

Abstract
β-Lactamases confer resistance to β-lactam-based antibiotics. There is great interest in understanding their mechanisms to enable the development of β-lactamase-specific inhibitors. The mechanism of class A β-lactamases has been studied extensively, revealing Lys-73 and Glu-166 as general bases that assist the catalytic residue Ser-70. However, the specific roles of these two residues within the catalytic cycle remain not fully understood. To help resolve this, we first identified an E166H mutant that is functional but is kinetically slow. We then carried out time-resolved crystallographic study of a full cycle of the catalytic reaction. We obtained structures that represent apo, ES*-acylation, and ES*-deacylation states and analyzed the conformational changes of His-166. The “in” conformation in the apo structure allows His-166 to form a hydrogen bond with Lys-73. The unexpected “flipped-out” conformation of His-166 in the ES*-acylation structure was further examined by molecular dynamics simulations, which suggested deprotonated Lys-73 serving as the general base for acylation. The “revert-in” conformation in the ES*-deacylation structure aligns His-166 toward the water molecule that hydrolyzes the acyl adduct. Finally, when the acyl adduct is fully hydrolyzed, His-166 rotates back to the “in” conformation of the apo-state, restoring the Lys-73/His-166 interaction. Using His-166 as surrogate, our study identifies distinct conformational changes within the active site during catalysis. We suggest that the native Glu-166 executes similar changes in a less constricted way. Taken together, this structural series improves our understanding of β-lactam hydrolysis in this important class of enzymes.
Keywords: antibiotic resistance, enzyme catalysis, enzyme kinetics, structure-function, X-ray crystallography
Introduction
β-Lactamase is the one of the major mechanisms of antibiotic resistance. Enzymes of this family have evolved to become highly efficient in hydrolyzing and inactivating a wide spectrum of β-lactam antibiotics (1). Most alarmingly, the widespread usage of antibiotics has led to the emergence of new β-lactamases that can hydrolyze even the “last resort” β-lactam antibiotics like meropenem and ertapenem (2). Pathogenic bacteria strains harboring these enzymes either in their chromosomes or on transmittable plasmids readily acquire super-resistance and pose serious threats to public health (3).
To overcome the antibiotic resistance mediated by β-lactamases, extensive studies of their catalytic mechanism have been carried out. The large number of over 1,000 reported β-lactamases have been grouped into four classes (A to D) based on sequence alignment (1, 4, 5). Although class B is unique by employing zinc ions for β-lactam hydrolysis, class A, C, and D all utilize a serine-based catalytic process that consists of two distinct steps. The first step is the acylation reaction when the catalytic serine residue carries out nucleophilic attack on the β-lactam substrate and forms the enzyme-substrate (ES*) acyl adduct (Fig. 1a). The second step is the subsequent deacylation step when a water molecule hydrolyzes the ES* acyl adduct so that the inactivated antibiotic is released with its β-lactam ring opened, and the active site is regenerated (Fig. 1a). For each step, a general base is needed to activate the normally non-reactive serine residue or the deacylation water molecule (Fig. 1a). Although class A, C, and D β-lactamases share the same two-step process, they have evolved substantially different strategies in terms of how to arrange the general bases for the acylation and deacylation reactions (1, 4, 5). The catalytic prowess of these β-lactamases depends critically on the effectiveness of these general bases.
FIGURE 1.

Reaction scheme of class A β-lactamase. a, reaction scheme for hydrolysis of β-lactam antibiotics in the active site of class A β-lactamases. B represents the general base. Base1 and Base2 represent different residues for the acylation and deacylation step respectively. b, concerted base model for the acylation step of the catalytic cycle with substrate-induced proton transfer from Lys-73 to Glu-166 so that Lys-73 serves as the general base. The deacylation step employs Glu-166 as the general base. The dashed arrow represents intermediate steps between the pre- and post-deacylation complex.
