

Abstract
Streptococcus pyogenes secretes various virulence factors for evasion from complement-mediated bacteriolysis. However, full understanding of the molecules possessed by this organism that interact with complement C1q, an initiator of the classical complement pathway, remains elusive. In this study, we identified an endopeptidase of S. pyogenes, PepO, as an interacting molecule, and investigated its effects on complement immunity and pathogenesis. Enzyme-linked immunosorbent assay and surface plasmon resonance analysis findings revealed that S. pyogenes recombinant PepO bound to human C1q in a concentration-dependent manner under physiological conditions. Sites of inflammation are known to have decreased pH levels, thus the effects of PepO on bacterial evasion from complement immunity was analyzed in a low pH condition. Notably, under low pH conditions, PepO exhibited a higher affinity for C1q as compared with IgG, and PepO inhibited the binding of IgG to C1q. In addition, pepO deletion rendered S. pyogenes more susceptible to the bacteriocidal activity of human serum. Also, observations of the morphological features of the pepO mutant strain (ΔpepO) showed damaged irregular surfaces as compared with the wild-type strain (WT). WT-infected tissues exhibited greater severity and lower complement activity as compared with those infected by ΔpepO in a mouse skin infection model. Furthermore, WT infection resulted in a larger accumulation of C1q than that with ΔpepO. Our results suggest that interaction of S. pyogenes PepO with C1q interferes with the complement pathway, which enables S. pyogenes to evade complement-mediated bacteriolysis under acidic conditions, such as seen in inflammatory sites.
Keywords: C1Q complex (C1QA), complement system, innate immunity, peptidase, Streptococcus pyogenes (S. pyogenes)
Introduction
The human pathogen Streptococcus pyogenes is a β-hemolytic Gram-positive bacterium that causes a wide variety of diseases in various anatomical sites. Superficial infection of S. pyogenes generally leads to local suppurative lesions, such as seen in pharyngitis and impetigo cases. Such mucosal and skin infections are occasionally followed by onset of immune sequelae, including acute rheumatic fever and acute glomerulonephritis. Furthermore, S. pyogenes infection can also cause invasive diseases, such as necrotizing fasciitis, cellulitis, bacteremia, and streptococcal toxic shock syndrome (STSS),2 with a high mortality rate, which has been a serious problem throughout the world (1–3).
Host possesses various immune systems such as the complement system, one of the most important components of innate immunity, that contributes to host defense against pathogens, especially in the early stage of infection (4). Recognition of an invading pathogen is the trigger for complement pathway activation, and then each complement factor is activated in a sequential manner with precise control (5). Together with opsonization, complement-mediated direct killing of pathogens is attributable to formation of the membrane attack complex (MAC), which consists of late complement factors and induces bacteriolysis by pore formation on the surface of bacterial cells. During the course of infection, S. pyogenes also produces virulence factors that are involved in evasion from complement immunity. The serotype determinant M protein, encoded by the emm gene, binds to C4b-binding protein, resulting in inhibition of classical pathway activation. Furthermore, a variety of M protein types enable escape from C3b opsonization by binding to Factor H (6, 7), whereas the streptococcal inhibitor of complement-mediated lysis (SIC) was reported to suppress bacteriolysis by MAC (8). In addition, the streptococcal C5a peptidase ScpA has been found to be associated with evasion from bacterial clearance (9).
Modulation of activation of the complement cascade is finely tuned by both activating and inhibitory functions. Consequently, stoichiometric imbalance of complement components, such as excess consumption or proteolysis of each component, allows discordant regulation of the complement cascade. In fact, deficiencies of several complement factors render the host more susceptible to bacterial infections (10). In other studies, the C1 esterase inhibitor (C1-INH), a complement regulating factor that inactivates the C1 complex, has been utilized as a therapeutic agent for sepsis (11, 12). In our previous examination, we focused on the similarity of symptoms between STSS and C1-INH deficiency, and noted that a decrease in C1-INH level caused by S. pyogenes infection is associated with the pathogenesis (13, 14). In that study, we also demonstrated that streptococcal pyrogenic exotoxin B (SpeB), a representative cysteine protease from S. pyogenes, cleaves C1-INH and complement factors, including C3 and late complement factors, allowing bacterial evasion from complement-mediated killing. It is considered likely that SpeB-mediated degradation of C1-INH leads to overconsumption of complement factors downstream of the cascade. Furthermore, SpeB also degrades C2, C4, and C3b, as well as late complement factors, thereby inhibiting MAC formation (15). Thus, we consider that an imbalanced consumption of complement factors caused by SpeB enables S. pyogenes to evade host innate immune responses.
Strains with a relatively low level of SpeB activity have been reported to efficiently interrupt complement immunity, thus we speculate that S. pyogenes is able to hamper the classical pathway in an SpeB-independent manner. Recently, endopeptidase O (PepO) secreted from Streptococcus pneumoniae has been shown to bind to C1q and affect opsonophagocytosis (16). S. pyogenes possesses a homologue with a 68% homology to S. pneumoniae PepO on the amino acid level. Although it was reported that cytoplasmic S. pyogenes PepO disrupts quorum sensing, it is unclear whether that is also secreted or has biological functions (17). In this study, we analyzed the interaction between S. pyogenes PepO and human C1q, and investigated whether PepO is involved in evasion from complement-mediated immunity and pathogenesis.
Results
S. pyogenes Endopeptidase PepO Is Conserved and Expressed in Various Strains
To examine the subcellular localization of PepO, we prepared culture supernatant, cell wall, and cytoplasmic fractions from batch cultures of S. pyogenes clinical isolates obtained from patients with invasive and non-invasive diseases, as well as a laboratory strain (Table 1), then subjected them to Western blot analysis using anti-PepO antiserum. The majority of strains secreted PepO that was detected as a 71.2-kDa band with anti-PepO antiserum into the culture supernatants (Fig. 1A), whereas it was scarcely detected in cell wall fractions (data not shown). Also, PepO was detected in the cytoplasmic fractions of all tested strains (Fig. 1B). Furthermore, PepO produced by TW3358 exhibited different band patterns in the culture supernatant and cytoplasmic fractions as compared with the other strains. In addition, the expression level of PepO was not related to disease severity, and there was no obvious correlation between PepO production and the emm type of the S. pyogenes strains (Fig. 1, A and B).
TABLE 1.
Streptococcal strains used in this study
| emm type | Strain | Origin |
|---|---|---|
| emm1 | SSI-9 | STSS |
| emm1 | SF370 | Wound infection |
| emm2 | NIH2 | STSS |
| emm2 | SE1013 | Pharyngitis |
| emm3 | SSI-1 | STSS |
| emm3 | TW3358 | Pharyngitis |
| emm4 | TW3392 | Pharyngitis |
| emm4 | TW3398 | Pharyngitis |
| emm6 | JRS4 | Laboratory strain |
| emm6 | SE1303 | Pharyngitis |
| emm12 | NY-5 | Scarlet fever |
| emm12 | TW3337 | Pharyngitis |
| emm18 | T18 | Rheumatic fever |
| emm18 | TW3338 | Pharyngitis |
| emm22 | NIH32 | STSS |
| emm22 | TW3340 | Pharyngitis |
| emm28 | NIH35 | STSS |
| emm28 | TW3339 | Pharyngitis |
| emm49 | CS101 | Impetigo |
| emm49 | NZ131 | Acute post-streptococcal glomerulonephritis |
FIGURE 1.

Subcellular localization of PepO. Culture supernatant (A) and cytoplasmic fractions (B) were prepared from a series of S. pyogenes strains. All fractions were subjected to Western blot analysis with anti-PepO antiserum. Black arrowheads indicate bands representing PepO. S. pyogenes strains isolated from invasive disease tissues are underlined. Data shown are representative of at least three independent experiments.
In a previous study, we found that SpeB has an ability to interrupt the host complement system via degradation of various host complement component factors. Despite low SpeB production, the SSI-1 strain is able to avoid complement immunity, indicating that alternative factors may be involved in the evasion mechanism (15). In addition, C1q was not cleaved by SpeB (data not shown). To examine the potential involvement of PepO in complement system dysfunction, the SSI-1 strain was used as the parental strain in the following experiments.
