

Abstract
Neutrophil myeloperoxidase (MPO) catalyzes the H2O2-dependent oxidation of chloride anion to generate hypochlorous acid, a potent antimicrobial agent. Besides its well defined role in innate immunity, aberrant degranulation of neutrophils in several inflammatory diseases leads to redistribution of MPO to the extracellular space, where it can mediate tissue damage by promoting the oxidation of several additional substrates. Here, we demonstrate that mannose 6-phosphate receptor-mediated cellular uptake and delivery of MPO to lysosomes of retinal pigmented epithelial (RPE) cells acts to clear this harmful enzyme from the extracellular space, with lysosomal-delivered MPO exhibiting a half-life of 10 h. Lysosomal-targeted MPO exerts both cell-protective and cytotoxic functions. From a therapeutic standpoint, MPO catalyzes the in vitro degradation of N-retinylidene-N-retinylethanolamine, a toxic form of retinal lipofuscin that accumulates in RPE lysosomes and drives the pathogenesis of Stargardt macular degeneration. Furthermore, chronic cellular uptake and accumulation of MPO in lysosomes coincides with N-retinylidene-N-retinylethanolamine elimination in a cell-based model of macular degeneration. However, lysosomal-delivered MPO also disrupts lysosomal acidification in RPE cells, which coincides with nuclear translocation of the lysosomal stress-sensing transcription factor EB and, eventually, cell death. Based on these findings we predict that under periods of acute exposure, cellular uptake and lysosomal degradation of MPO mediates elimination of this harmful enzyme, whereas chronic exposure results in progressive accumulation of MPO in lysosomes. Lysosomal-accumulated MPO can be both cell-protective, by promoting the degradation of toxic retinal lipofuscin deposits, and cytotoxic, by triggering lysosomal stress and cell death.
Keywords: ABC transporter, lysosomal storage disease, lysosome, mannose 6-phosphate (M6P), myeloperoxidase, retina, retinal degeneration
Introduction
MPO2 is a heme-containing peroxidase synthesized during myeloid differentiation that constitutes a major component of neutrophil azurophilic granules (1). Produced as a single chain precursor, MPO is post-translationally glycosylated in the Golgi, then cleaved into light and heavy chains in secretory azurophilic granules. The mature form of MPO is a dimer composed of two light chains and two heavy chains (1). In circulating neutrophils MPO catalyzes the H2O2-mediated oxidation of halide ions to predominantly form hypochlorous acid, a strong oxidant used to kill internalized bacteria and other pathogens. However, in some disease conditions degranulation of neutrophils leads to a redistribution of MPO to the extracellular space, where MPO can oxidize halide ions as well as other substrates to mediate tissue damage and pathogenesis of several inflammatory diseases, including ocular inflammation, Parkinson's disease, and Alzheimer's disease (2–4). How MPO exerts tissue damage in the retina is for the most part unknown.
Retinal pigmented epithelium (RPE) cells play an essential phagocytic role in preserving the overall health of the retina by removing rod outer segments shed from the photoreceptor layer. Although the majority of rod outer segment material delivered to RPE lysosomes can be degraded and eliminated from RPE lysosomes by the natural cohort of lysosomal hydrolases, some material remains resistant to further lysosomal degradation and gradually accumulates inside lysosomes as lipofuscin. Later in life lysosomal accumulation of retinal lipofuscin can contribute to the dry form of age-related macular degeneration, which is a complex disease with many causes (5).
In Stargardt disease (SD), the leading cause of macular degeneration and vision loss in children, the main component of lysosomal retinal lipofuscin is N-retinylidene-N-retinylethanolamine (A2E), a fluorescent derivative of vitamin A (6). A2E normally arises at a very slow rate as a byproduct of the visual cycle from the reaction of two molecules of all-trans-retinal with phosphatidylethanolamine in rod outer segments to form N-retinylidene-N-retinyl phosphatidylethanolamine (A2-PE; Ref. 7). In healthy photoreceptors ABCA4 flippase activity present in the rod outer segment membrane transfers the majority of all-trans-retinal from rod outer segments to RPE cells for conversion back to 11-cis-retinal and further phototransduction. Consequently, all-trans-retinal levels are maintained at low levels in rod outer segments with normal ABCA4 transporter activity, resulting in only very small amounts of A2-PE formation over time. In contrast, a genetic deficiency of the ABCA4 transporter in SD patients accelerates the formation of A2-PE in rod outer segments. These A2-PE-filled rod outer segments are eventually delivered to RPE lysosomes by phagocytosis (8).
In RPE lysosomes, a lysosomal-specific phosphatase converts newly delivered A2-PE to A2E (9). A2E then remains resistant to further degradation in RPE lysosomes (9). The accelerated lysosomal accumulation of A2E in SD promotes RPE cell death, inflammation, and the onset of retinal degeneration by several mechanisms; A2E is a major photo-sensitive chromophore for visible blue light, resulting in the generation of singlet oxygen, superoxide anion, and hydrogen peroxide, all of which may act as direct injury stimuli and the onset of chronic inflammation to the RPE (10–12). Oxidative damage arising from lysosomal A2E accumulation in RPE cells also compromises the phagocytic function of these cells (13). Accumulation of lysosomal lipofuscin also raises lysosomal pH and exerts a cationic detergent-like effect on cellular membranes (14).
Currently, no effective treatment is available for SD. Ongoing clinical trials to prevent further accumulation of A2E include gene therapy (STARGEN), stem cell-based therapy (Ocata Therapeutics Inc.), and oral inhibitors of A2E formation (Alkeus Pharmaceuticals Inc.). However, none of these approaches will help to eliminate existing A2E that would have accumulated before diagnosis and therapeutic intervention.
The peroxidase activity of horseradish peroxidase (HRP) was previously shown to catalyze the H2O2-mediated oxidation, destabilization, and subsequent degradation of A2E in a cell-free system and in cells loaded with A2E (15). Given that A2E accumulation in lysosomes gives rise to the generation of H2O2 (10–12), we reasoned that lysosomes filled with A2E in SD RPE cells may liberate sufficient H2O2 to permit the H2O2-dependent oxidation and degradation of this storage material by a lysosomal-targeted mammalian peroxidase therapeutic.
Of the five known mammalian peroxidases, only neutrophil MPO is reported to be synthesized as a precursor molecule with phosphorylated glycans, a property that permits binding with the cell surface mannose 6-phosphate (M6P) receptor, internalization into clathrin-coated vesicles, and delivery to lysosomes (16, 17). Here, we confirm by capillary zone electrophoresis that MPO contains low but sufficient amounts of phosphorylated oligomannose to permit its clearance from the extracellular space and delivery to lysosomes of RPE cells in a M6P receptor-dependent manner. We also identify A2E as a substrate for MPO, showing that MPO, like HRP, can degrade A2E both in vitro and after cellular uptake and delivery to RPE lysosomes preloaded with A2E. However, our results also reveal insight into how exogenous MPO can mediate cellular damage in inflammatory diseases; chronic cellular uptake and delivery of MPO to lysosomes disrupts lysosomal acidification and triggers the translocation of the lysosomal stress-sensing transcription factor EB (TFEB) from the cytosol to the nucleus, an indicator of lysosomal stress, which coincides with RPE cell death.