The active site of class A β-lactamases contains two residues, Lys-73 and Glu-166, as general bases, and both are positioned in close proximity to the catalytic residue Ser-70 (1, 4, 6). Decades of comprehensive studies have confirmed distinct catalytic roles for these two residues. Lys-73 is assigned as the critical general base for the acylation step because mutating this residue completely abolishes this reaction. In contrast, Glu-166 is regarded as the exclusive general base for the deacylation reaction because Glu-166 mutations significantly reduce its kinetic rate. However, some crucial mechanistic details regarding the roles of these two general bases are still not well understood. For example, multiple structural studies have revealed that when the active site is at the apo-state, Lys-73 is protonated, and Glu-166 is deprotonated, thus forming an electrostatically favorable Lys-73–Glu-166 salt bridge within the active site (Fig. 1b) (7–9). Under such setup, Lys-73 is not ready to serve as a general base for the acylation reaction because only its deprotonated form can abstract a proton from Ser-70 to promote its nucleophilic attack on the substrate. It is reasonable to expect that when the active site proceeds from the apo-state to the acylation state, some changes may occur, likely triggered by the event of substrate binding, so that Lys-73 can become deprotonated and proceed to fulfill its catalytic role (Fig. 1b). Although quantum mechanics/molecular mechanics (QM/MM)2 studies have suggested such a possibility, no experimental evidence for such a transfer in a functional active site while turning over a natural substrate has been reported (10). Furthermore, as the active site completes the acylation reaction and proceeds into the deacylation reaction, how the role of general base is switched from Lys-73 to Glu-166 is not known. These mechanistic details can be highly valuable in further improving our understanding of this important class of enzymes.
To gain in-depth understanding of how the active site of class A β-lactamases coordinates key catalytic residues such as Lys-73 and Glu-166 to facilitate efficient catalysis, it would be ideal if a complete catalytic cycle of hydrolyzing a natural substrate can be tracked and individual functional intermediates along the reaction pathway can be analyzed at atomic resolution. However, it is experimentally challenging to obtain such information given the ultrafast kinetic rates of wild-type class A β-lactamases that usually approach the diffusion limit for natural substrates (kcat/Km ∼108 m−1 s−1). Here, we report our work on a mutant class A β-lactamase with slowed kinetics so that the catalysis of natural substrates like cephaloridine can be tracked by in crystallo catalysis. These structures reveal novel conformational changes within the active site in sync with distinct catalytic steps and offer fresh perspective on mechanistic understanding of β-lactamase catalysis.
Results
E166H Substitution Yields a Functional Class A β-Lactamase with Slowed Kinetics Suitable for in Crystallo Catalysis
PenP from Bacillus licheniformis has been used in our previous studies as a model enzyme for class A β-lactamases because of its high sequence identity (>80%) to the clinically significant TEM and SHV series and its amenability to mutational perturbations (11–13). It shows high catalytic efficiency toward first-generation β-lactam antibiotics like penicillin and cephaloridine with kcat/Km in the order of 106 m−1 s−1, comparable with that for the TEM and SHV β-lactamases.
In crystallo catalysis followed by time-resolved X-ray crystallography has been a powerful tool to capture functional intermediates within the active site of an enzyme to understand its catalytic mechanism. For example, this approach has been successfully applied on a time scale of minutes to visualize the formation of phosphodiester bond and to investigate novel catalytic mechanism mediated by a third Mg2+ ion not included in the classical model (14, 15). This method has also been employed at a femtosecond time scale to analyze ultrafast motions in CO myoglobin (16). However, no such study has been done for β-lactamases probably because the turnover rate of the wild-type enzyme is so fast that reaction intermediates cannot be readily captured. To enable our investigation, we first set out to generate PenP derivatives that are kinetically slow but nonetheless functional. We targeted the general base Glu-166 as it is located on the structurally flexible Ω-loop, and mutations at this site would lead to minimal perturbation to the other residues within the active site.
The E166H mutant was identified as a suitable candidate. As shown by electrospray ionization-mass spectroscopy (ESI-MS) experiments, the rapid formation of the enzyme-substrate acyl adduct (ES*) in the acylation step and the subsequent hydrolysis of ES* back to enzyme only (E) during the deacylation step occur over a prolonged period of up to 2 min (Fig. 2a). Furthermore, the pH-dependent activity profile of E166H was measured by nitrocefin assay and showed a large shift from ∼7.0 to ∼8.5 in terms of optimal activity as compared with wild-type PenP (Fig. 2b) (17). This shift suggests that His-166 probably replaces Glu-166 and directly participates in the catalytic reactions. Furthermore, the kinetic parameters of E166H turning over natural β-lactam antibiotics were measured using standard UV spectroscopy method (18). E166H shows ∼103-fold reduction in terms of kcat/Km value for hydrolysis of penicillin G at pH 7.0, in agreement with a previous report on the slowed kinetic profile of TEM-1 E166H mutant (19). However, such reduction is only 100-fold for the hydrolysis of cephaloridine (Table 1). For subsequent in crystallo catalysis, we will thus choose cephaloridine as the suitable substrate for investigation.
