding to substrate-bound states, and arrows in violet and blue indicate metal binding to apo states. B and C, simulated current evoked by 30 μm dopamine stimulated by 10 μm Ni2+ (B) or inhibited by 10 μm Cu2+ (C). D, simulated concentration dependence of dopamine-induced currents in the absence and presence of 10 μm Zn2+, Ni2+, and Cu2+, respectively. E, simulated Vmax of dopamine uptake by DAT in the absence and presence of 1 and 10 μm Zn2+, assuming 25 mm [Na+]i. F, simulated currents induced by 30 μm dopamine with [Na+]i set to 25 mm. Under this condition, the model predicts current stimulation by Ni2+ and inhibition by Zn2+. G, simulated peak current recovery after application of 10 μm Ni2+ (green circles) or Zn2+ (blue circles). The simulations evidently account for the measured data.
The affinity of Mn2+ for the Zn2+-binding site of DAT was so low that there was no appreciable occupancy up to 100 μm Mn2+. This low affinity is consistent with the Irving-Williams series (5, 8, 9). Based on this low affinity, it appears unlikely that mismetallation of the Zn2+-binding site by manganese is relevant to the mechanistic understanding of manganese-induced dopamine deficiency (10–15). In contrast, in the low micromolar range, Cu2+ caused a profound inhibition of DAT. Hence, this inhibition may be relevant to understand symptoms that occur in the course of Wilson's disease for the following reasons. 1) Although it is difficult to estimate the free concentration of Cu2+ in the brain, the serum concentration of free Cu2+ (i.e. not bound to coeruloplasmin) is in the micromolar range (30). The concentration of Cu2+ at the site of dopaminergic projections is thought to exceed that in serum because Cu2+ accumulates in several brain regions, including the corpus striatum. Thus, Cu2+ levels in the brain are likely to reach concentrations that impair retrieval of dopamine by DAT and thus lead to a hypodopaminergic state. 2) Imaging studies suggest that a presynaptic dopaminergic deficit contributes to the neurological symptoms from which patients with Wilson's disease suffer (31–33). 3) In addition, patients with Wilson's disease are very susceptible to side effects of neuroleptic drugs that arise from dopamine receptor blockage in the caudate nucleus and putamen. When erroneously treated with both typical and atypical neuroleptic/antipsychotic drugs, these patients are prone to develop drug-induced extrapyramidal symptoms (34). This enhanced susceptibility presumably arises from low D2 receptor occupancy by endogenous dopamine because of impaired synaptic release. 4) Finally, administration of zinc (e.g. 150 mg of zinc acetate/day) is an effective treatment for Wilson's disease and can be used as an alternative to chelation therapy with d-penicillamine or trientine (16, 30). The effect relies on the Zn2+-dependent induction of metallothionein into the gut epithelium and the resulting reduced systemic bioavailability of dietary copper (16). Interestingly, Zn2+ has been found to be particularly effective in improving neurological deficits (30, 35) even when prior chelation therapy failed (36, 37). Based on this circumstantial evidence and on our observations, we propose that displacement of Cu2+ from the Zn2+-binding site of DAT may contribute to the therapeutic efficacy of Zn2+ in Wilson's disease.
Occupancy of the allosteric Zn2+-binding site allows for a wide range of effects, i.e. stimulation and inhibition of the forward transport mode and enhancing the substrate exchange mode. It is likely that this binding site can be addressed by compounds other than transition metals. Allosteric activators of the forward transport mode are of particular interest. There are several loss-of-function mutations in the coding sequence of human DAT that lead to a syndrome of infantile or juvenile dystonia and parkinsonism (14). The vast majority of these mutations result in defective folding and, hence, retention of the mutated proteins in the endoplasmic reticulum. However, there are some mutants that do reach the cell surface but fail to transport dopamine with adequate turnover rates. A search for allosteric activators is therefore justified because they may act as correctors and restore the forward transport mode to a velocity that is commensurate with normal dopaminergic transmission (40). In fact, it appears worthwhile to test whether Ni2+ (or any other transition metal) accelerates transport in these mutants.