Results
MPO Is a Fully Processed Dimer That Exhibits M6P Receptor-dependent Uptake into ARPE-19 Cells
Given that HRP has been reported to degrade A2E in vitro (15), we set out to determine if a mammalian peroxidase could be used as a targeted enzyme therapeutic to promote the degradation of retinal lipofuscin in SD. SDS-PAGE and Western blotting analysis showed that commercially available MPO purified from human neutrophils was predominantly composed of the fully mature heavy and light chains under reducing conditions and as a >100-kDa dimer under non-reducing conditions (Fig. 1, A–C). The peroxidase-specific activity of MPO was confirmed using ABTS substrate and shown to be comparable with that of HRP (MPO, 181 ± 2.4 units/mg; HRP, 196 ± 1.4 units/mg).
FIGURE 1.

Glycan analysis of MPO. A, SDS-PAGE analysis of MPO purified from human neutrophils under reducing and non-reducing conditions. B and C, Western blotting analysis of MPO as described in A using heavy chain MPO (B) and light chain MPO (C) antibodies. The bands corresponding to MPO monomer, dimer, mature HC and mature LC are indicated with arrows. D, oligosaccharide analysis of MPO by capillary zone electrophoresis. Phosphorylated peaks were digested to non-phosphorylated glycans with alkaline phosphatase (+AP). Indicated tentative peak IDs were assigned from co-migration with a known reference lysosomal enzyme standard included in the run. BPM7; bis-phosphorylated oligomannose 7; MPM5, monophosphorylated oligomannose 5; MPM6, monophosphorylated oligomannose 6. Results in A–C are representative of three separate experiments. Results in D are representative of two separate experiments. RFU, relative fluorescence units.
A phosphorylated MPO precursor has previously been detected in 32P-labeled HL60 myeloid cells (16), prompting us to directly analyze the glycan composition of this enzyme and its lysosomal targeting in RPE cells. Five consensus N-linked glycosylation sites on MPO have been reported, all of which are located on the heavy subunit of MPO (18). N-Linked oligosaccharide profiling of peptide N-glycosidase F-digested glycans by capillary zone electrophoresis demonstrated that MPO contains multiple glycan moieties (Fig. 1D). Some of the glycans present on MPO were digested to non-phosphorylated glycans after treatment with alkaline phosphatase (Fig. 1D). Compared with a reference lysosomal enzyme known to contain glycans with phosphorylated oligomannose, MPO contained a small amount of bis-phosphorylated oligomannose (BPM7) and mono-phosphorylated oligomannose (MPM5 and MPM6; Fig. 1D). However, the overall amount of phosphorylated oligomannose present on MPO corresponded to only 6.2% of the total oligosaccharide content. As a comparison, a well characterized reference lysosomal enzyme control included in the analysis contained 35–40% of its total glycan content as phosphorylated oligomannose using the same oligosaccharide profiling method (data not shown). Collectively, these results suggest that MPO contains a relatively low amount of phosphorylated oligomannose, the preferred glycan moiety for M6P receptor-dependent cellular uptake and lysosomal targeting.
Despite the presence of only low amounts of phosphorylated oligomannose, MPO exhibited M6P receptor-dependent uptake in immortalized human RPE-derived ARPE-19 cells; MPO was not detected by activity (Fig. 2A) or by Western blotting (Fig. 2B) in untreated control ARPE-19 cells. Incubation of ARPE-19 cells with MPO resulted in a dose-dependent increase in MPO activity detected in cell lysates (Fig. 2A). MPO uptake was found to approximate Michaelis-Menten kinetics in ARPE-19 cells, with an average Kuptake of 33 nm ± 4.4 nm (n = 3) after 24 h of uptake (Fig. 2A). Uptake of MPO was also monitored by Western blotting, with the mature heavy (HC) and light (LC) chains of MPO readily detected in cell lysates after 24 h of exposure to 25 nm neutrophil-derived MPO (Fig. 2B). Importantly, uptake of MPO heavy- and light-chain isoforms into ARPE-19 cells was completely inhibited when ARPE-19 cells were treated with 5 mm M6P to block M6P receptor-mediated cellular uptake and lysosomal targeting (Fig. 2B). The internalized enzyme exhibited a short half-life of 10 h, as measured by activity (Fig. 2C) and coincided with reduced amounts of MPO protein detected in cell lysates by Western blotting over the post-uptake (chase) time (Fig. 2D). After 24 h of uptake into ARPE-19 cells MPO conjugated to AF488 partially co-localized with LysoTracker Red+-acidified organelles, which is suggestive of successful delivery of MPO to lysosomes (Fig. 2E). Collectively, these results confirm that MPO purified from neutrophils contains low but sufficient amounts of phosphorylated oligomannose to permit M6P receptor-dependent cellular uptake and delivery to RPE lysosomes, the site of A2E accumulation in SD.
FIGURE 2.

Extracellular MPO is targeted to RPE lysosomes in a M6P receptor-dependent manner for degradation. A, representative Kuptake determination experiment for MPO in ARPE-19 cells. See “Experimental Procedures” for details. B, Western blotting of cell lysates prepared from ARPE-19 cells incubated with either control growth medium, 25 nm MPO, or 25 nm MPO in the presence of 5 mm M6P for 24 h. The presence of mature heavy chain MPO (HC) and light chain MPO (LC) are indicated. An enolase-specific antibody was used as a loading control. MPO present in the uptake medium (input) is included as a comparison on each gel. C, representative MPO half-life determination in ARPE-19 cells. See “Experimental Procedures” for details. D, analysis of MPO stability after uptake into ARPE-19 cells by Western blotting. E, representative images of PFA-fixed ARPE-19 cells after a 24-h incubation with 25 nm Alexa Fluor 488 MPO (green channel). Before fixation, cells were incubated with LysoTracker Red (red channel). An example of MPO co-localization with a LysoTracker Red+-acidified organelle is indicated with an arrow. Results in A–E are representative of three separate experiments.
MPO Degrades A2E in Vitro
To determine if MPO can degrade A2E in vitro, in a similar manner as reported for HRP (15), chemically synthesized A2E was incubated for 4 h with HRP or MPO in the presence of H2O2. A2E was also incubated with bovine serum albumin (BSA) in the presence of H2O2 as a control protein that lacked peroxidase activity. By UPLC, chemically synthesized A2E incubated with BSA was predominantly composed of the intact parent molecule after 4 h (Fig. 3, A and quantified in Fig. 3D). As previously reported by Wu et al. (15), incubation of A2E with HRP for 4 h resulted in a significant reduction in the amount of A2E parent molecule detected by UPLC and the appearance of several smaller degradation products to the left of the peak corresponding to the A2E parent molecule (Fig. 3, B and D). Wu et al. (15) performed a detailed characterization to show that these smaller products corresponded to oxidized forms of the long and short arms of A2E, suggesting that HRP-mediated oxidation of A2E promotes its fragmentation and instability (15). Similar to HRP, incubation of A2E with MPO also resulted in reduced amounts of the A2E parent molecule (Fig. 3, C and D). These results suggest that like HRP, the peroxidase activity of mammalian MPO can catalyze the in vitro degradation of A2E, a toxic bis-retinoid that accumulates in RPE lysosomes and drives the pathogenesis of SD.