S. pyogenes PepO Interacts with Human Complement Factor C1q
S. pneumoniae PepO has been shown to bind to human complement factor C1q, which leads to bacterial evasion from complement immunity via a classical pathway (16). Thus, we examined the direct interaction between S. pyogenes PepO and human C1q by SPR analysis at pH 7.4 (Fig. 2A and Table 2). Surprisingly, C1q was found to bind to rPepO with high affinity (KD = 1.09 × 10−9 m), which is comparable with the affinity of C1q for IgG (KD = 2.45 × 10−9 m)(Table 2). Next, rPepO was incubated with C1q following serial dilution with binding buffer containing a physiological concentration of NaCl (150 mm), then, binding of C1q to PepO was determined by ELISA (Fig. 2B). Our ELISA results showed that rPepO exhibited a lower level of binding activity to C1q as compared with that of human IgG, yet C1q was shown to bind to rPepO in a concentration-dependent manner, similar to IgG. On the other hand, BSA showed little or no capacity for binding with C1q. Although PepO released from S. pyogenes exhibits a lower capacity to bind to human C1q as compared with IgG, PepO possesses a high affinity for C1q, similar to the affinity of IgG for C1q under physiological conditions.
FIGURE 2.
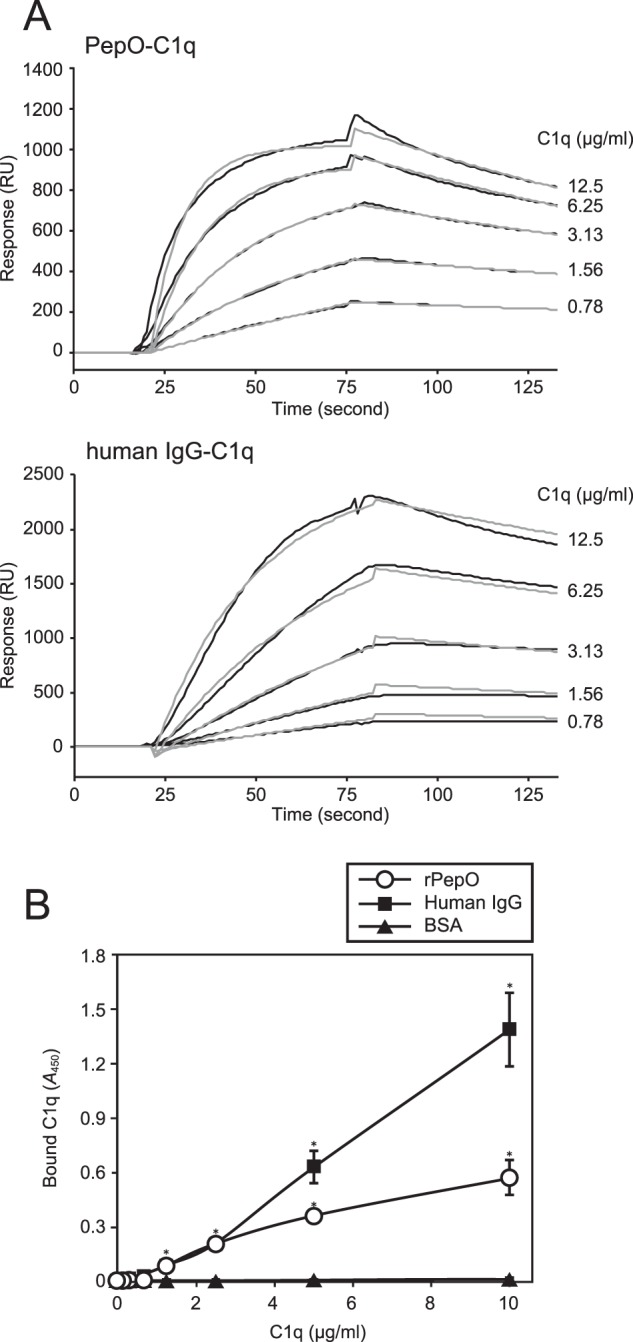
Interaction between rPepO and human C1q at pH 7.4 with physiological levels of salt concentration. A, the binding of C1q to immobilized rPepO (upper panel) and human IgG (lower panel) was examined using SPR analysis. Increasing concentrations of C1q (0.78–12.5 μg/ml) were applied as an analyte. Black and gray curves represent sensorgrams obtained from SPR analysis and fitting curves, respectively. B, the binding activity of rPepO to C1q was assessed by ELISA. Microtiter plates were coated with rPepO, human IgG, or BSA, and incubated with human C1q. C1q binding was detected with the anti-C1q antibody and a secondary antibody. Statistical significance was calculated using a two-way ANOVA. *, p < 0.01 versus BSA. Error bars indicate S.D. Representative data obtained from at least three independent experiments are shown.
TABLE 2.
Kinetics and affinity of human C1q for rPepO and IgG
| Ligands | ka | kd | KD |
|---|---|---|---|
| m−1s−1 | s−1 | m | |
| PepO | 3.37 × 106 | 3.69 × 10−3 | 1.09 × 10−9 |
| IgG | 1.23 × 106 | 3.01 × 10−3 | 2.45 × 10−9 |
Effects of NaCl and pH on Interaction of PepO with C1q
C1q consists of both collagen-like stalk and globular head domains (18). At the beginning of the classical complement pathway, an antigen-antibody complex binds to the C-terminal globular head of C1q via lysine residues in the Fc region of IgG in an electrostatic manner. Accordingly, it has been shown that the interaction of IgG to C1q is inhibited under conditions that include high concentrations of NaCl (19). To examine whether electrostatic interaction is involved in the binding of PepO with C1q, ELISA was performed in the presence of various concentrations of NaCl (Fig. 3A). We found that rPepO bound to C1q, peaking at 37.5 mm NaCl, and then the binding capacity was decreased in an NaCl concentration-dependent manner, with a similar tendency observed in regard to the binding of IgG to C1q. This result indicated that the association of PepO with C1q is achieved by electrostatic interaction. However, unlike IgG, the binding of PepO to C1q was strongly inhibited by higher concentrations of NaCl.
FIGURE 3.
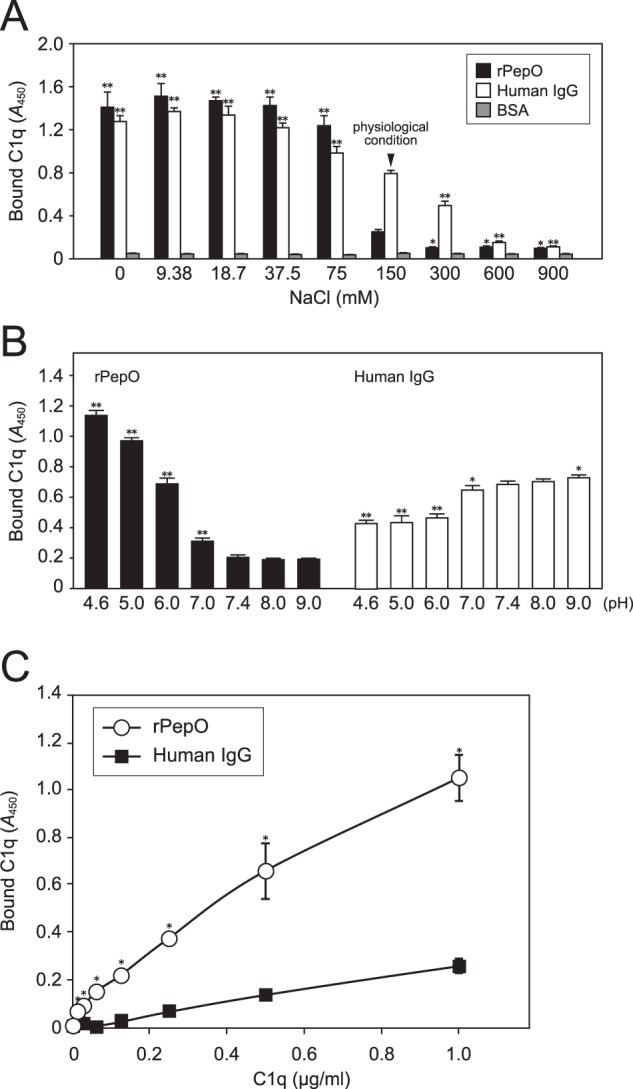
Effects of NaCl and pH on binding of rPepO to human C1q. A, the binding activity of rPepO to C1q in the presence of various concentrations of NaCl was assessed by ELISA. IgG and BSA were used as positive and negative controls, respectively. Statistical significance was determined by comparing to results obtained with 150 mm NaCl using two-way ANOVA. **, p < 0.01; *, p < 0.05. B, microtiter plates were coated with rPepO or human IgG, then 1 μg of C1q was added at various pH levels with 150 mm NaCl. Statistical analyses were performed using two-way ANOVA. **, p < 0.01; *, p < 0.05 versus pH 7.4 condition. C, the binding activities of rPepO and IgG to C1q at pH 5.0 with 150 mm NaCl were analyzed using ELISA. Bound C1q was detected with anti-C1q and secondary antibodies. Statistical analyses were performed using two-way ANOVA. *, p < 0.01 versus IgG. Error bars indicate S.D. Representative data obtained from three independent experiments are shown.