Experimental Procedures
Whole-cell Patch Clamp
HEK293 cells stably expressing hDAT or transiently expressing hDAT H193K were seeded at low density 24 h before recordings. Currents by the transporter were measured in the whole-cell patch clamp configuration. The electrode resistances were between 2–5 megohms. For the majority of the recordings, we used the following internal solution: 133 mm potassium gluconate, 6 mm NaCl, 1 mm CaCl2, 0.7 mm MgCl2, 10 mm EGTA, and 10 mm HEPES adjusted to pH 7.2 with KOH. For some experiments we used an internal solution containing 25 mm Na+, 25 mm NaCl, 125 mm KCl, 1 mm CaCl2, 0.7 mm MgCl2, 10 mm EGTA, and 10 mm HEPES (pH 7.2 with KOH). The external solution in all experiments was 140 mm NaCl, 3 mm KCl, 2.5 mm CaCl2, 2 mm MgCl2, 20 mm glucose, and 10 mm HEPES adjusted to pH 7.4 with NaOH. For rapid solution exchange, we used an OctaFlow superfusion device (ALA Scientific, Farmingdale, NY). Cells were continuously superfused either with blank external solution or an external solution containing dopamine, respectively. For the solutions containing Mn2+, Co2+, Ni2+, Zn2+, or Cu2+, we used the respective chloride salts (Sigma-Aldrich, St. Louis, MO). For current acquisition, we employed an Axopatch 200B amplifier and pClamp 10.2 software (Molecular Devices, Sunnyvale, CA). The cells were clamped to −60 mV, and the washout period following substrate application was 30 s in all cases. Current traces were filtered at 1 kHz and digitized at 10 kHz using a Digidata 1550 (Molecular Devices). The currents induced by dopamine were quantified using Clampfit 10.2 software. Passive holding currents were subtracted, and the traces were filtered using a 100-Hz digital Gaussian low-pass filter.
Uptake Assay
HEK293 cells stably expressing hDAT were seeded on 48-well plates precoated with poly-d-lysine (0.5 × 105 cells/well) 24 h before the assay. Cells were first washed with 500 μl/well Krebs-HEPES buffer (KHP): 130 mm NaCl, 10 mm HEPES, 1.3 mm KH2PO4, 1.5 mm CaCl2, and 0.5 mm MgSO4 (pH 7.4) with NaOH. The cells were then incubated in 200 μl of KHP buffer containing 0.1 μm [3H]dopamine and diluted with the respective cold substrates to reach final concentrations ranging from 0.1–100 μm, both in the absence and presence of 1 and 10 μm ZnCl2 or NiCl2, respectively. The cells were incubated at room temperature for 1 min with [3H]dopamine. A subsequent wash with 500 μl/well of ice-cold KHP buffer terminated uptake. Cells were finally lysed with 500 μl of 1% SDS, transferred into 2 ml of scintillation mixture (Rotiszint Eco Plus LSC, Art. 0016.3, Carl Roth GmbH + Co. KG, Karlsruhe, Germany), and counted in a Packard 2300TR TriCarb liquid scintillation analyzer (PerkinElmer Life Sciences).
Superfusion Experiments
HEK293 cells stably expressing hDAT (2.5 × 104 cells/coverslip) were preloaded with 0.2 μm [3H]MPP+ for 30 min. The coverslips were transferred to superfusion chambers and perfused with KHP buffer (0.7 ml/min at 22 °C). After an initial wash period of 45 min, fractions were collected every 2 min. The collection of the first fraction defined time point zero. Cells were exposed 10 μm ZnCl2 or 10 μm NiCl2 after two baseline fractions (4 min). 3 μm d-amphetamine was added to the superfusion buffer after 12 min. All fractions were analyzed by liquid scintillation counting (see above).