FIGURE 3.

MPO degrades A2E in vitro. Representative chromatographs showing the profile of chemically synthesized A2E incubated with BSA control (A, upper panel), HRP (B, middle panel), or MPO (C, bottom panel) in the presence of H2O2 for 4 h and then analyzed by UPLC. The peak corresponding to the A2E parent molecule (Parish et al., Ref. 5) is indicated with an arrow. D, quantification of the A2E parent molecule. Results are expressed as the mean (n = 4) ± S.D. Chromatographic peaks corresponding to the A2E parent molecule were measured by comparing to a calibration plot spiked with known amounts of A2E. The amount of A2E parent molecule was expressed as a percentage of the total amount of retinal pigment present after 4 h. AU, arbitrary units.
Uptake of Low Doses of MPO into ARPE-19 Cells Promotes A2E Clearance in the Absence of Cytotoxicity
We next examined the potential of exploiting MPO as a targeted enzyme therapy to degrade A2E in SD macular degeneration. To model macular degeneration in cultured cells, post-confluent ARPE-19 cells were loaded with chemically synthesized A2E using a previously published method, where the inherent autofluorescence of A2E is utilized to monitor its uptake and lysosomal delivery by phagocytosis (19). A2E uptake was monitored by A2E autofluorescence at an emission wavelength of 488 nm, a property also exploited to monitor disease progression in SD patients (19, 20). After 6 h of uptake followed by an overnight chase in growth medium, A2E autofluorescence was found to dose-dependently accumulate inside ARPE-19 cells by high content imaging, with the maximal A2E granular autofluorescence at 488 nm observed at 7.5 μm A2E in the absence of cytotoxicity (Fig. 4, A and C), as previously observed by Sparrow et al. (19). The amount of A2E parent molecule was also dose-dependently detected in cell lysates prepared from A2E-loaded ARPE-19 cells by UPLC (Fig. 4B), further supporting the high content imaging results (Fig. 4, A–C).
FIGURE 4.

MPO promotes A2E clearance in a cell-based model of macular degeneration in the absence of cytotoxicity. A, representative high content images showing A2E autofluorescence in ARPE-19 cells preloaded with A2E for 6 h. B, detection of A2E parent molecule by UPLC in ARPE-19 cells preloaded with A2E for 6 h. C, quantification of high content images: cells preloaded with A2E, as in A, were incubated without (solid boxes) or with (open boxes) 25 nm MPO for 48 h. Top, A2E+ granules per cell: green. Bottom, nuclei number (cell number) per image: blue. D, Western blots of TFEB levels in nuclear and cytosol-enriched fractions from ARPE-19 cells preloaded with increasing concentrations of A2E as indicated or treated with 2 μm Torin-1. Blots were subsequently probed with nuclear and cytosolic marker proteins to confirm subcellular enrichment. Data are representative of three separate experiments. E, MPO dose-dependently promotes clearance of A2E in ARPE-19 cells. ARPE-19 cells were preloaded with 5 μm A2E for 6 h and then incubated with up to 25 nm MPO as indicated for 21 days. A2E autofluorescence was quantified by high content imaging. F, uptake of 25 nm MPO time-dependently promotes A2E clearance over 21 days in ARPE-19 cells preloaded with 5 μm A2E. Results in B, C, E, and F are expressed as the mean (n = 3 cultures) ± S.D. See “Experimental Procedures” for further details. Significant differences (p < 0.05) to control A2E groups.
Incubation of ARPE-19 cells with A2E at higher concentrations (≥20 μm) did not further increase granularity but did promote a reduction in nuclei number by high content imaging in some experiments (results not shown), suggestive of cytotoxicity. Various forms of lysosomal stress, including lysosomal accumulation of storage material in multiple lysosomal storage diseases, have previously been shown to trigger a lysosomal stress response pathway, which involves the translocation of TFEB from the cytosol to the nucleus (21). We reasoned that the lysosomal accumulation of A2E in RPE cells, which is known to neutralize lysosomes and exert a cationic detergent-like effect on membranes (13, 14), may also be promoting translocation of TFEB from the cytosol to the nucleus, which could potentially be used as a direct read-out of A2E-induced lysosomal stress in our cell-based model of macular degeneration.
To test this, we first set out to determine if ARPE-19 cells are responsive to lysosomal stress-induced TFEB translocation by inhibiting mTOR, a regulator of nutrient sensing at lysosomes (22). Under control conditions, the majority of TFEB in control-treated ARPE-19 cells was present in the cytosol-enriched fraction of ARPE-19 cells by Western blotting (Fig. 4D). Torin-1, an mTOR inhibitor and known inducer of TFEB dephosphorylation and nuclear translocation (22), promoted a robust translocation of TFEB from the cytosol-enriched fraction to the nuclear-enriched fraction, suggesting that ARPE-19 cells are responsive to Torin-1-induced lysosomal stress and TFEB nuclear translocation (Fig. 4D). Like Torin-1, incubation of ARPE-19 cells with A2E for 6 h also promoted a dose-dependent translocation of TFEB from the cytosol to the nucleus in ARPE-19 cells, indicative of A2E-induced lysosomal stress (Fig. 4D). These results are in agreement with previous studies, demonstrating that A2E accumulation in RPE lysosomes compromises the function of these organelles and promotes cytotoxicity (10–14).
We used this cell-based model of macular degeneration to determine if cellular uptake and delivery of MPO to lysosomes could promote A2E clearance. ARPE-19 lysosomes were preloaded with 2.5, 5, or 7.5 μm A2E for 6 h, concentrations that promoted some translocation of TFEB to the nucleus (Fig. 4D) in the absence of cytotoxicity (Fig. 4C). Incubation of A2E-loaded cells with 25 nm MPO for 48 h promoted a significant reduction in A2E granularity when compared with control A2E-loaded cells in the absence of cytotoxicity (Fig. 4C). To further evaluate the kinetics of MPO-mediated A2E clearance, we loaded ARPE-19 cells with a constant dose of 5 μm A2E and then incubated cells for up to 21 days in the presence of MPO. Cells were replenished with MPO every 2 days. Incubation of A2E-loaded ARPE-19 cells with MPO for 21 days promoted a dose-dependent (Fig. 4E) and time-dependent (Fig. 4F) reduction in A2E+ granules, as measured by high content imaging. Furthermore, repeated low dose administration of MPO promoted A2E clearance in ARPE-19 cells in the absence of cytotoxicity, as measured by high content imaging of nuclei numbers (results not shown). Collectively, these proof-of-principle experiments demonstrate a rationale for a targeted enzymatic degradation approach to eliminate retinal A2E using low doses of a lysosomal-targeted mammalian peroxidase such as MPO.