FIGURE 2.
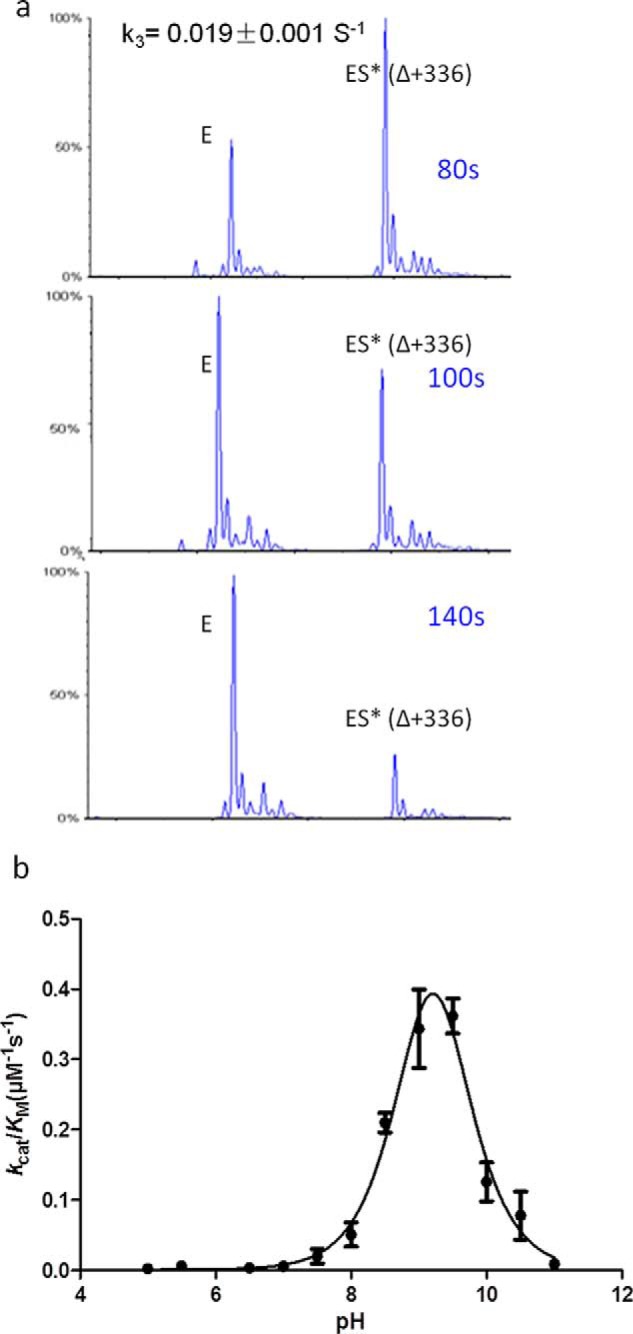
E166H shows slow deacylation rate and has a distinct pH-dependent profile. a, time-dependent deacylation for E166H-cephaloridine acyl adduct. b, pH dependence of kcat/Km for nitrocefin hydrolysis by E166H mutant β-lactamase.
TABLE 1.
Kinetics parameters for E166H hydrolyzing penicillin G and cephaloridine
| β-Lactamase | Antibiotics | Km | kcat | kcat/Km |
|---|---|---|---|---|
| μm | s−1 | m−1s−1 | ||
| WT | Penicillin G | 60.9 ± 5.3 | 393.8 ± 18.5 | (6.5 ± 0.9) × 106 |
| Cephaloridine | 20.7 ± 4.1 | 8.2 ± 0.8 | (4.0 ± 1.0) × 105 | |
| E166H | Penicillin G | 149.4 ± 38.2 | 0.46 ± 0.07 | (3.1 ± 1.0) × 103 |
| Cephaloridine | 4.7 ± 0.8 | 0.022 ± 0.001 | (4.7 ± 1.1) × 103 |
With kinetically slowed E166H PenP β-lactamase we set out to crystallize this enzyme and tracked in crystallo catalysis of cephaloridine within its active site by time-resolved X-ray crystallography. Three structures were determined, representing the apo, ES*-acylation, and ES*-deacylation states, respectively (Table 2). We also tracked the progression of the slowed deacylation reaction until the cephaloridine substrate departed from the active site. Collectively, our structures represent for the first time a full cycle of β-lactam hydrolysis within the active site of a class A β-lactamase. Detailed analysis of these structures and their mechanistic implications are discussed below.