Modeling
The recorded currents were emulated with a previously published kinetic model of the transport cycle of DAT (2). This model was extended to account for the binding of Zn2+, Ni2+, and Cu2+. The time-dependent changes in state occupancies were evaluated by numerical integration of the resulting system of differential equations using Systems Biology Toolbox (38) and Matlab 2012a (Mathworks, Natick, MA). The voltage dependence of individual rates was modeled according to Laeuger (39), assuming a symmetric barrier as kij = k0ijexp(−zQijFV / 2RT), with F = 96,485 Cmol−1, r = 8.314 JK−1mol−1, V the membrane voltage in volts, and T = 293 K. The extra- and intracellular ion concentrations were set to the values used in the experiments. Substrate uptake was modeled as (TiClS × kSioff − TiCl × Si × kSion + TiClSZn × kSioff − TiClZn × Si × kSion) × NC/NA, where NC is the number of transporters and NA is the Avogadro constant. Currents and substrate uptake were simulated assuming a transporter density of 25 × 106/cell.
Author Contributions
Y. L., F. P. M., P. S. H., K. S., M. F., and W. S. conceptualized the study and designed the experiments. P. S. H., K. S., and W. S. designed the model. Y. L., F. P. M., and V. B. performed all experiments and analyzed the data. Y. L., F. P. M., H. H. S., M. F., and W. S. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Gary Rudnick for discussions and comments on the data.
This work was supported by the Austrian Science Fund/FWF Project P28090 (to W. S.), Project Program Grant SFB35 (Grants F3506 to H. H. S. and F3510 to M. F.), Project W1232 (to H. H. S.), and Chinese Scholarship Council Grant 201308310446 (to Y. L.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table 1.
- DAT
- dopamine transporter
- hDAT
- human dopamine transporter
- AMPH
- d-amphetamine
- EL
- extracellular loop
- DA
- dopamine
- EC50
- half-maximal effective concentration.
References
- 1. Kristensen A. S., Andersen J., Jørgensen T. N., Sørensen L., Eriksen J., Loland C. J., Strømgaard K., and Gether U. (2011) SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 63, 585–640 [DOI] [PubMed] [Google Scholar]
- 2. Li Y., Hasenhuetl P. S., Schicker K., Sitte H. H., Freissmuth M., and Sandtner W. (2015) Dual action of Zn2+ on the transport cycle of the dopamine transporter. J. Biol. Chem. 290, 31069–31076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sensi S. L., Paoletti P., Bush A. I., and Sekler I. (2009) Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 10, 780–791 [DOI] [PubMed] [Google Scholar]
- 4. Andreini C., Banci L., Bertini I., and Rosato A. (2006) Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 5, 196–201 [DOI] [PubMed] [Google Scholar]
- 5. Waldron K. J., Rutherford J. C., Ford D., and Robinson N. J. (2009) Metalloproteins and metal sensing. Nature 460, 823–830 [DOI] [PubMed] [Google Scholar]
- 6. Maret W. (2012) New perspectives of zinc coordination environments in proteins. J. Inorg. Biochem. 111, 110–116 [DOI] [PubMed] [Google Scholar]
- 7. Auld D. S. (2009) The ins and outs of biological zinc sites. BioMetals 22, 141–148 [DOI] [PubMed] [Google Scholar]
- 8. Foster A. W., Osman D., and Robinson N. J. (2014) Metal preferences and metallation. J. Biol. Chem. 289, 28095–28103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Irving H., and Williams R. J. P. (1948) Order of stability of metal complexes. Nature 162, 746–747 [Google Scholar]
- 10. Couper J. (1837) On the effects of black oxide of manganese when inhaled into the lungs. Br. Ann. Med. Pharm. Vital Stat. Gen. Sci. 1, 41–42 [Google Scholar]