Prolonged Exposure to MPO Promotes Cytotoxicity in RPE Cells at High Nanomolar Concentrations
Our results suggest that low nm doses of MPO can potentially be exploited therapeutically to degrade lysosomal A2E associated with macular degeneration (Fig. 4). However, MPO has also been implicated in the pathogenesis of several inflammatory diseases (2–4). To investigate potential MPO toxicity in RPE cells, we next tested for cytotoxicity in RPE cells after exposure to MPO. Exposure of RPE cells to MPO promoted a dose-dependent cytotoxicity, as measured by reduced nuclei numbers by high content imaging, with cytotoxicity only being observed at MPO concentrations ≥200 nm (Fig. 5A). Furthermore, MPO-induced cytotoxicity only became noticeable after prolonged exposure to MPO for 8 days or greater (Fig. 5B). In a similar manner to A2E accumulation in lysosomes, we reasoned that chronic cellular uptake and accumulation of MPO in lysosomes of ARPE-19 cells may also induce lysosomal stress, which over a period of several days, ultimately leads to cell death.
FIGURE 5.
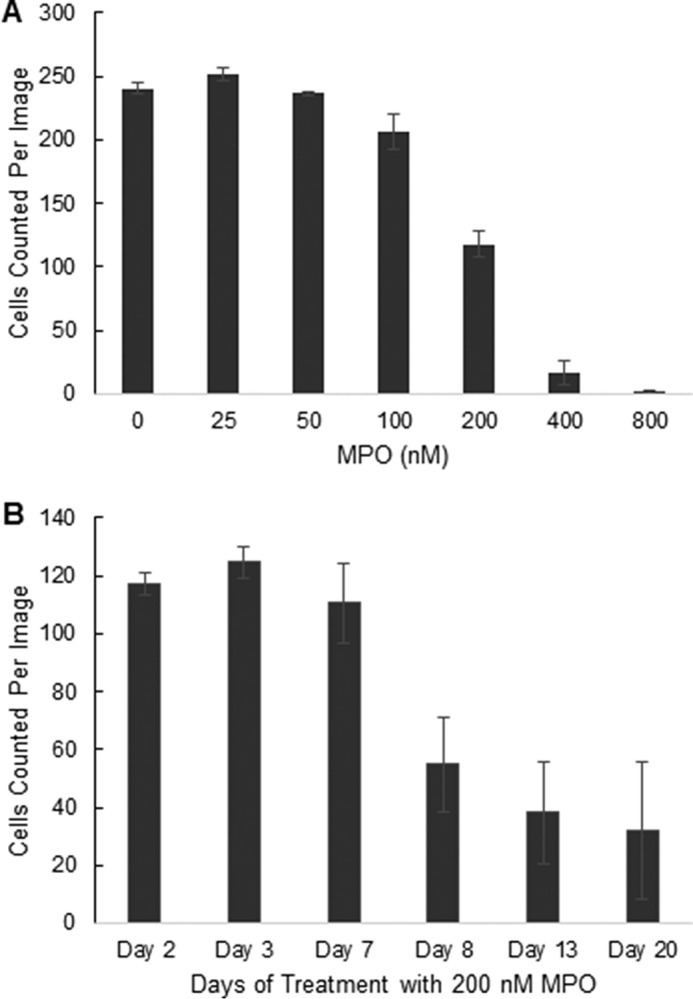
Chronic exposure to MPO promotes a dose- and time-dependent cytotoxicity. A, MPO promotes a dose-dependent cytotoxicity in ARPE-19 cells. Cells were incubated with increasing doses of MPO for 8 days (3 cultures per concentration), then analyzed for cell viability by high content imaging. B, MPO promotes a time-dependent cytotoxicity in ARPE-19 cells. Cells were incubated with MPO for up to 20 days (3 cultures per time point) then analyzed for cell viability by high content imaging. In A and B, cells were fixed and stained with Hoechst. High content images were acquired at 40× magnification using the blue channel to visualize nuclei as an indication of cell numbers per image (four sites per well). Results in A and B are representative of three repeat experiments.
We, therefore, tested for MPO-induced lysosomal stress in RPE cells at an earlier time point, after 3 days of continuous uptake. As in Fig. 4D, ARPE-19 cells were responsive to lysosomal stress, with inhibition of mTOR triggering a robust nuclear translocation of TFEB (Fig. 6A). After 3 days of continuous uptake, only high doses (>200 nm) of MPO promoted a robust translocation of TFEB from the cytosol to the nucleus in ARPE-19 cells by Western blot analysis (Fig. 6B) and by high content imaging (Fig. 6, C and D). Collectively, these results suggest that RPE cells exposed to higher concentrations of MPO undergo lysosomal stress, which triggers TFEB nuclear translocation, an indicator of lysosomal stress (22).
FIGURE 6.

Before cell death MPO triggers a loss of lysosomal acidification and TFEB translocation to the nucleus. A, TFEB Western blot analysis of nuclear and cytosol-enriched fractions isolated from DMSO (control) or Torin-1 treated ARPE-19 cells. Antibodies to poly(ADP-ribose) polymerase (PARP) and enolase were used as nuclear and cytosol loading controls, respectively. B, Western blot analysis of TFEB protein levels in cytosol and nuclear-enriched fractions from ARPE-19 cells treated with increasing concentrations of MPO for 3 days. C, representative high content images showing subcellular TFEB distribution by immunofluorescence (green channel) in ARPE-19 cells after 3 days of continuous exposure to control growth medium or 800 nm MPO. In each well (n = 3 wells per concentration) 4 individual images were acquired. D, quantification of TFEB nuclear translocation events (mean ± S.D.) in images described in C. The nuclei in corresponding images acquired in the blue channel were masked and used for analyzing the extent of TFEB translocation from the cytosol to the nucleus in the green channel. The number of nuclei that co-localized with TFEB were scored as translocation events and expressed as a percentage of the total number of nuclei imaged in each field. Note that nuclei number were not reduced after only 3 days of exposure to MPO (results not shown here; see also Fig. 5B). E and F, MPO promotes loss of lysosomal acidification. ARPE-19 cells were treated with increasing concentrations of MPO for 3 days, then treated with the lysosomotropic LysoTracker Red DND-99 probe, as described under “Experimental Procedures.” E, representative image of control untreated ARPE-19 cells showing LysoTracker Red+ granules alongside a representative image of ARPE-19 cells treated with 800 nm MPO for 3 days, with mostly red diffuse staining. F, quantification of high content imaging (three wells per treatment, with images acquired at four sites per well) of LysoTracker Red-treated cells. Data are the mean ± S.D. Results representative of two (panels E and F) and three repeats (panels A–D) of the experiment.