It is known that pH levels are decreased in inflamed sites (20). Therefore, we examined the effects of pH level on the binding of PepO to C1q with phosphate buffer, the pH of which ranges from 4.6 to 9 as binding buffer for use with ELISA (Fig. 3B). rPepO at less than pH 7.0 showed a significantly higher affinity for C1q as compared with that at pH 7.4, 8.0, and 9.0 (Fig. 3B, left). On the other hand, lower pH levels seemed to have an inhibitory effect on the affinity of IgG for C1q (Fig. 3B, right). The pH level in inflamed tissues is substantially reduced to a range of 5.0–6.0 (20–22). Thus the binding activities of PepO and IgG to C1q at pH 5.0 were further examined using ELISA. As expected, the binding of PepO to C1q showed a higher affinity, as compared with that of IgG at pH 5.0 (Fig. 3C). We speculated that PepO highly exerts its activity in low pH conditions.
PepO Inhibits the Binding of Human IgG to C1q under Low pH Conditions
To examine whether PepO and IgG bind to C1q in a mutually antagonistic manner under acidic conditions, immobilized rPepO was incubated with C1q in the presence or absence of increasing concentrations of human IgG as a competitor at pH 5.0. IgG did not significantly inhibit the binding of rPepO to C1q (Fig. 4A). On the other hand, immobilized human IgG showed a lower affinity with C1q in the presence of higher concentrations of rPepO at pH 5.0 (Fig. 4B). Taken together, our results demonstrated that PepO possesses an ability to suppress the binding of human IgG to C1q in an acidic pH condition.
FIGURE 4.
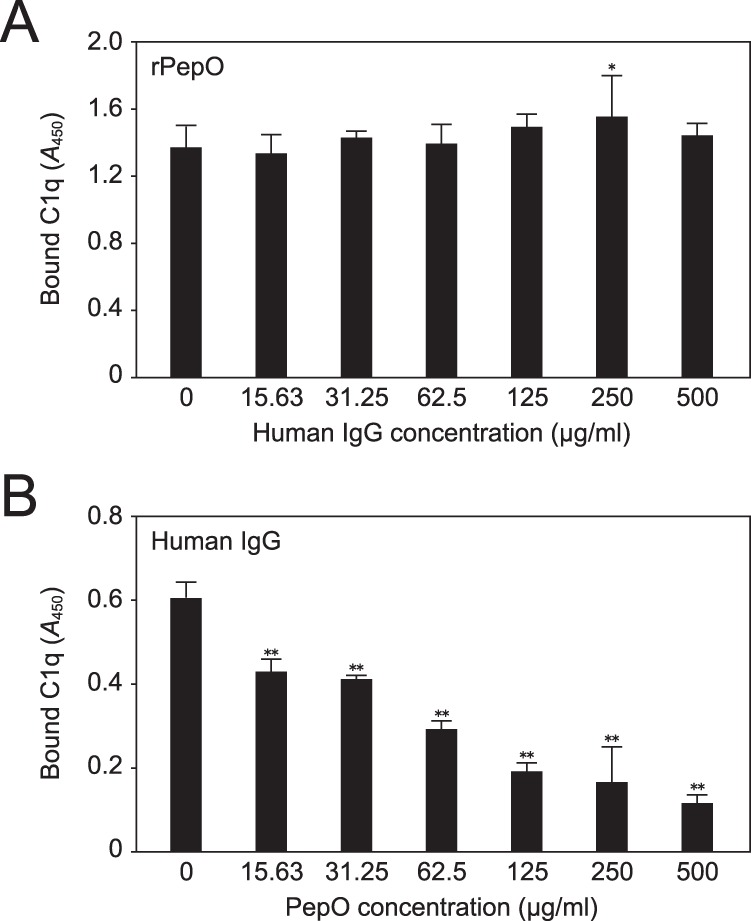
Inhibitory effect of PepO on binding of IgG to C1q at pH 5.0 with the physiological level of salt concentration. A, C1q was preincubated with increasing concentrations of human IgG as a competitor, then reacted with immobilized rPepO. B, C1q was preincubated with increasing concentrations of rPepO as a competitor, then reacted with immobilized human IgG. Bound C1q was detected using an anti-C1q antibody and secondary antibody. Statistical significance was calculated using a two-way ANOVA. **, p < 0.01; *, p < 0.05 versus the condition without competitor. Error bars indicate S.D. Data shown are representative of three independent experiments.
PepO Is Involved in S. pyogenes Resistance to Human Complement System under Low pH Conditions
Complement C1q is an initiator of the classical pathway, indicating the possibility that PepO disturbs the complement cascade via binding to C1q. Because it was previously reported that complement factors infiltrate from blood vessels to inflamed tissues (23), we examined the survival of S. pyogenes WT and pepO deletion mutant (ΔpepO) strains when grown in normal and complement-inactivated human serum at pH 5.0. As shown in Fig. 5A, when incubated in normal serum, the WT strain exhibited a significantly higher growth rate as compared with the ΔpepO strain. Although there was no significant difference between normal and complement-inactivated serum in regard to the survival rate of WT, the growth rate of the ΔpepO strain incubated in complement-inactivated serum was significantly higher as compared with that in normal serum. The survival rates of bacteria incubated in normal and complement-inactivated serum with 20 mm phosphate buffer at pH 5.0 were similar to the rate of bacterial survival in 10 mm phosphate buffer (data not shown). We also examined the survival rates of these strains in serum and complement-inactivated serum at pH 6.0 and 7.0. WT showed significantly higher growth rate in normal serum than ΔpepO at pH 6.0, whereas the growth of ΔpepO in complement-inactivated serum was not significantly high as compared with the growth of that strain in normal serum (Fig. 5B). In addition, the survival rate of WT at pH 7.0 was equivalent to that of ΔpepO (Fig. 5C). The effect of PepO for survival of S. pyogenes in serum was decreased under higher pH conditions. These results indicated that PepO renders S. pyogenes more resistant to the bacteriostatic activity of human serum under lower pH conditions.
FIGURE 5.

Function of PepO in evasion from complement-mediated bacterial killing under various pH conditions. WT and ΔpepO strains were resuspended in 10 mm phosphate buffer (A, pH 5.0; B, pH 6.0; C, pH 7.0), then incubated with normal or complement-inactivated human serum for 3 h at 37 °C. To determine cfu values, each sample was plated on THY agar plates. Representative results from three independent experiments are shown. The cfu of each strain before incubation with serum was set at a value of 1. Black and white bars indicate mean values, whereas error bars indicate the S.D. of three samples from a representative experiment. **, p < 0.01; *, p < 0.05, Mann-Whitney U test. D, S. pyogenes strains were resuspended in 10 mm phosphate buffer (pH 5.0), then incubated with normal or complement-inactivated human serum for 30 min at 37 °C. Bacterial surfaces following incubation in serum were observed with an SEM. Bar = 1 μm. Data shown are representative of at least three independent experiments.
To examine the effect of PepO on bacteriolysis, the bacterial strains were incubated with normal or complement-inactivated human serum at pH 5.0 in 10 mm phosphate buffer, then their surfaces were observed using a scanning electron microscope (SEM) (Fig. 5D). Following 30 min of incubation in normal serum, the ΔpepO strain displayed damaged bacterial surfaces as compared with non-treated ΔpepO cells. Although the WT also showed irregular surfaces after growth in normal serum as compared with the non-treated WT cells, the ΔpepO grown in normal serum showed more severe morphological changes. In contrast, when grown in inactivated serum, both strains exhibited only slightly irregular bacterial surfaces as compared with when grown in normal serum. When 20 mm phosphate buffer at pH 5.0 was added to normal serum, both the WT and ΔpepO strains showed the same tendency as bacteria incubated in 10 mm phosphate buffer at pH 5.0 (data not shown). These findings suggest that human serum-induced morphological changes of the bacterial surface under a low pH condition are inhibited by PepO.
Effects of PepO on Development of Mouse Skin Lesions
A mouse skin infection model was used to investigate the influence of PepO on S. pyogenes virulence in vivo. The WT strain formed significantly larger lesions as compared with those formed in mice infected with the ΔpepO strain until 3 days after infection (Fig. 6, A and B). Therefore, lesions at 4 days postinfection exhibited a size similar to those seen at 3 days, and lesions formed by both strains began to show a decrease in size at 5 days (data not shown). Furthermore, the complement activity in infected tissues was analyzed (Fig. 6C). Lesions formed following infection with WT exhibited lower complement activity as compared with that with ΔpepO. To verify the effect of the interaction between PepO and C1q on S. pyogenes virulence, C1q levels in the lesions were semiquantified. Those levels were increased in skin lesions of mice infected with WT as compared with those infected with the ΔpepO strain (Fig. 6D). These findings indicate that the PepO-C1q interaction participates in development of inflammatory lesions via suppression of complement activity.