- 11. Rodier J. (1955) Manganese poisoning in Moroccan miners. Br. J. Ind. Med. 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guilarte T. R., Burton N. C., McGlothan J. L., Verina T., Zhou Y., Alexander M., Pham L., Griswold M., Wong D. F., Syversen T., and Schneider J. S. (2008) Impairment of nigrostriatal dopamine neurotransmission by manganese is mediated by pre-synaptic mechanism(s): implications to manganese-induced parkinsonism. J. Neurochem. 107, 1236–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tuschl K., Meyer E., Valdivia L. E., Zhao N., Dadswell C., Abdul-Sada A., Hung C. Y., Simpson M. A., Chong W. K., Jacques T. S., Woltjer R. L., Eaton S., Gregory A., Sanford L., Kara E., et al. (2016) Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat. Commun. 7, 11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ng J., Zhen J., Meyer E., Erreger K., Li Y., Kakar N., Ahmad J., Thiele H., Kubisch C., Rider N. L., Holmes Morton D. H., Strauss K. A., Puffenberger E. G., D'Agnano D., Anikster Y., et al. (2014) Dopamine transporter deficiency syndrome: phenotypic spectrum from infancy to adulthood. Brain 137, 1107–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leyva-Illades D., Chen P., Zogzas C. E., Hutchens S., Mercado J. M., Swaim C. D., Morrisett R. A., Bowman A. B., Aschner M., and Mukhopadhyay S. (2014) SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J. Neurosci. 34, 14079–14095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bandmann O., Weiss K. H., and Kaler S. G. (2015) Wilson's disease and other neurological copper disorders. Lancet Neurol. 14, 103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barbeau A., Murphy G. F., and Sourkes T. L. (1961) Excretion of dopamine in diseases of basal ganglia. Science. 133, 1706–1707 [DOI] [PubMed] [Google Scholar]
- 18. Schicker K., Uzelac Z., Gesmonde J., Bulling S., Stockner T., Freissmuth M., Boehm S., Rudnick G., Sitte H. H., and Sandtner W. (2012) Unifying concept of serotonin transporter-associated currents. J. Biol. Chem. 287, 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hasenhuetl P. S., Schicker K., Koenig X., Li Y., Sarker S., Stockner T., Sucic S., Sitte H. H., Freissmuth M., and Sandtner W. (2015) Ligand selectivity among the dopamine and the serotonin transporter specified by the forward binding reaction. Mol. Pharmacol. 10.1124/mol.115.099036 [DOI] [PubMed] [Google Scholar]
- 20. Norregaard L., Frederiksen D., Nielsen E. O., and Gether U. (1998) Delineation of an endogenous zinc-binding site in the human dopamine transporter. EMBO J. 17, 4266–4273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scholze P., Nørregaard L., Singer E. A., Freissmuth M., Gether U., and Sitte H. H. (2002) The role of zinc ions in reverse transport mediated by monoamine transporters. J. Biol. Chem. 277, 21505–21513 [DOI] [PubMed] [Google Scholar]
- 22. Pifl C., Wolf A., Rebernik P., Reither H., and Berger M. L. (2009) Zinc regulates the dopamine transporter in a membrane potential and chloride dependent manner. Neuropharmacology 56, 531–540 [DOI] [PubMed] [Google Scholar]
- 23. Meinild A. K., Sitte H. H., and Gether U. (2004) Zinc potentiates an uncoupled anion conductance associated with the dopamine transporter. J. Biol. Chem. 279, 49671–49679 [DOI] [PubMed] [Google Scholar]
- 24. Bjorklund N. L., Volz T. J., and Schenk J. O. (2007) Differential effects of Zn2+ on the kinetics and cocaine inhibition of dopamine transport by the human and rat dopamine transporters. Eur. J. Pharmacol. 565, 17–25 [DOI] [PubMed] [Google Scholar]