We next set out to determine the mechanism by which lysosomal-delivered MPO exerts lysosomal stress. Hasilik et al. (16) previously demonstrated that the cationic nature of MPO can partially mediate its cellular uptake and partitioning into lysosomes of fibroblasts. These observations by Hasilik et al. (16) prompted us to speculate that lysosomal-delivered MPO exerts lysosomal stress in RPE cells by neutralizing lysosomal pH. To assess the effects of MPO delivery to lysosomes on the health and integrity of these organelles, we monitored lysosomal pH using the fluorescent, pH-sensitive probe, LysoTracker Red DND-99 (24). Control, untreated ARPE-19 cells contained numerous LysoTracker Red+ organelles by high content imaging, reflecting the presence of intact, fully acidified lysosomes under basal conditions (Fig. 6E, quantified in Fig. 6F). In contrast, continuous cellular uptake of MPO for 3 days promoted a dose-dependent reduction in the number of LysoTracker Red+ granules by high content imaging, suggestive of MPO-induced disruption of lysosomal acidification (Fig. 6, E and F). Collectively, these results suggest that chronic exposure to high doses of MPO leads to cellular uptake and accumulation of this cationic enzyme in RPE lysosomes, where it promotes the neutralization of these organelles, which coincides with nuclear translocation of the lysosomal stress-sensing transcription factor, TFEB, and eventually cell death.
Discussion
Our results uncover a novel mechanism whereby MPO can be cleared from the extracellular space and targeted to RPE lysosomes in a M6P receptor-dependent manner, with both therapeutic and cytotoxic consequences.
Lysosomal accumulation of A2E promotes macular degeneration in SD, making the removal of this highly reactive storage material an urgent un-met medical need for these patients. A seminal report from the Sparrow laboratory (15) has demonstrated rationale for using an enzymatic degradation approach to remove lysosomal A2E from RPE lysosomes with HRP. HRP was shown to catalyze the H2O2-mediated oxidation of A2E in vitro and also in ARPE-19 cells loaded with chemically synthesized A2E to promote its fragmentation and elimination (15). Wu et al. (15) prompted us to explore the potential of using a mammalian peroxidase as a targeted enzyme therapy in SD. We focused on testing MPO for lysosomal degradation of A2E because a previous study showed that MPO uptake in fibroblasts can be partially inhibited with M6P addition to the medium and inhibited further with the addition of protamine sulfate to the medium, in combination with M6P (16). The authors concluded that MPO can be targeted to lysosomes of fibroblasts by both M6P receptor-dependent and -independent mechanisms, with the latter arising from the inherent cationic properties of MPO (16). We show here that a mature form of MPO purified from neutrophils can degrade A2E in vitro and contains low but sufficient amounts of phosphorylated oligomannose to permit M6P receptor-dependent cellular uptake and targeting of this non-lysosomal enzyme to lysosomes of RPE cells. It is possible that the dimeric neutrophil MPO may behave as a multivalent ligand for cell surface M6P-receptor, with each monomer of neutrophil MPO containing M6P, thus enabling more efficient uptake. This phenomenon has been reported for the tetrameric lysosomal hydrolase, β-glucuronidase (25).
After cellular uptake, mature neutrophil MPO activity was readily detected in ARPE-19 cells, albeit with a short half-life of 10 h. Although the mechanism of lysosomal MPO activation in ARPE-19 lysosomes was not investigated in this study, it is possible that local levels of H2O2 present in RPE lysosomes could potentially limit the activation of newly delivered MPO. H2O2 is required for the activation of MPO by reacting with the heme component of MPO, converting the iron group from a ferric form to an oxy-ferryl form (26). Presumably lysosomal H2O2, which is critical for MPO activation, was present in sufficient amounts in RPE lysosomes to transiently activate MPO. It is likely that lysosomal A2E itself could provide a local source of H2O2 as it is a major photosensitive chromophore for visible blue light, resulting in the generation of H2O2 (10–12). H2O2-mediated activation of MPO in RPE lysosomes could, therefore, consume lysosomal H2O2 liberated by A2E and at the same time oxidize, destabilize, and subsequently degrade lysosomal A2E. In support of this, cellular uptake of neutrophil MPO was sufficient to promote A2E clearance in ARPE-19 cells. The relatively short half-life of neutrophil MPO after uptake likely contributed to the need for repeated additions of the enzyme to the medium to promote A2E clearance, highlighting a limitation of using neutrophil MPO as a therapeutic.
Interestingly, the low abundance of phosphorylated oligosaccharides on MPO does not appear to be required for the correct targeting of newly synthesized MPO to neutrophil azurophilic granules, as I-cell patients, who lack mannose 6-phosphorylated lysosomal enzymes, exhibit normal levels of neutrophil MPO (27, 28). Our oligosaccharide profiling results suggest that the mature form of MPO contains a small amount of phosphorylated oligomannose, which coincides with M6P receptor-dependent uptake of MPO into ARPE-19 cells. If newly synthesized MPO does not rely on the mannose 6-phosphate receptor pathway for trafficking to azurophilic granules, the question remains as to why the mature form of this enzyme is stored in azurophilic granules with phosphorylated glycans? One possibility is that M6P receptor-dependent cellular uptake of MPO and targeting to lysosomes may serve to clear this potentially toxic enzyme from the extracellular space after neutrophil degranulation, thereby limiting tissue damage at inflammatory lesions.
Our results in RPE cells suggest that M6P receptor-mediated cellular uptake of MPO and targeting to lysosomes for degradation can be both cell-protective and cytotoxic, depending on the dose and duration of exposure to MPO (Fig. 7). At low nm concentrations, MPO can be efficiently cleared from the extracellular space and targeted to lysosomes for degradation. Lysosomal-delivered MPO exhibits a half-life of ∼10 h, suggesting that short periods of low level exposure to degranulated MPO can be cleared from the extracellular space by this pathway. Chronic exposure of RPE cells to higher concentrations of MPO results in MPO accumulation in lysosomes, as the recycling of the M6P receptor back to the cell surface for further rounds of MPO targeting to lysosomes occurs at a much faster rate (∼15 min; Ref. 29) when compared with the relatively slow rate of MPO degradation in lysosomes (t½ of ∼10 h). Low levels of MPO accumulation in lysosomes may be therapeutically beneficial by promoting the turnover of A2E (Fig. 7). Progressively higher levels of MPO accumulation in lysosomes triggers a loss of lysosomal acidification, which coincides with nuclear translocation of the lysosomal stress-sensing transcription factor, TFEB, and eventually, cell death (Fig. 7). MPO may also promote additional lysosomal stress by directly oxidizing other lysosomal components in addition to A2E or indirectly via the generation of hypochlorous acid, a strong prooxidant.
FIGURE 7.