TABLE 2.
Statistics of X-ray crystallography data collection and structure refinement
| Apo | ES*-acylation | ES*-deacylation | |
|---|---|---|---|
| Data collection | |||
| PDB code | 5GHX | 5GHY | 5GHZ |
| Space group | P1 | P1 | P1 |
| Unit cell parameters | |||
| a, b, and c (Å) | 43.331, 45.849, 66.139 | 43.240, 45.940, 65.990 | 43.290, 45.490, 66.100 |
| α, β, γ (°) | 77.84, 75.65, 69.09 | 76.84, 75.58, 69.07 | 77.81, 75.48, 68.76 |
| Resolution range (Å) | 50.00–1.24 (1.28–1.24) | 42.40–2.01 (2.11–2.01) | 42.02–1.93 (2.03–1.93) |
| No. of total reflections | 483,575 | 111,449 | 121,668 |
| No. of unique reflections | 118,377 | 28,415 | 31,135 |
| I/σ | 14.7 (8.0) | 14.2 (5.3) | 18.0 (9.8) |
| Completeness (%) | 91.5 (88.3) | 93.3 (89.1) | 91.2 (86.3) |
| Rmerge (%) | 5.2 (17.2) | 6.8 (23.9) | 5.5 (12.8) |
| Structure refinement | |||
| Resolution (Å) | 33.27–1.24 | 38.00–2.01 | 37.4–1.93 |
| Rcryst/Rfree (%) | 13.9/16.2 | 19.5/24.4 | 15.6/20.9 |
| r.m.s.d. bonds (Å)/angles (°) | 0.030/2.666 | 0.017/1.930 | 0.018/1.869 |
| No. of reflections | |||
| Working set | 112,438 | 26,977 | 29,548 |
| Test set | 5930 | 1434 | 1580 |
| No. of atoms | |||
| Protein atoms | 4143 | 4097 | 4112 |
| Ligand/ion atoms | 16 | 44 | 44 |
| Water molecules | 868 | 145 | 485 |
| Average B-factor (Å2) | |||
| Main chain | 9.396 | 19.141 | 13.254 |
| Side chain | 17.450 | 23.271 | 18.363 |
Apo Structure Reveals an Active Site with Steady Lys-73/His-166 Interaction but Is Not Catalytically Ready
The apo structure of E166H solved at 1.24 Å resolution is nearly identical to the wild-type structure with overall root mean square deviation (r.m.s.d.) of only ∼0.29 Å. The mutant His-166 side chain occupies a position largely identical to that of Glu-166 in the wild-type structure and is engaged in two H-bond interactions, one with Lys-73 via the Nϵ2 atom of the imidazole ring (distance 2.87 Å) and the other with a solvent water molecule via the Nδ1 atom (distance 2.84 Å) (Fig. 3, a and b). Beyond His-166, the rest of the E166H active site is identical to that of the wild type. The only notable difference is the absence of a water molecule (DW) that would be strategically positioned between Ser-70 and Glu-166 in the wild-type structure and has been speculated to serve as a bridging entity to facilitate the substrate-induced proton transfer from Lys-73 to Glu-166 (Fig. 3b).
FIGURE 3.

Active site in the apo structure of E166H reveals a stable Lys-73–His-166 H-bond. a, H-bond interactions involving Lys-73 and His-166 in the active site. Fo − Fc omit map (purple) is drawn in mesh format and contoured at 2.0 σ. The dashed lines indicate hydrogen bonds. b, superposition of E166H active site with that of the wild-type. c, inferred protonation states for Lys-73 and His-166 to sustain the Lys-73–His-166 H-bond.