- 25. Sitte H. H., and Freissmuth M. (2015) Amphetamines, new psychoactive drugs and the monoamine transporter cycle. Trends Pharmacol. Sci. 36, 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sandtner W., Schmid D., Schicker K., Gerstbrein K., Koenig X., Mayer F. P., Boehm S., Freissmuth M., and Sitte H. H. (2014) A quantitative model of amphetamine action on the 5-HT transporter. Br. J. Pharmacol. 171, 1007–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmid D., Koenig X., Bulusu S., Schicker K., Freissmuth M., Sitte H. H., and Sandtner W. (2015) The conservative view: is it necessary to implant a stent into the dopamine transporter? Br. J. Pharmacol. 172, 4775–4778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Loland C. J., Norregaard L., and Gether U. (1999) Defining proximity relationships in the tertiary structure of the dopamine transporter: identification of a conserved glutamic acid as a third coordinate in the endogenous Zn2+ binding site. J. Biol. Chem. 274, 36928–36934 [DOI] [PubMed] [Google Scholar]
- 29. Stockner T., Montgomery T. R., Kudlacek O., Weissensteiner R., Ecker G. F., Freissmuth M., and Sitte H. H. (2013) Mutational analysis of the high-affinity zinc binding site validates a refined human dopamine transporter homology model. PLoS Comput. Biol. 9, e1002909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Linn F. H., Houwen R. H., van Hattum J., van der Kleij S., and van Erpecum K. J. (2009) Long-term exclusive zinc monotherapy in symptomatic Wilson disease: experience in 17 patients. Hepatology 50, 1442–1452 [DOI] [PubMed] [Google Scholar]
- 31. Jeon B., Kim J. M., Jeong J. M., Kim K. M., Chang Y. S., Lee D. S., and Lee M. C. (1998) Dopamine transporter imaging with [123I]-β-CIT demonstrates presynaptic nigrostriatal dopaminergic damage in Wilson's disease. J. Neurol. Neurosurg. Psychiatry 65, 60–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hermann W., Eggers B., Barthel H., Clark D., Villmann T., Hesse S., Grahmann F., Kühn H. J., Sabri O., and Wagner A. (2002) Correlation between automated writing movements and striatal dopaminergic innervation in patients with Wilson's disease. J. Neurol. 249, 1082–1087 [DOI] [PubMed] [Google Scholar]
- 33. Wang P., Hu P., Yue D.-C., Liang H., and Xu J.-H. (2007) The clinical value of tc-99m TRODAT-1 SPECT for evaluating disease severity in young patients with symptomatic and asymptomatic Wilson disease. Clin. Nucl. Med. 32, 844–849 [DOI] [PubMed] [Google Scholar]
- 34. Srinivas K., Sinha S., Taly A. B., Prashanth L. K., Arunodaya G. R., Janardhana Reddy Y. C., and Khanna S. (2008) Dominant psychiatric manifestations in Wilson's disease: a diagnostic and therapeutic challenge. J. Neurol. Sci. 266, 104–108 [DOI] [PubMed] [Google Scholar]
- 35. Schilsky M. (2009) Zinc treatment for symptomatic Wilson disease: moving forward by looking back. Hepatology 50, 1341–1343 [DOI] [PubMed] [Google Scholar]
- 36. Heckmann J. M., Eastman R. W., De Villiers J. C., and Hewlett R. (1994) Wilson's disease: neurological and magnetic resonance imaging improvement on zinc treatment. J. Neurol. Neurosurg. Psychiatry 57, 1273–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ishida S., Doi Y., Yamane K., Sugino M., Kimura F., Hanafusa T., Fukui H., and Tamai H. (2012) Resolution of cranial MRI and SPECT abnormalities in a patient with Wilson's disease following oral zinc monotherapy. Intern. Med. 51, 1759–1763 [DOI] [PubMed] [Google Scholar]
- 38. Schmidt H., and Jirstrand M. (2006) Systems biology toolbox for MATLAB: a computational platform for research in systems biology. Bioinformatics 22, 514–515 [DOI] [PubMed] [Google Scholar]
- 39. Laeuger P. (1991) Electrogenic Ion Pumps, Sinauer Associates, Sunderland, MA [Google Scholar]
- 40. Hamilton P. J., Shekar A., Belovich A. N., Christianson N. B., Campbell N. G., Sutcliffe J. S., Galli A., Matthies H. J., and Erreger K. (2015) Zn2+ reverses functional deficits in a de novo dopamine transporter variant associated with autism spectrum disorder. Mol. Autism 6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.