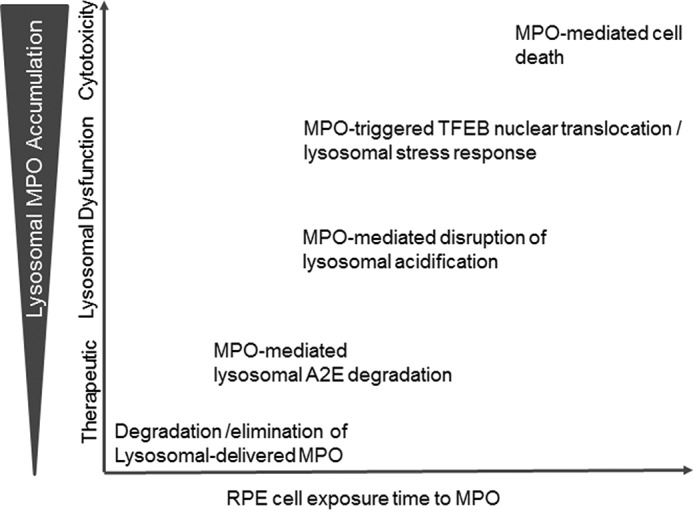
Schematic model summarizing the dose-dependent and time-dependent effects observed in RPE cells after cellular uptake and delivery of MPO to RPE lysosomes. At low concentrations over a short period of time, MPO, a known mediator of tissue damage in many inflammatory diseases, can be efficiently cleared from the extracellular space by M6P receptor-dependent cellular uptake and targeted to lysosomes for degradation. After delivery of MPO to lysosomes, the recycling time for the M6P receptor back to the cell surface for subsequent rounds of lysosomal targeting of MPO was ∼15 min (29), whereas the rate of lysosomal MPO degradation was much slower (t½ = 10 h; Fig. 2C). Therefore, with continuous exposure of RPE cells to MPO over prolonged periods of time, the rate of MPO cellular uptake and delivery to lysosomes exceeded the rate of MPO degradation, resulting in a net overall accumulation of MPO in lysosomes. Low amounts of lysosomal MPO accumulation in RPE cells may be beneficial by promoting the degradation of toxic retinal bis-retinoids such as A2E. Progressively higher amounts of cationic MPO accumulation in lysosomes disrupted lysosomal acidification, which in turn promoted nuclear translocation of the lysosomal stress-sensing transcription factor, TFEB, and eventually cell death.
Like MPO, we show that A2E itself can also trigger TFEB nuclear translocation at high concentrations most likely due to its cationic detergent-like properties (14). These results raise the possibility that both MPO and A2E accumulation in RPE lysosomes may act synergistically to promote lysosomal stress and disease progression in inflammatory macular degenerative diseases associated with A2E accumulation. From a more global standpoint, our cytotoxicity findings are consistent with the cellular uptake of neutrophil MPO and its delivery to lysosomes being implicated in the pathogenesis of several inflammatory disease conditions, including ocular inflammation, atherosclerosis, Parkinson's disease, and Alzheimer's disease (2–4, 10, 22) and highlight for the first time the importance of lysosomes in this tissue-damaging process.
In conclusion, we demonstrate that M6P receptor-dependent cellular uptake and lysosomal targeting of MPO helps to clear this potentially harmful enzyme from the extracellular space, with both cell-protective and cytotoxic consequences.
Experimental Procedures
Materials and Reagents
A2E was synthesized by Acme Bioscience, Inc. Palo Alto CA as previously described (30). The lyophilized powder was dissolved in DMSO to a final concentration of 10 mm and stored in aliquots at −80 °C. Human neutrophil MPO (180–220 units/mg; lot# MP2015–04) was from Athens Research and Technology (Athens, GA). HRP (type II, 150–250 units/mg; MLD: MFCD00071339) was from Sigma.
Analysis of A2E by UPLC
A2E was analyzed by UPLC using a reverse phase column (Zorbax 300SB-C18, Rapid Resolution HD 2.1 × 100 mm, 1.8 μm) and eluted with a gradient of methanol in water (85–96% methanol + 0.1% TFA, 0.2 ml/min, Waters UPLC system) over 25 min. A2E was detected using a UV detector set to 430 nm wavelength. For in vitro degradation assays, A2E (200 μm from a 10 mm stock in DMSO) was incubated with 0.15% (v/v) H2O2, 400 nm neutrophil MPO or 400 nm HRP at 37 °C for 4 h in phosphate-buffered saline, pH 7. Mixtures were analyzed by UPLC. The amount of A2E parent molecule present in each sample was determined by calculating the area under the A2E peak and then expressing this value as a percentage of the total area for all peaks. A calibration curve was included with known amounts of A2E analyzed to ensure that A2E measurements in test samples were in the linear range of the A2E standard curve.
UPLC and High Content Imaging of A2E-loaded ARPE-19 Cells
ARPE-19 cells were obtained from ATCC (CRL-2302) and passaged in DMEM/F-12 supplemented with fetal bovine serum (10% v/v). A2E was loaded into ARPE-19 cells using a modified method from Sparrow et al. (19). Briefly, ARPE-19 cells were seeded in 96-well plates, grown to 2 days post-confluence, and then incubated with varying concentrations of A2E (1.25 to 20 μm) directly diluted into growth medium for 6 h, at which time cells were washed twice with phosphate-buffered saline (PBS) and then allowed to recover overnight in growth medium.
For UPLC analysis of A2E levels after uptake into ARPE-19 cells, lysates were prepared in M-PER (mammalian protein extraction reagent; Thermo Fisher Scientific, Waltham, MA #78501). Before UPLC analysis, protein was removed from each cell extract by slow precipitation with acetonitrile increasing in concentration up to 80% (v/v) over 16 h at 4 °C. For high content image analysis of A2E autofluorescence, A2E-loaded cells were washed, fixed in PBS containing 4% (v/v) paraformaldehyde (Santa Cruz Biotechnology, sc-281692), stained with Hoechst (Life Technologies, H3570) to visualize nuclei and then imaged using a ImageXpress MicroXLS Widefield high content imaging system (Molecular Devices, Sunnyvale, CA) at 40× magnification at four different fields per well. For each field A2E autofluorescence was calculated using MetaXpress high content image acquisition and analysis software (Molecular Devices), with the number of A2E+ granules per cell being calculated as the total granular area (TGA) multiplied by the average granular intensity (AGI) divided by the total number of nuclei per field (TGA × AGI/# of cells).
Analysis of TFEB Translocation
Nuclear and cytosol-enriched fractions were prepared from ARPE-19 cells using the Pierce NE-PER nuclear and cytoplasmic reagents (Thermo Fisher Scientific) and analyzed by Western blotting with a rabbit polyclonal antibody to TFEB (Cell Signaling #4240). Alternatively, TFEB distribution was analyzed by immunofluorescence using a rabbit polyclonal anti-TFEB antibody (Cell Signaling #4240). Stained cells were analyzed by high content imaging using the ImageXpress. In each well four individual images were acquired at 40× magnification. Images were acquired in the green (TFEB) and blue (nuclei) channels, and the number of nuclei was recorded as an indicator of cell viability. The nuclei in the blue channel were masked and used for analyzing the extent of TFEB translocation from the cytosol to the nucleus in the green channel. The number of nuclei that co-localized with endogenous TFEB were scored as translocation events and expressed as a percentage of the total number of cells imaged in each field. Torin-1 (2 μm), a known inducer of TFEB translocation (23), was used as a positive control for TFEB translocation.