Although it is not possible to visualize protons at the resolution of our structure, we managed to derive the protonation states of Lys-73 and His-166 using the observed Lys-73/His-166 interaction as the critical clue. First of all, the possibility of protonated His-166 is ruled out because Lys-73 must also be protonated in this case given its stronger basicity. However, the resulting electrostatic repulsion between these two residues would render the Lys-73/His-166 interaction unsustainable. Second, Lys-73 is likely protonated given its predicted pKa value of ∼8.0–8.5 according to a previous study and our crystallization condition at pH 7.5 (9). With protonated Lys-73 and deprotonated His-166, the Lys-73/His-166 interaction is underpinned by the terminal −NH3+ group of Lys-73 as the H-donor and the deprotonated Nϵ2 atom of His-166 as the H-acceptor (Fig. 3c).
With this setup neither Lys-73 nor His-166 is at a suitable protonation state to serve as the general base for initiating acylation. For Lys-73, it needs to be deprotonated first to activate the catalytic Ser-70 by proton abstraction. For His-166, a critical deacylation water molecule must emerge between His-166 and Ser-70 so that the neutral His-166 can deprotonated the water and trigger its nucleophilic attack on Ser-70. Thus, in absence of a substrate, the E166H active site at apo-state is not catalytically ready, similar to what has been observed for other Class A β-lactamases (7, 8).
ES* Acylation Structure Captures Unexpected “Flipped-out” Conformation of His-166 Likely Caused by Substrate-induced Proton Transfer from Lys-73
The ES* acylation structure was obtained when cephaloridine-soaked E166H crystals were flash frozen and immediately loaded onto X-ray source for data collection. This protocol ensures that the active site is “trapped” at a functional intermediate in which the ES* acyl adduct is already formed but the deacylation process hasn't started. For the ES* acylation structure, the Fo − Fc map unambiguously reveals an acyl-enzyme adduct with covalent linkage between C8 of cephaloridine and the Oγ atom of Ser-70 (Fig. 4a). The binding mode of cephaloridine is nearly identical to that seen in our previous report and that by Chen et al. (13, 20).
FIGURE 4.

ES*-acylation structure shows His-166 adopting flipped-out conformation and confirms Lys-73 as general base for acylation. a, active site in the ES*-acylation structure. Fo − Fc omit map (purple) is drawn in mesh format and contoured at 2.0 σ. The dashed lines indicate hydrogen bonds. b, superposition of ES*-acylation (cyan) and apo structures (green). Dashed lines mark the H-bonds. The flipped-out conformation of E166H is marked with an arrow. The tilting of Lys-73 is marked by Lys-73–Ser-130 H-bond. c, scheme of the proposed proton transfer from Lys-73 to His-166 during the acylation step and the resulting “flipped-out” conformation of His-166.
Unexpectedly, the imidazole side chain group of His-166 adopts an alternative conformation and is completely flipped out of the active site in the ES*-acylation structure (Fig. 4, a and b). This motion by His-166 displaces the side chains of Asn-104 and Tyr-105, leading to a new H-bond formed between the flipped-out His-166 and Asn-104 (supplemental Fig. S1). Two water molecules emerge within the active site near the area vacated by the His-166 side chain. One water molecule (WAT1) is located within 2.77 Å from the carbonyl carbon of the acyl adduct, poised to act as the deacylation water to carry out nucleophilic attack on the ES* moiety for hydrolysis of the acyl adduct (Fig. 4b). The other water molecule (WAT2) is located between WAT1 and His-166, forming hydrogen bonds with both of them as well as the nearby residue Asn-132 (Fig. 4b). Furthermore, Lys-73 adopts a slightly altered conformation with its side chain tilted away from the Ser-70 Oγ atom (distance 3.21 Å) and engaged in stable H-bonds with the backbone carbonyl and hydroxyl side chain of Ser-130 (distance 2.97 Å and 2.94 Å, respectively) while maintaining H-bond with Asn-132 (distance 2.70 Å) (Fig. 4b).