FIGURE 6.
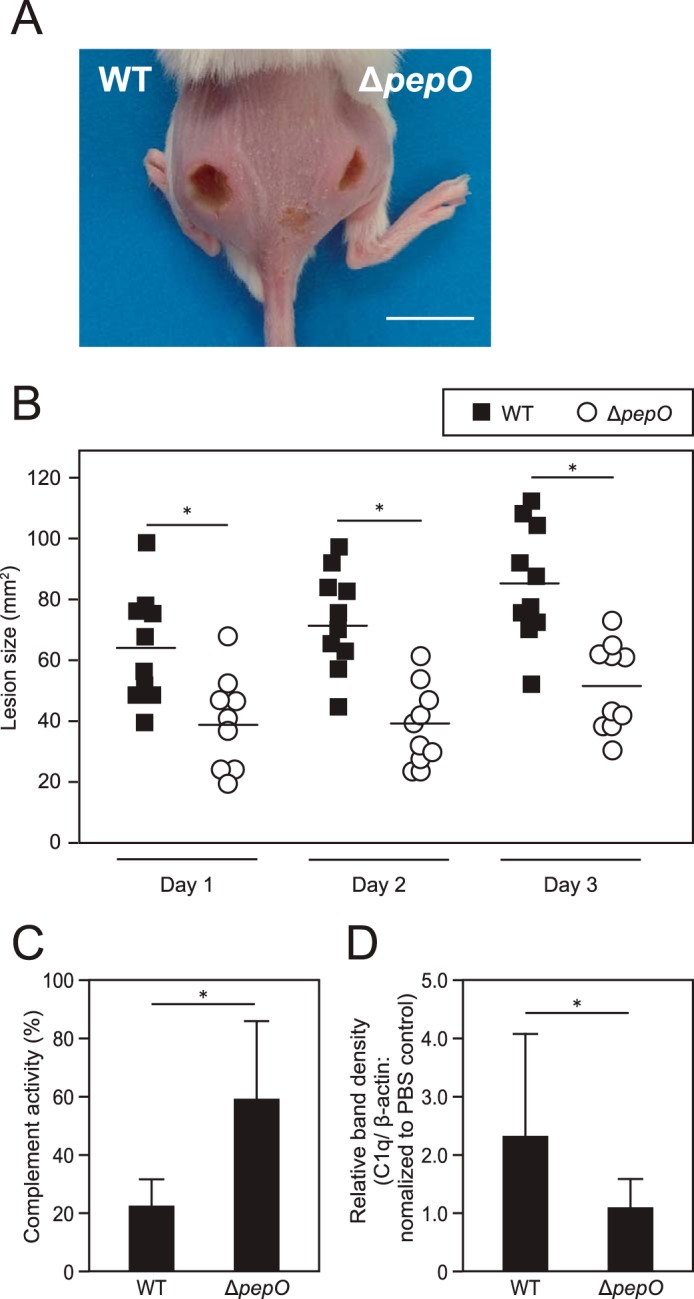
Effects of PepO on development of mouse skin lesions. A, WT (left) and ΔpepO (right) were separately injected in an intradermal manner into depilated mice skin. A representative image at 3 days after infection is shown (bar = 10 mm). B, lesion size was examined daily for up to 3 days after infection (bar represents mean of lesion size). Statistical significance was calculated using a Mann-Whitney U test. *, p < 0.01. C, hemolytic activity in tissues infected with WT or ΔpepO was measured using antibody-coated erythrocytes. The degree of hemolysis was determined by measuring of hemoglobin released from lysed erythrocytes by the membrane attack complex. The value for absorbance of the sample obtained from the PBS-injected tissue in each mouse was set as 100% complement activity. Statistical significance was calculated using a Mann-Whitney U test. *, p < 0.05. Error bars represent S.D. D, the C1q levels in lesions formed following infection with WT or ΔpepO were determined using Western blot analysis. The obtained band density of C1q in each tissue sample was normalized to that of β-actin as a loading control, then the relative amount of C1q to that in PBS-injected tissue was calculated. Statistical significance was calculated using a Mann-Whitney U test. *, p < 0.05. Error bars represent S.D. Representative data obtained from three independent experiments are shown.
Discussion
Endopeptidase O, a zinc metalloendopeptidase, belongs to the M13 peptidase family (24). A number of bacterial species, including Streptococcus dysgalactiae, Streptococcus equi, Streptococcus gallolyticus, Streptococcus parasanguis, Streptococcus thermophilus, Lactococcus lactis, and Lactococcus helveticus, possess its homologue and the active sites of these metalloproteases are all conserved. At the time of writing, the multifaceted functions of S. pneumoniae PepO have received the most attention in research investigations. Pneumococcal PepO binds to human plasminogen and fibronectin, thus facilitating bacterial internalization into host cells and evasion from innate immunity (25). Also, in vivo analysis using a mouse model revealed that S. pneumoniae PepO induces a strong innate immune response via the Toll-like receptor (TLR) 2 and TLR4 signaling pathways (26). Furthermore, secretory PepO of S. pneumoniae has an affinity to human C1q, thereby inhibiting C3b opsonization (16). On the other hand, surface-associated PepO contributes to bacterial adherence to host cells via its interaction with C1q. The present results revealed that PepO produced by S. pyogenes is an extracellular protein and involved in complement evasion via binding to C1q. In addition, there is a possibility that S. pyogenes-produced PepO also possesses an immunomodulatory function or host cell adhesion activity, as previously reported for pneumococcal PepO.
The pepO gene is ubiquitously conserved in S. pyogenes strains. There is no typical signal sequence in S. pyogenes PepO, thus PepO is accordingly speculated to be a cytoplasmic protein. However, our results revealed that S. pyogenes PepO localizes not only in cytoplasm but also in culture supernatant, whereas it was scarcely detected in cell wall fractions, unlike S. pneumoniae PepO (Fig. 1, A and B). In addition to its previously reported functions, our findings suggest that PepO is multifunctional in both cytoplasm and the extracellular milieu, similar to the properties of GAPDH and enolase, which lack a signal sequence but are found in culture supernatant (27–31). The present study provides clues about the variable ability of S. pyogenes strains to express and secrete PepO. On the other hand, the relationships among disease severity, emm type, and PepO localization were not revealed, and future studies of the secretion mechanism and its contribution to virulence are required.
C1q consists of three subunits, each of which possesses globular head and collagen-like domains. For C1q-IgG interaction, acidic and basic amino residues of the IgG and C1q globular head domains constitute salt bridges that provide connection (32). These ionic bonds indicate that the interaction of IgG with C1q is inhibited under a high salt concentration condition. Our findings suggested that the ionic bond between acidic and basic amino acid residues is also involved in the association of PepO with C1q. In addition, the effect of PepO to inhibit the association of IgG with C1q (Fig. 4B) indicates the possibility that PepO and IgG share either the same or proximate site for C1q binding. On the other hand, a previous study noted that a high pH level of ∼8.5 enhances the binding of IgG to C1q (33), which is consistent with our findings regarding the effect of pH level. The three subunits of C1q are linked to each other by disulfide bonds. It is known that an alkaline environment promotes formation of a disulfide bond that plays a prominent role in strengthening the C1q-IgG association (33). C-reactive protein (CRP) is also able to interact with C1q and activates the classical complement pathway. The reported crystal structure of the C1q-CRP complex shows that lysine residues at the top of C1q globular head have an electrostatic contact with Asp112 and Tyr175 of CRP (32). Furthermore, it has been demonstrated that a low pH of ∼6.0 induces conformational changes of aromatic residues in CRP, allowing the tyrosine residue of CRP to interact with the lysine residue on C1q, which consequently activates the classical complement pathway (34). Our results showed that the interaction between PepO and C1q is enhanced under an acidic condition, and not attributable to the disulfide bond-dependent morphology of C1q. Furthermore, the influence of pH on binding of PepO to C1q is similar to the property of CRP. Structural analysis of S. pyogenes PepO is required to further elucidate the interaction between PepO and C1q.