Oligosaccharide Profiling by Capillary Zone Electrophoresis
Oligosaccharides were released from neutrophil-derived MPO enzymatically using peptide N-glycosidase F. MPO samples were exchanged into 50 mm Tris, pH 7.3, via 10,000 molecular weight cutoff membrane spin filtration (Amicon Ultra, #UFC501096) to 2.0 mg/ml. Each digestion reaction contained 60 μg of sample in 50 mm Tris buffer, pH 7.3, with 0.1% SDS, 50 mm β mercaptoethanol. Reaction tubes were incubated at 95 °C for 10 min, then Nonidet P-40 was added to 0.75%. Digestion of oligosaccharides was initiated by the addition of 10 milliunits of peptide N-glycosidase F (QA-bio, #E-PNG05) followed by incubation at 37 °C for 17 h. Reactions were dried in a vacuum concentrator centrifuge (Thermo Scientific). The dried released oligosaccharides were derivatized by reductive amination with 8-aminopyrene-1,3,6-trisulfonic acid (APTS). Oligosaccharide pellets were labeled in 3 μl of 50 mg/ml APTS (Beckman Coulter, Brea, CA, #M408389) in 0.9 m citric acid and 5 μl of sodium cyanoborohydride (1 m in tetrahydrofuran, Sigma). The labeling reaction was incubated at 37 °C overnight. Before electrophoresis, the samples were applied to G10 Sepharose (GE Healthcare, #17-0010-01) in a Micro Biospin column (Bio-Rad, #644015212) to remove excess APTS dye.
Capillary electrophoresis was performed on the P800 CE (Beckman Coulter) using laser-induced fluorescence (excitation 488 nm). A 40-cm, 50-μm inner diameter with ProteomeLab N-CAP buffer (Beckman Coulter, #477623). Separation voltage was set to 21 kV for 14 min in reverse polarity. Analysis of profiles was performed using 32 Karat software (Beckman Coulter). Phosphorylated glycans were identified by digestion of the APTS labeled glycans using 4 μl of alkaline phosphatase (Roche Applied Science, #10713023001) and monitoring phosphorylated peak shifts in the profile.
Analysis of Neutrophil-MPO Uptake and Delivery to Lysosomes in ARPE-19 Cells
ARPE-19 cells were grown to 2 days post-confluence in 96-well plates, then incubated with increasing concentrations of neutrophil-derived MPO for 24 h. Uptake of MPO into ARPE-19 cells was monitored by Western blotting cell lysates with anti-MPO antibodies specific for the MPO heavy chain (Santa Cruz Biotechnology, sc16128) and light chain (Abcam, 45977) subunits. Alternatively, MPO uptake was monitored by measuring MPO activity using ABTS substrate. Briefly cell lysates or input medium were incubated with 2.5 mm ABTS and 0.1 mm H2O2 in assay buffer (20 mm potassium phosphate, 5 mm NaCl, pH 5) for up to 1 h at 37 °C in a Spectramax i3 plate reader (Molecular Devices) with absorbance at 405 nm measured every 5 min using Softmax Pro 6.3 software. A standard curve containing known amounts of neutrophil-derived MPO activity was included in each assay and used to calculate MPO activity levels in samples. Neutrophil MPO uptake in ARPE-19 cells was also monitored by imaging, where both proteins were directly conjugated with Alexa Fluor 488 before uptake. Proteins were reacted with a 5-fold molar excess of Alexa Fluor 488 sulfodichlorophenol ester (Thermo Scientific A30052) and purified as described by the manufacturer, yielding ∼3 fluorophores per molecule for both preparations. MPO activity was tested before and after labeling to ensure that AF488 conjugation did not affect activity toward ABTS substrate (data not shown). ARPE-19 cells were incubated with 0.2 mm LysoTracker Red before fixation to permit identification of acidified lysosomes.
To determine kuptake, ARPE-19 cells were incubated with varying concentrations of enzyme over a 24-h period (dose range 0.195–100 nm) at which time the enzyme activity present in cells was plotted into a Michaelis-Menten curve using GraphPad Prism software, La Jolla, CA. To determine the half-life of neutrophil MPO after uptake, ARPE-19 cells were incubated with 50 nm neutrophil-derived MPO for 24 h then washed several times to remove non-internalized enzyme. MPO-loaded cells were then incubated in growth medium and assayed at varying chase times for MPO activity, as described above. The MPO activity present in cells at each chase time point was plotted into a nonlinear regression exponential decay curve using GraphPad Prism software (La Jolla, CA) to determine enzymatic half-life.
Author Contributions
G. Y. led the project, wrote the paper, and coordinated the experiments. A. R. L. performed all the Western blots, MPO activity assays, K-uptake, and half-life studies and cytotoxicity studies shown in the paper. D. S. M., S. B., and T. C. contributed to cell-based assay development and provided feedback for manuscript preparation. H. P., T. J. M., and A. R. L. performed the A2E clearance assays. A. R. designed and performed the UPLC analysis of A2E. G. Y. B. conjugated AF488 to purified MPO. C. H. designed and performed the glycan analysis. J. L. H. provided guidance with the project and helped with the design of Kuptake studies.
Acknowledgments
We thank Sherry Bullens and Gordon Vehar for support and encouragement with this exploratory project.
The authors declare that they have no conflicts of interest with the contents of this article.
- MPO
- myeloperoxidase
- RPE
- retinal pigmented epithelium
- A2E
- N-retinylidene-N-retinylethanolamine
- SD
- Stargardt disease
- M6P
- mannose 6-phosphate
- TFEB
- transcription factor EB
- A2-PE
- N-retinylidene-N-retinyl phosphatidylethanolamine
- ABTS
- 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid)
- HC
- mature heavy chain
- LC
- light chain
- UPLC
- ultra performance liquid chromatography
- APTS
- 8-aminopyrene-1,3,6-trisulfonic acid.