We are intrigued by this “flipped-out” conformation of His-166, which has never been reported in any other β-lactamase structures. Apparently it is tied to the acylation reaction. As the protonated Lys-73 at the apo-state needs to be deprotonated when serving as general base for the acylation reaction, one speculation is for a proton to be transferred from the −NH3+ group of Lys-73 side chain to the Nϵ2 atom of His-166 via the Lys-73/His-166 interaction. Simultaneously the deprotonated Lys-73 can abstract a proton from the Ser-70-OH group, and the activated Ser-70 becomes ready to carry out nucleophilic attack on the cephaloridine substrate (Fig. 4c). As all these events may occur in concurrent manner, the Lys-73/His-166 interaction thus becomes unsustainable when both residues become protonated. As a result, the protonated His-166 would adopt a “flipped-out” conformation to avoid electrostatic repulsion with Lys-73 (Fig. 4c).
To test whether substrate-induced proton transfer from Lys-73 to His-166 would render the active site unsuitable for catalysis unless His-166 flips out, we carried out molecular dynamics (MD) analysis. Two models of the enzyme-substrate complex, one with His-166 adopting the “in” conformation and the other with flipped-out conformation, were constructed using the ES*-acylation structure as the starting conformation (Fig. 5, a and b). In both models the ES* acyl adduct was replaced with Ser-70 and cephaloridine. Lys-73 was set as deprotonated, and His-166 was protonated. In the MD trajectories with His-166 at the “in” conformation, the distance between the Hγ atom of the Ser-70 side chain (HG) and the Nϵ atom of Lys-73 side chain (NZ) is always larger than 3.5 Å after equilibration, suggesting that no H-bond is formed between them, and thus proton abstraction to activate Ser-70 could not occur under this conformation (Fig. 5a). In contrast, in the MD trajectories with His-166 at the flipped-out conformation, the distance between Ser-70:Hγ and Lys-73:Nϵ is stable at ∼1.8 Å, within the range for ready proton transfer (Fig. 5b). The distance between Ser-70:Oγ (OG) and C3 of cephaloridine is also steady at ∼3.0 Å, suitable to establish the covalent linkage for acyl adduct (Fig. 5b). Thus, our MD simulations suggest that the active site is only suitable for the acylation reaction if the protonated His-166 adopts the flipped-out conformation, in support of our crystal structure.
FIGURE 5.

MD simulations to compare two models of the enzyme-substrate complex. Two models of the enzyme-substrate complex were constructed, one with His-166 adopting the “in” conformation as seen in the apo structure and the other with His-166 adopting the “flipped-out” conformation of the ES*-acylation structure. a and c, time course of the distance of Ser-70HG–Lys-73NZ (red) and Ser-70OG-C3 (black) with His-166 adopting “in” conformation. b and d, time course of the distance of Ser-70HG–Lys-73NZ (red) and Ser-70OG-C3 (black) with His-166 adopting flipped-out conformation.
ES* Deacylation Structure Shows His-166 Adopting “Revert-in” Conformation to Serve as the General Base for Deacylation
The ES*-deacylation structure was obtained when cephaloridine-soaked E166H crystals were equilibrated in cephaloridine-free solution for 1 min first and then flash-frozen and mounted onto an X-ray source. This additional equilibration step is designed to capture the E166H active site at a state when the deacylation state is engaged, while the ES* acyl adduct is still intact. This state can be tracked by X-ray crystallography because the E166H mutation has significantly slowed down the kinetic rate of deacylation to the order of minutes.
The ES*-deacylation structure is essentially the same as that for the ES*-acylation state with overall r.m.s.d. of only ∼0.3 Å. The Fo − Fc map reveals the full cephaloridine acyl adduct covalently linked to Ser-70 in an orientation identical to that in the ES*-acylation structure (Fig. 6a). Interestingly, His-166 side chain is reverted back into the active site and occupies the same position as seen in the apo structure (Fig. 6a). With this “revert-in” conformation, His-166 displaces the water molecule WAT2 seen in ES*-acylation structure, while the deacylation water WAT1 remains at the same position. This way His-166 is well positioned to serve as general base for deacylation by abstracting a proton from WAT1 (Fig. 6a). Intriguingly, close inspection reveals that this “revert-in” conformation of His-166 is not exactly the same as the “in” conformation in the apo structure because the imidazole ring is flipped 180° so that the Nϵ2 atom can form an H-bond with WAT1 (distance 3.0 Å) for proton abstraction (Fig. 6a).
FIGURE 6.