WT strain exhibited a significantly higher survival rate in human serum at pH 5.0, as compared with the ΔpepO strain. On the other hand, the survival rate of WT in complement-inactivated serum was not significantly different as compared with that in normal serum, whereas ΔpepO showed a significantly higher growth rate when incubated in complement-inactivated serum as compared with normal serum. Also, in accordance with the increase in pH, the difference between the WT and ΔpepO strains in regard to survival rate when grown in normal serum was reduced. Our findings indicated that PepO induces dysfunction of the classical pathway under low pH conditions (Fig. 5, A–C). This speculation is also explained by the higher binding activity of PepO for C1q as compared with that of IgG (Fig. 3C), as well as the activity of PepO to suppress the interaction between IgG and C1q under low pH conditions (Fig. 4B). Thus, our in vitro findings suggest that PepO functions as a virulence factor to inhibit the interaction between IgG and C1q in the initial step of the classical complement pathway in inflammatory tissue. In addition, SEM observation of bacterial surfaces revealed that the severe morphological changes on the bacterial surface were evoked by deletion of PepO. A previous study also reported that C1q-deficient serum demonstrated low complement activity (35). Although another group found that complement immunity associates with C1q for S. pyogenes infection was independent of opsonophagocytosis, the S. pyogenes molecule that interacts with C1q was not identified (36). It has also been speculated that some M proteins have an interaction with C1q, although that was not analyzed in detail (37). Our results showing that PepO enabled escape from complement-mediated bacteriolysis (Fig. 5), indicate that the interaction of PepO with C1q may lead to inhibition of opsonophagocytosis and depression of the late complement pathway.
At sites of infection or inflammation, infiltration and activation of inflammatory cells including phagocytes increase the energy demand for mobility and oxygen (38), after which glucose and oxygen consumption is accelerated via glycolysis by phagocytes (22). Thus, local hypoxia induces metabolism conversion. Accordingly, accumulation of lactic acid by fermentation induces a significant decrease in pH level at the inflammatory locus (39). On the other hand, lack of neutrophil migration to sites of infection with S. pyogenes in the early phase of STSS has been shown (40, 41). Notably, the SSI-9 strain was shown in previous studies to produce SpeB, which degrades C3b and enables evasion from opsonophagocytosis by neutrophils (41). Consequently, no pH decrease may occur in inflammatory tissues because of a lack of neutrophil migration. In the present study, we utilized an SSI-1 strain that produces a small amount of SpeB. Accordingly, it is possible that a pH decrease was evoked at the site of infection in our mouse experiments, because of the reduced effect of SpeB on neutrophils. Several studies have found that low pH activates the complement system (42–44). Our results showed that PepO is involved in the pathological mechanism (Fig. 6, A and B) and associated with dysfunction of the complement system (Fig. 5) in low pH conditions. In addition, tissues infected with WT displayed a significantly lower level of complement activity as compared with those infected with the mutant strain (Fig. 6C). On the other hand, the C1q level in lesions formed by WT infection was significantly higher than that in lesions formed by the ΔpepO strain (Fig. 6D). Based on these results, we speculated that secreted PepO binds to C1q, thus the interaction of IgG with C1q is inhibited in an acidic environment (Figs. 3C and 4). Although PepO-bound C1q remains in inflammatory lesions, C1q is prevented from participating in the complement cascade. Therefore, the interaction of PepO with C1q inhibits complement activation by IgG in a low pH condition, indicating that PepO is involved in the pathological mechanism (Fig. 7).
FIGURE 7.

Schematic diagram of evasion of S. pyogenes from complement-mediated bacteriolysis via PepO-C1q interaction. At the site of inflammation, secreted PepO binds to C1q, thus the binding of C1q to IgG is inhibited. Although PepO-bound C1q accumulates, C1q is not able to participate in the complement cascade. Consequently, complement activation via the classical pathway is suppressed and S. pyogenes escapes from complement-mediated bacteriolysis.
Nevertheless, the direct relationship between virulence in vivo and the interaction of PepO with C1q remains unclear. The expression of various virulence factors produced by S. pyogenes is enhanced by the CovRS two-component system signal transduction pathway under various stress conditions, including the low level of pH, the host antimicrobial peptide LL-37, and a low concentration of extracellular Mg2+ (45–49). In addition, pepO gene expression has been demonstrated to be negatively regulated by CovRS (17). Although the secretion pathway of PepO remains unknown, it might also be influenced by environmental stress factors, such as found in vivo. Thus, it is possible that PepO expression in infected lesions in vivo is facilitated by the two-component system under a low pH condition. In addition, a previous study reported an alternative function of S. pyogenes cytoplasmic PepO that degrades mature short hydrophobic peptides (peptide pheromones) and blocks its Rgg2/Rgg3 quorum-sensing responses (17). Although additional detailed investigations are required to elucidate the mechanism of PepO as a virulence factor, the present findings revealed that S. pyogenes attenuates complement activation by PepO-mediated interaction with C1q, which contributes to its pathogenicity under a low pH condition, such as in inflamed tissues.
Experimental Procedures
Bacterial Strains and Culture Conditions
S. pyogenes strain SSI-1 (serotype M3) was isolated from a Japanese patient with STSS in 1994 (50, 51). S. pyogenes strains obtained from pharyngitis, impetigo, scarlet fever, and STSS were kindly provided by Dr. T. Murai (Graduate School of Japanese Red Cross Akita College of Nursing, Akita, Japan), Dr. K. Kikuchi (Tokyo Women's Medical University, Tokyo, Japan), and Dr. K. Moriya (Saga Prefectural Institute of Public Health and Pharmaceutical Research, Saga, Japan) (52–54). The laboratory strain JRS4 was kindly provided by Dr. E. Hanski (The Hebrew University, Hadassah Medical School, Jerusalem, Israel) (55) (Table 1). Escherichia coli strain XL-10 Gold (Agilent Technologies, Santa Clara, CA) was used as a host for derivatives of plasmids pSET4s (56) and pQE30 (Qiagen, Hilden, Germany). All streptococcal strains were grown at 37 °C in Todd-Hewitt broth (BD Bioscience) supplemented with 0.2% yeast extract (BD Bioscience) (THY medium) or on THY agar plates. E. coli strains were cultured in Luria-Bertani (LB; Sigma) medium at 37 °C with agitation. For selection and maintenance of mutant strains, antibiotics were added to the medium at the following concentrations: spectinomycin, 100 μg/ml for S. pyogenes and E. coli: ampicillin, 100 μg/ml for E. coli.
Construction of S. pyogenes Mutant Strain and Recombinant Vectors
Construction of the SSI-1 pepO in-frame deletion mutant strain (ΔpepO) was performed using pSET4s and primers, as previously described (57). For construction of plasmids expressing recombinant PepO from SSI-1, mature genes were amplified using primers pepOFW/BamHI (5′-CGGGATCCATGACAACTTATCAAGATGATTTTTACCAG-3′) and pepORV/PstI (5′-TTCTGCAGTTACCAAATAATCACACGGTCTTTCGGAGC-3′), then each PCR product was cloned into pQE30 and transformed into E. coli XL10-Gold, as previously described (58).
Preparation of His-tagged Recombinant PepO and Antiserum
N-terminal His-tagged recombinant PepO (rPepO) was hyperexpressed in E. coli and then purified using a QIAexpress protein purification system (Qiagen), as previously reported (58).
Mouse polyclonal antiserum against rPepO was acquired by immunizing 5-week-old female BALB/c mice (Japan SLC, Inc., Shizuoka, Japan) with purified rPepO, as previously reported (58). All mouse experiments were conducted under a protocol approved by the Animal Care and Use Committee of Osaka University Graduate School of Dentistry (authorization number 26-014-0).
Preparation of Culture Supernatant, Cell Wall, and Cytoplasm Fractions, and Western Blot Analysis
To examine the expression of PepO, various S. pyogenes strains were cultured at 37 °C to the late-exponential phase (A600 = 1.2) in THY broth, then culture supernatants were obtained by centrifugation at 5,800 × g for 10 min. Proteins in the culture supernatant samples were precipitated with 20% trichloroacetic acid at 4 °C overnight, followed by centrifugation at 13,000 × g for 20 min. The resulting pellets were washed several times with ice-cold ethanol and resuspended in 25 μl of 1 m Tris buffer (pH 8.5). Bacterial cells thus obtained were resuspended in 200 μl of protoplasting buffer (0.1 m KPO4, pH 6.2, 40% sucrose, 10 mm MgCl2) containing Complete EDTA-free Protease Inhibitor tablets (Roche Applied Science, Basel, Switzerland) and 150 units/ml of N-acetylmuramidase (Sigma), then incubated for 1 h at 37 °C. The resulting protoplasts underwent sedimentation at 16,000 × g for 10 min, then the supernatants and pellets were subjected to Western blot analysis as cell wall and cytoplasmic fractions, respectively.