References
- 1. McCormick S., Nelson A., and Nauseef W. M. (2012) Proconvertase proteolytic processing of an enzymatically active myeloperoxidase precursor. Arch. Biochem. Biophys. 527, 31–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Soubhye J., Aldib I., Delporte C., Prévost M., Dufrasne F., and Antwerpen P. V. (2016) Myeloperoxidase as a target for the treatment of inflammatory syndromes: mechanisms and structure activity relationships of inhibition. Curr. Med. Chem. 23, 3975–4008 [DOI] [PubMed] [Google Scholar]
- 3. Ray R. S., and Katyal A. (2016) Myeloperoxidase: bridging the gap in neurodegeneration. Neurosci. Biobehav. Rev. 68, 611–620 [DOI] [PubMed] [Google Scholar]
- 4. Arafat S. N., Suelves A. M., Spurr-Michaud S., Chodosh J., Foster C. S., Dohlman C. H., and Gipson I. K. (2014) Neutrophil collagenase, gelatinase, and myeloperoxidase in tears of patients with stevens-johnson syndrome and ocular cicatricial pemphigoid. Ophthalmology 121, 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parish C. A., Hashimoto M., Nakanishi K., Dillon J., and Sparrow J. (1998) Isolation and one-step prepapration of A2E and iso-A2E, fluorphores from human retinal pigment epithelum. Proc. Natl. Acad. Sci. U.S.A. 95, 14609–14613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kennedy C. J., Rakoczy P. E., and Constable I. J. (1995) Lipofuscin of the retinal pigment epithelium: a review. Eye 9, 763–771 [DOI] [PubMed] [Google Scholar]
- 7. Sparrow J. R., Fishkin N., Zhou J., Cai B., Jang Y. P., Krane S., Itagaki Y., and Nakanishi K. (2003) A2E, a byproduct of the visual cycle. Vis. Res. 43, 2983–2990 [DOI] [PubMed] [Google Scholar]
- 8. Molday R. S. (2007) ATP-binding cassette transporter ABCA4: molecular properties and role in vision and macular degeneration. J. Bioenerg. Biomembr. 39, 507–517 [DOI] [PubMed] [Google Scholar]
- 9. Sparrow J. R., Kim S. R., Cuervo A. M., and Bandhyopadhyayand U. (2008) A2E, a pigment of RPE lipofuscin is generated from the precursor, A2PE by a lysosomal enzyme activity. Adv. Exp. Med. Biol. 613, 393–398 [DOI] [PubMed] [Google Scholar]
- 10. Radu R. A., Hu J., Yuan Q., Welch D. L., Makshanoff J., Lloyd M., McMullen S., Travis G. H., and Bok D. (2011) Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J. Biol. Chem. 286, 18593–18601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beatty S., Koh H., Phil M., Henson D., and Boulton M. (2000) The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 45, 115–134 [DOI] [PubMed] [Google Scholar]
- 12. Winkler B. S., Boulton M. E., Gottsch J. D., and Sternberg P. (1999) Oxidative damage and age-related macular degeneration. Mol. Vis. 5, 32. [PMC free article] [PubMed] [Google Scholar]
- 13. Vives-Bauza C., Anand M., Shiraz A. K., Shirazi A. K., Magrane J., Gao J., Vollmer-Snarr H. R., Manfredi G., and Finnemann S. C. (2008) The age lipid A2E and mitochondrial dysfunction syngergistically impair phagoctyosis by retinal pigment epithelial cells. J. Biol. Chem. 283, 24770–24780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De S., and Sakmar T. P. (2002) Interaction of A2E with model membranes: implications to the pathogenesis of age-related macular degeneration. J. Gen. Physiol. 120, 147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu Y., Zhou J., Fishkin N., Rittmann B. E., and Sparrow J. R. (2011) Enzymatic degradation of A2E, a retinal pigment epithelial lipofuscin bisretinoid. J. Am. Chem. Soc. 133, 849–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hasilik A., Pohlmann R., Olsen R. L., and von Figura K. (1984) Myeloperoxidase is synthesized as larger phosphorylated precursor. EMBO J. 3, 2671–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cieutat A. M., Lobel P., August J. T., Kjeldsen L., Sengeløv H., Borregaard N., and Bainton D. F. (1998) Azurophilic granules of human neutrophilic leukocytes are deficient in lysosome-associated membrane proteins but retain the mannose 6-phosphate recognition marker. Blood 91, 1044–1058 [PubMed] [Google Scholar]
- 18. Van Antwerpen P., Slomianny M. C., Boudjeltia K. Z., Delporte C., Faid V., Calay D., Rousseau A., Moguilevsky N., Raes M., Vanhamme L., Furtmüller P. G., Obinger C., Vanhaeverbeek M., Nève J., and Michalski J. C. (2010) Glycosylation pattern of mature dimeric leukocyte and recombinant monomeric myeloperoxidase. J. Biol. Chem. 285, 16351–16359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sparrow J. R., Parish C. A., Hashimoto M., and Nakanishi K. (1999) A2E, a lipofuscin fluorophore, in human retinal pigmented epithelial cells in culture. Invest. Ophthalmol. Vis. Sci. 40, 2988–2995 [PubMed] [Google Scholar]
- 20. Cukras C. A., Wong W. T., Caruso R., Cunningham D., Zein W., and Sieving P. A. (2012) Centrifugal expansion of fundus autofluorescence patterns in Stargardt disease over time. Arch. Ophthalmol. 130, 171–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sardiello M., Palmieri M., di Ronza A., Medina D. L., Valenza M., Gennarino V. A., Di Malta C., Donaudy F., Embrione V., Polishchuk R. S., Banfi S., Parenti G., Cattaneo E., and Ballabio A. (2009) A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 [DOI] [PubMed] [Google Scholar]
- 22. Roczniak-Ferguson A., Petit C. S., Froehlich F., Qian S., Ky J., Angarola B., Walther T. C., and Ferguson S. M. (2012) The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 5, ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klebanoff S. J. (2005) Myeloperoxidase: friend or foe. J. Leukoc Biol. 77, 598–625 [DOI] [PubMed] [Google Scholar]
- 24. Oberle C., Huai J., Reinheckel T., Tacke M., Rassner M., Ekert P. G., Buellesbach J., and Borner C. (2010) Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ. 17, 1167–1178 [DOI] [PubMed] [Google Scholar]
- 25. York S. J., Arneson L. S., Gregory W. T., Dahms N. M., and Kornfeld S. (1999) The rate of internalization of the mannose-6-phosphate/insulin-like growth factor II receptor is enhanced by multivalent ligand binding. J. Biol. Chem. 274, 1164–1171 [DOI] [PubMed] [Google Scholar]
- 26. Prokopowicz Z., Marcinkiewicz J., Katz D. R., and Chain B. M. (2012) Neutrophil myeloperoxidase: soldier and statesman. Arch. Immunol. Ther. Exp. (Warsz) 60, 43–54 ( 10.1007/s00005-011-0156-8) [DOI] [PubMed] [Google Scholar]
- 27. Lemansky P., Gieselmann V., Hasilik A., and von Figura K. (1985) Synthesis and transport of lysosomal acid phosphatase in normal and I-cell fibroblasts. J. Biol. Chem. 260, 9023–9030 [PubMed] [Google Scholar]
- 28. Owada M., and Neufeld E. F. (1982) Is there a mechanism for introducing acid hydrolases into liver lysosomes that is independent of mannose 6-phosphate recognition: evidence from I-cell disease. Biochem. Biophys. Res. Commun. 105, 814–820 [DOI] [PubMed] [Google Scholar]
- 29. Cardone M., Porto C., Tarallo A., Vicinanza M., Rossi B., Polishchuk E., Donaudy F., Andria G., De Matteis M. A., and Parenti G. (2008) Abnormal mannose-6-phosphate receptor trafficking impairs recombinant α-glucosidase uptake in Pompe disease fibroblasts. Pathogenetics 1, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sparrow J. R., Kim S. R., and Wu Y. (2010) Experimental approaches to the study of A2E, a bisretinoid lipofuscin chromophore of retinal pigment epithelium. Methods Mol. Biol. 652, 315–327 [DOI] [PMC free article] [PubMed] [Google Scholar]