To examine PepO expression levels in the obtained samples, Western blot analysis was performed, as previously described, with minor modifications (41, 58). Briefly, PepO in each sample was detected using mouse anti-PepO antiserum diluted to a concentration of 1:2000. Immunoreactive bands were detected using horseradish peroxidase (HRP)-labeled goat anti-mouse IgG (Cell Signaling Technologies, Danvers, MA) diluted to 1:2000 and chemiluminescent HRP substrate (Thermo Scientific).
Surface Plasmon Resonance Measurements
SPR analysis was performed using a BIACORE X (GE Healthcare UK Ltd., Buckinghamshire, UK), as previously described (59). rPepO and human purified IgG (Merck Millipore, Darmstadt, Germany) were diluted to 50 μg/ml in 10 mm sodium acetate (pH 4.0 and 5.5, respectively), and immobilized on a CM5 sensor chip using an amine coupling kit (GE Healthcare). A blank was used as the reference. Human purified C1q (Merck Millipore) at concentrations of 0.7813, 1.563, 3.125, 6.25, and 12.5 μg/ml was prepared with HBS-P buffer (0.01 m HEPES, pH 7.4, 0.15 m NaCl, 0.005% surfactant P20; GE Healthcare). C1q was injected as the analyte at a flow rate of 30 μl/min at 37 °C followed by injection of 1 m NaCl for regeneration. Obtained sensorgrams were analyzed using Bio-evaluation software (version 3.0.2), with application of the 1:1 Langmuir binding model.
Direct Binding Assay
Direct binding assays were performed as previously described (16). Briefly, 96-well plates (Sumitomo Bakelite Co., Ltd., Tokyo, Japan) were coated with 500 ng of rPepO and human IgG (IgG) in 0.1 m bicarbonate buffer (pH 9.8) overnight at 4 °C. As a negative control, 500 ng of BSA (Sigma) was coated onto each well. After blocking overnight at 4 °C, rPepO, IgG, or BSA was incubated with 100 μl of 2-fold serial dilutions of C1q (0–10 μg/ml) in HEPES-based binding buffer (50 mm HEPES, pH 7.4, 150 mm NaCl, 2 mm CaCl2, 50 μg/ml of BSA) or phosphate-based binding buffer (50 mm phosphate buffer at pH 5.0, 150 mm NaCl, 50 μg/ml of BSA) for 2 h at room temperature. To examine the effect of NaCl on the C1q-PepO interaction, 500 ng of rPepO, IgG, or BSA in bicarbonate buffer was coated onto the plates. Following a blocking step, C1q in binding buffer containing NaCl at a final concentration ranging from 0 to 900 mm was reacted with rPepO or IgG. To identify the effect of pH on the binding of rPepO to C1q, C1q was prepared in phosphate-based binding buffer (50 mm phosphate buffer, pH 4.6 to 9.0, 150 mm NaCl, 50 μg/ml of BSA), and then added to plates coated with rPepO or IgG. To obtain competition ELISA findings, C1q (10 μg/ml) was reacted with serially diluted IgG or rPepO (0–500 μg/ml) adjusted with phosphate-based binding buffer (50 mm phosphate buffer, pH 5.0, 150 mm NaCl, 50 μg/ml of BSA) as competitors for 1 h at 37 °C. Then, plates coated with 500 ng of rPepO or human IgG were incubated with the above-mentioned mixture containing 500 ng of C1q for 2 h at room temperature. After blocking, bound C1q was detected using a rabbit anti-human C1q polyclonal antibody (DakoCytometry, Glostrup, Denmark) at a concentration of 1:2000. Furthermore, immunoreactive protein on the plates was reacted with an HRP-labeled goat anti-rabbit IgG polyclonal antibody, then detected with an HRP substrate prepared for enzyme-linked immunosorbent assays (Moss, Pasadena, MD). Absorbance was measured at 450 nm.
Serum Bactericidal Test
Human serum bactericidal assays were performed in vitro according to a protocol reported by Lancefield (60), with some modifications (15). S. pyogenes strains SSI-1 and ΔpepO were cultured until the mid-exponential phase (A600 = 0.4), then cell concentrations were adjusted to 2–3 × 103 cfu/ml with 10 mm phosphate buffer (pH 5.0, 6.0, or 7.0). Bacteria were incubated with a 10% volume of human complement serum (Sigma) or human complement-inactivated serum at 37 °C for 3 h, then the suspensions were plated on THY agar plates, and incubated overnight and quantified. The cfu value obtained after 3 h of incubation was divided by that obtained before incubation, then the fold-change of bacterial increase was determined (% inoculum).
Scanning Electron Microscopic Analysis
WT and ΔpepO strains were cultured until the mid-exponential phase (A600 = 0.4), then adjusted using 10 mm phosphate buffer (pH 5.0) to 1.5 × 107 cfu/ml. Bacterial suspensions (90 μl) for each sample were incubated with 10 μl of human complement serum or human complement-inactivated serum at 37 °C for 30 min, then fixed with 2.5% glutaraldehyde on cover glasses coated with Matrigel (BD bioscience) at room temperature for 1 h, and washed with distilled water. The samples were dehydrated with 100% t-butyl alcohol and freeze-dried. Finally, they were coated with platinum and examined with an SEM (JSM-6390LVZ, JEOL Ltd, Tokyo, Japan), as previously described (15).
Mouse Experiments
One day before infection, the dorsal skin of 6-week-old female BALB/c mice (Japan SLC, Inc.) was depilated with Veet® hair removal cream (Reckitt Benckiser Group plc, Slough, UK). Next, SSI-1 WT and ΔpepO strains were cultured until the mid-exponential phase (A600 = 0.4), and adjusted with PBS to 1.0 × 109 cfu/ml, then bacterial suspensions (1 × 108 cfu) were injected in an intradermal manner into depilated mice. The WT and ΔpepO strains were separately injected into the flank on both sides of individual mice. Areas with alteration in skin color were defined as lesions including an ulcer. Lesion size was semiquantified using a computer-assisted densitometer (ImageJ, version 8.8.7, National Institutes of Health, Bethesda, MD) for up to 3 days after infection.
Complement activity in infected tissues was analyzed using a CH50 complement activity measuring kit (Denka Seiken Co., Ltd., Tokyo, Japan), as previously described (16, 41). Briefly, PBS was injected into the flank on one side and the WT (n = 7 mice) or ΔpepO (n = 6 mice) strain into the flank on the other side of individual mice. Skin lesions were dissected at 3 days after infection and placed in round-bottomed microcentrifuge tubes filled with 300 μl of ice-cold RIPA buffer (150 mm NaCl, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mm Tris, pH 8.0) containing EDTA-free protease inhibitor. Following homogenization with an electric homogenizer (MagNA Lyser, Roche Applied Science), the samples were maintained with constant agitation for 2 h at 4 °C. Next, lysed tissues were centrifuged at 11,000 × g for 20 min at 4 °C. Utilizing a BCA protein assay kit (Thermo Scientific), the amount of protein in each supernatant was adjusted to 300 μg/ml with veronal buffer, then 130-μl samples were incubated with 1 × 107 sensitized sheep erythrocytes adjusted with 20 μl of veronal buffer in 96-well round-bottomed microtiter plates (Iwaki & Co., Ltd., Tokyo, Japan) for 1 h at 37 °C. Instead of tissue samples, veronal buffer and distilled water were used as negative and positive controls, respectively, for hemolysis. When the period of incubation was finished, the reaction was stopped by placing the samples on ice, then they were centrifuged in microtiter plates at 500 × g to pellet intact cells. The resulting supernatants were subjected to measurement of hemolysis as complement activity at a wavelength of 550 nm using an absorption spectrometer. Absorbance of samples obtained from the PBS-injected tissues in each mouse was set at 100% for calculation of the complement activity of the samples from infected tissues.
For quantification of C1q in infected tissues, the total amount of protein in each sample was adjusted to 3 mg/ml and C1q was detected by Western blot analysis. The immunoblot membrane was incubated with mouse anti-mouse C1q antibody (Abcam, Cambridge, UK) diluted to a concentration of 1:1000, followed by incubation with HRP-labeled goat anti-mouse IgG diluted to 1:2000. Immunoreactive bands were detected with a chemiluminescent HRP substrate. After detection of C1q, primary and secondary antibodies were removed using stripping solution (Wako Pure Chemical Industries, Ltd., Osaka, Japan), then a rabbit anti-β-actin antibody (Cell Signaling Technologies) diluted to 1:2000 was reacted as a loading control, followed by reaction with HRP-labeled goat anti-rabbit IgG diluted to 1:2000. Semiquantitative densitometric analysis of protein bands was conducted using ImageJ software (version 8.8.7). The obtained density of C1q in each sample was normalized to that of β-actin. Finally, the relative amount of C1q in the infected lesion to that in PBS-injected tissue obtained from the same mouse was calculated.
Statistical Analysis
For direct binding assays, the statistical significance of difference was calculated using two-way analysis of variance (ANOVA) with Bonferroni post-test correction. For the bactericidal tests and mouse experiments, the significance of the difference between the mean of each group was evaluated using a nonparametric Mann-Whitney U test. All results were analyzed using StatMate III software (ATMS, Tokyo, Japan). Statistical differences were considered to be significant at p < 0.05.
Author Contributions
M. H. O. and S. K. conceived and designed the experiments. M. H. O., T. S., Y. M., and D. H. performed experiments and analyzed data. M. H. O., T. S., Y. M., T. O., M. Y., M. N., and S. K. contributed to the writing of the manuscript. All the authors participated in discussions of the research and reviewed the manuscript.
Acknowledgments
We thank T. Murai, K. Kikuchi, K. Moriya, and E. Hanski for providing S. pyogenes strains. We gratefully acknowledge T. Sekizaki and D. Takamatsu for the pSET4s plasmid.
This work was supported in part by Grant-in-aid for Scientific Research (B) A15H05012, Grant-in-aid for Scientific Research (B) A16H058470, Grant-in-Aid for challenging Exploratory Research T266708000, and Grant-in-Aid for Young Scientists (Start-up) A15H06380 from the Japan Society for the Promotion of Science (JSPS), and the Naito Foundation. The authors declare that they have no conflicts of interest with the contents of this article.
- STSS
- streptococcal toxic shock syndrome
- MAC
- membrane attack complex
- C1-INH
- C1 esterase inhibitor
- SpeB
- streptococcal pyrogenic exotoxin B
- THY
- Todd-Hewitt broth supplemented with 0.2% yeast extract
- PepO
- endopeptidase O
- IgG
- immunoglobulin G
- SPR
- surface plasmon resonance
- CRP
- C-reactive protein
- TLR
- Toll-like receptor
- ANOVA
- analysis of variance
- SEM
- scanning electron microscope.
References
- 1. Cunningham M. W. (2000) Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13, 470–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carapetis J. R., Steer A. C., Mulholland E. K., and Weber M. (2005) The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5, 685–694 [DOI] [PubMed] [Google Scholar]
- 3. Ralph A. P., and Carapetis J. R. (2013) Group A streptococcal diseases and their global burden. Curr. Top. Microbiol. Immunol. 368, 1–27 [DOI] [PubMed] [Google Scholar]
- 4. Sakamoto M., Fujisawa Y., and Nishioka K. (1998) Physiologic role of the complement system in host defense, disease, and malnutrition. Nutrition 14, 391–398 [DOI] [PubMed] [Google Scholar]
- 5. Merle N. S., Church S. E., Fremeaux-Bacchi V., and Roumenina L. T. (2015) Complement system part I: molecular mechanisms of activation and regulation. Front. Immunol. 6, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thern A., Stenberg L., Dahlbäck B., and Lindahl G. (1995) Ig-binding surface proteins of Streptococcus pyogenes also bind human C4b-binding protein (C4BP), a regulatory component of the complement system. J. Immunol. 154, 375–386 [PubMed] [Google Scholar]
- 7. Horstmann R. D., Sievertsen H. J., Knobloch J., and Fischetti V. A. (1988) Antiphagocytic activity of streptococcal M protein: selective binding of complement control protein factor H. Proc. Natl. Acad. Sci. U.S.A. 85, 1657–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Akesson P., Sjöholm A. G., and Björck L. (1996) Protein SIC, a novel extracellular protein of Streptococcus pyogenes interfering with complement function. J. Biol. Chem. 271, 1081–1088 [DOI] [PubMed] [Google Scholar]
- 9. Ji Y., McLandsborough L., Kondagunta A., and Cleary P. P. (1996) C5a peptidase alters clearance and trafficking of group A streptococci by infected mice. Infect. Immun. 64, 503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Skattum L., van Deuren M., van der Poll T., and Truedsson L. (2011) Complement deficiency states and associated infections. Mol. Immunol. 48, 1643–1655 [DOI] [PubMed] [Google Scholar]
- 11. Igonin A. A., Protsenko D. N., Galstyan G. M., Vlasenko A. V., Khachatryan N. N., Nekhaev I. V., Shlyapnikov S. A., Lazareva N. B., and Herscu P. (2012) C1-esterase inhibitor infusion increases survival rates for patients with sepsis. Crit. Care Med. 40, 770–777 [DOI] [PubMed] [Google Scholar]
- 12. Liu D., Lu F., Qin G., Fernandes S. M., Li J., and Davis A. E. 3rd. (2007) C1 inhibitor-mediated protection from sepsis. J. Immunol. 179, 3966–3972 [DOI] [PubMed] [Google Scholar]
- 13. Brosnahan A. J., and Schlievert P. M. (2011) Gram-positive bacterial superantigen outside-in signaling causes toxic shock syndrome. FEBS J. 278, 4649–4667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Castelli R., Zanichelli A., and Cugno M. (2013) Therapeutic options for patients with angioedema due to C1-inhibitor deficiencies: from pathophysiology to the clinic. Immunopharmacol. Immunotoxicol. 35, 181–190 [DOI] [PubMed] [Google Scholar]
- 15. Honda-Ogawa M., Ogawa T., Terao Y., Sumitomo T., Nakata M., Ikebe K., Maeda Y., and Kawabata S. (2013) Cysteine proteinase from Streptococcus pyogenes enables evasion of innate immunity via degradation of complement factors. J. Biol. Chem. 288, 15854–15864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Agarwal V., Sroka M., Fulde M., Bergmann S., Riesbeck K., and Blom A. M. (2014) Binding of Streptococcus pneumoniae endopeptidase O (PepO) to complement component C1q modulates the complement attack and promotes host cell adherence. J. Biol. Chem. 289, 15833–15844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilkening R. V., Chang J. C., and Federle M. J. (2016) PepO, a CovRS-controlled endopeptidase, disrupts Streptococcus pyogenes quorum sensing. Mol. Microbiol. 99, 71–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kishore U., and Reid K. B. (2000) C1q: structure, function, and receptors. Immunopharmacology 49, 159–170 [DOI] [PubMed] [Google Scholar]
- 19. Lin T. Y., and Fletcher D. S. (1978) Interaction of human Clq with insoluble immunoglobulin aggregates. Immunochemistry 15, 107–117 [DOI] [PubMed] [Google Scholar]
- 20. Lardner A. (2001) The effects of extracellular pH on immune function. J. Leukoc. Biol. 69, 522–530 [PubMed] [Google Scholar]
- 21. Steen K. H., Steen A. E., and Reeh P. W. (1995) A dominant role of acid pH in inflammatory excitation and sensitization of nociceptors in rat skin, in vitro. J. Neurosci. 15, 3982–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Menkin V. (1956) Biology of inflammation; chemical mediators and cellular injury. Science 123, 527–534 [DOI] [PubMed] [Google Scholar]
- 23. auf dem Keller U., Prudova A., Eckhard U., Fingleton B., and Overall C. M. (2013) Systems-level analysis of proteolytic events in increased vascular permeability and complement activation in skin inflammation. Sci. Signal. 6, rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oetjen J., Fives-Taylor P., and Froeliger E. (2001) Characterization of a streptococcal endopeptidase with homology to human endothelin-converting enzyme. Infect. Immun. 69, 58–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Agarwal V., Kuchipudi A., Fulde M., Riesbeck K., Bergmann S., and Blom A. M. (2013) Streptococcus pneumoniae endopeptidase O (PepO) is a multifunctional plasminogen- and fibronectin-binding protein, facilitating evasion of innate immunity and invasion of host cells. J. Biol. Chem. 288, 6849–6863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang H., Kang L., Yao H., He Y., Wang X., Xu W., Song Z., Yin Y., and Zhang X. (2016) Streptococcus pneumoniae endopeptidase O (PepO) elicits a strong innate immune response in mice via TLR2 and TLR4 signaling pathways. Front. Cell Infect. Microbiol. 6, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bergmann S., Rohde M., Chhatwal G. S., and Hammerschmidt S. (2001) α-Enolase of Streptococcus pneumoniae is a plasmin(ogen)-binding protein displayed on the bacterial cell surface. Mol. Microbiol. 40, 1273–1287 [DOI] [PubMed] [Google Scholar]
- 28. Bergmann S., Rohde M., and Hammerschmidt S. (2004) Glyceraldehyde-3-phosphate dehydrogenase of Streptococcus pneumoniae is a surface-displayed plasminogen-binding protein. Infect. Immun. 72, 2416–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Terao Y., Yamaguchi M., Hamada S., and Kawabata S. (2006) Multifunctional glyceraldehyde-3-phosphate dehydrogenase of Streptococcus pyogenes is essential for evasion from neutrophils. J. Biol. Chem. 281, 14215–14223 [DOI] [PubMed] [Google Scholar]
- 30. Mori Y., Yamaguchi M., Terao Y., Hamada S., Ooshima T., and Kawabata S. (2012) α-Enolase of Streptococcus pneumoniae induces formation of neutrophil extracellular traps. J. Biol. Chem. 287, 10472–10481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sumitomo T., Nakata M., Higashino M., Yamaguchi M., and Kawabata S. (2016) Group A Streptococcus exploits human plasminogen for bacterial translocation across epithelial barrier via tricellular tight junctions. Sci. Rep. 7, 20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gaboriaud C., Juanhuix J., Gruez A., Lacroix M., Darnault C., Pignol D., Verger D., Fontecilla-Camps J. C., and Arlaud G. J. (2003) The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. J. Biol. Chem. 278, 46974–46982 [DOI] [PubMed] [Google Scholar]
- 33. Kaul M., and Loos M. (1997) Dissection of C1q capability of interacting with IgG: time-dependent formation of a tight and only partly reversible association. J. Biol. Chem. 272, 33234–33244 [DOI] [PubMed] [Google Scholar]
- 34. Miyazawa K., and Inoue K. (1990) Complement activation induced by human C-reactive protein in mildly acidic conditions. J. Immunol. 145, 650–654 [PubMed] [Google Scholar]
- 35. Sjöholm A. G., Selander B., Ostenson S., Holmström E., and Söderström C. (1991) Normal human serum depleted of C1q, factor D and properdin: its use in studies of complement activation. APMIS 99, 1120–1128 [DOI] [PubMed] [Google Scholar]
- 36. Yuste J., Ali S., Sriskandan S., Hyams C., Botto M., and Brown J. S. (2006) Roles of the alternative complement pathway and C1q during innate immunity to Streptococcus pyogenes. J. Immunol. 176, 6112–6120 [DOI] [PubMed] [Google Scholar]
- 37. Koroleva I. V., Sjöholm A. G., and Schalén C. (1998) Binding of complement subcomponent C1q to Streptococcus pyogenes: evidence for interactions with the M5 and FcRA76 proteins. FEMS Immunol. Med. Microbiol. 20, 11–20 [DOI] [PubMed] [Google Scholar]
- 38. Lewis J. S., Lee J. A., Underwood J. C., Harris A. L., and Lewis C. E. (1999) Macrophage responses to hypoxia: relevance to disease mechanisms. J. Leukoc. Biol. 66, 889–900 [DOI] [PubMed] [Google Scholar]
- 39. Brahimi-Horn M. C., Chiche J., and Pouysségur J. (2007) Hypoxia signalling controls metabolic demand. Curr. Opin. Cell Biol. 19, 223–229 [DOI] [PubMed] [Google Scholar]
- 40. Hidalgo-Grass C., Mishalian I., Dan-Goor M., Belotserkovsky I., Eran Y., Nizet V., Peled A., and Hanski E. (2006) A streptococcal protease that degrades CXC chemokines and impairs bacterial clearance from infected tissues. EMBO J. 25, 4628–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Terao Y., Mori Y., Yamaguchi M., Shimizu Y., Ooe K., Hamada S., and Kawabata S. (2008) Group A streptococcal cysteine protease degrades C3 (C3b) and contributes to evasion of innate immunity. J. Biol. Chem. 283, 6253–6260 [DOI] [PubMed] [Google Scholar]
- 42. Fishelson Z., Horstmann R. D., and Müller-Eberhard H. J. (1987) Regulation of the alternative pathway of complement by pH. J. Immunol. 138, 3392–3395 [PubMed] [Google Scholar]
- 43. Emeis M., Sonntag J., Willam C., Strauss E., Walka M. M., and Obladen M. (1998) Acidosis activates complement system in vitro. Mediators Inflamm. 7, 417–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sonntag J., Emeis M., Strauss E., and Obladen M. (1998) In vitro activation of complement and contact system by lactic acidosis. Mediators Inflamm. 7, 49–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dalton T. L., and Scott J. R. (2004) CovS inactivates CovR and is required for growth under conditions of general stress in Streptococcus pyogenes. J. Bacteriol. 186, 3928–3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gryllos I., Tran-Winkler H. J., Cheng M. F., Chung H., Bolcome R 3rd, Lu W., Lehrer R. I., and Wessels M. R. (2008) Induction of group A Streptococcus virulence by a human antimicrobial peptide. Proc. Natl. Acad. Sci. U.S.A. 105, 16755–16760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gryllos I., Grifantini R., Colaprico A., Jiang S., Deforce E., Hakansson A., Telford J. L., Grandi G., and Wessels M. R. (2007) Mg2+ signalling defines the group A streptococcal CsrRS (CovRS) regulon. Mol. Microbiol. 65, 671–683 [DOI] [PubMed] [Google Scholar]
- 48. Sumby P., Whitney A. R., Graviss E. A., DeLeo F. R., and Musser J. M. (2006) Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Engleberg N. C., Heath A., Miller A., Rivera C., and DiRita V. J. (2001) Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J. Infect. Dis. 183, 1043–1054 [DOI] [PubMed] [Google Scholar]
- 50. Kawabata S., Kuwata H., Nakagawa I., Morimatsu S., Sano K., and Hamada S. (1999) Capsular hyaluronic acid of group A streptococci hampers their invasion into human pharyngeal epithelial cells. Microb. Pathog. 27, 71–80 [DOI] [PubMed] [Google Scholar]
- 51. Nakagawa I., Kurokawa K., Yamashita A., Nakata M., Tomiyasu Y., Okahashi N., Kawabata S., Yamazaki K., Shiba T., Yasunaga T., Hayashi H., Hattori M., and Hamada S. (2003) Genome sequence of an M3 strain of Streptococcus pyogenes reveals a large-scale genomic rearrangement in invasive strains and new insights into phage evolution. Genome Res. 13, 1042–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kunitomo E., Terao Y., Okamoto S., Rikimaru T., Hamada S., and Kawabata S. (2008) Molecular and biological characterization of histidine triad protein in group A streptococci. Microbes Infect. 10, 414–423 [DOI] [PubMed] [Google Scholar]
- 53. Murakami J., Kawabata S., Terao Y., Kikuchi K., Totsuka K., Tamaru A., Katsukawa C., Moriya K., Nakagawa I., Morisaki I., and Hamada S. (2002) Distribution of emm genotypes and superantigen genes of Streptococcus pyogenes isolated in Japan, 1994–9. Epidemiol. Infect. 128, 397–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Haanes E. J., and Cleary P. P. (1989) Identification of a divergent M protein gene and an M protein-related gene family in Streptococcus pyogenes serotype 49. J. Bacteriol. 171, 6397–6408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hanski E., Horwitz P. A., and Caparon M. G. (1992) Expression of protein F, the fibronectin-binding protein of Streptococcus pyogenes JRS4, in heterologous streptococcal and enterococcal strains promotes their adherence to respiratory epithelial cells. Infect. Immun. 60, 5119–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Takamatsu D., Osaki M., and Sekizaki T. (2001) Thermosensitive suicide vectors for gene replacement in Streptococcus suis. Plasmid 46, 140–148 [DOI] [PubMed] [Google Scholar]
- 57. Okahashi N., Nakata M., Sakurai A., Terao Y., Hoshino T., Yamaguchi M., Isoda R., Sumitomo T., Nakano K., Kawabata S., and Ooshima T. (2010) Pili of oral Streptococcus sanguinis bind to fibronectin and contribute to cell adhesion. Biochem. Biophys. Res. Commun. 391, 1192–1196 [DOI] [PubMed] [Google Scholar]
- 58. Nakata M., Köller T., Moritz K., Ribardo D., Jonas L., McIver K. S., Sumitomo T., Terao Y., Kawabata S., Podbielski A., and Kreikemeyer B. (2009) Mode of expression and functional characterization of FCT-3 pilus region-encoded proteins in Streptococcus pyogenes serotype M49. Infect. Immun. 77, 32–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yamaguchi M., Terao Y., Mori Y., Hamada S., and Kawabata S. (2008) PfbA, a novel plasmin- and fibronectin-binding protein of Streptococcus pneumoniae, contributes to fibronectin-dependent adhesion and antiphagocytosis. J. Biol. Chem. 283, 36272–36279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lancefield R. C. (1957) Differentiation of group A streptococci with a common R antigen into three serological types, with special reference to the bactericidal test. J. Exp. Med. 106, 525–544 [DOI] [PMC free article] [PubMed] [Google Scholar]