ES* deacylation structure shows “revert-back” conformation of His-166 to serve as exclusive general base for deacylation. The active site of the ES* deacylation structure with “revert-back” conformation for His-166 at the start of the deacylation reaction (a) and tracked by time-dependent X-ray crystallography over a period of 6 min focusing on the disappearance of the acyl adduct (b). Fo − Fc omit map (purple) is drawn in mesh format and contoured at 2.0 σ. The dashed lines indicate hydrogen bonds.
Except for the “revert-in” movement of His-166, the rest of the active site remains largely unchanged when the active site progresses from ES*-acylation to ES*-deacylation. Asn-170 side chain is positioned very close to WAT1 (distance 2.5 Å), probably to provide a structural role in coordinating the deacylation water molecule (Fig. 6b). Lys-73 remains tilted away from Ser-70 and forms H-bonds with Ser-130 and Asn-132 instead (Fig. 6b).
We tracked the progression of the deacylation reaction by equilibrating soaked crystals in cephaloridine-free buffer for longer, i.e. 5 and 6 min before mounting them to X-ray source. In the “deacylation 5-min” structure the electron density for acyl adduct starts to deteriorate, particularly for the pyridine ring and the dihydrothiazine group, suggesting active in crystallo deacylation (Fig. 6b). In the “deacylation 6-min” structure only a small piece of electron density is visible that extends beyond the Oγ moiety of Ser-70, possibly representing the carbonyl group of cephaloridine (Fig. 6b). No continuous electron density can be identified for the rest of the cephaloridine molecule, suggesting that deacylation is nearly complete and the hydrolyzed substrate has departed from the active site. Additionally, the deacylation water molecule WAT1 is no longer present in the deacylation 6-min structure, whereas Lys-73 tilts back toward Ser-70 and re-establishes the Ser-70/Lys-73 interaction. Notably, the His-166 side chain is again flipped 180° from its conformation in the ES*-deacylation state and adopts a rotamer conformation that is identical to that in the apo structure with its Nϵ2 atom now facing the Lys-73 side chain to restore the Lys-73/His-166 H-bond (Fig. 6b). In summary, the deacylation 6-min structure marks the end of a full catalytic cycle, and the active site is fully restored to the apo-state.
Discussion
Comprehensive understanding of the catalytic mechanism of class A β-lactamases has been gained through structural studies that capture the active sites of these enzymes at various reaction intermediates by either mutating key catalytic residues or using unnatural ligands. For example, the enzyme-substrate complex state can be obtained by S70G and K73A mutations, whereas mutations like E166A and E166N stall the active site at the acylation state (21–25). Furthermore, non-hydrolyzable boronic acid derivatives have been used as ligands in the active site of class A β-lactamases to mimic the tetrahedral intermediate of the deacylation reaction (26, 27). However, findings from these functional intermediates have not successfully reconstituted a full catalytic cycle for class A β-lactamases because it is not known how the active site transits from one intermediate to the next along the reaction pathway.
First of all, it is not fully understood how Lys-73, protonated and thus catalytically not ready in the apo-state, can fulfill its role as general base in the acylation reaction of the catalytic cycle. QM/MM studies by Meroueh et al. (10) suggest that substrate-induced proton transfer from Lys-73 to Glu-166 via Ser-70 and a water molecule is energetically favorable and can render Lys-73 deprotonated to initiate the acylation reaction. Additionally, the study by Nichols et al. (28) observed a proton transfer from Ser-70 to Glu-166 in the active site of class A β-lactamase CTX-M upon binding of a non-electrophilic ligand, lending experimental support to this model. Very recently, Vandavasi et al. (29) reported for the first time structures of CTX-M in the acyl-enzyme complex with a monobactam, with Lys-73 indeed deprotonated to serve as general base for the acylation reaction. All these results support the model that the protonated Lys-73 in the apo-state can become deprotonated upon substrate binding to initiate the acylation reaction. However, the exact mechanism of such change, i.e. the proton transfer route, is not clear.
Here in our study, the unexpected “flipped-out” conformation of His-166 at the ES*-acylation state serves as “surrogate readout” to offer additional support of substrate-induced change in Lys-73 protonation state. Our MD analysis shows that only the flipped-out conformation, but not the “in” conformation of the apo structure, is compatible with deprotonated Lys-73 to serve as general base for the acylation reaction. Furthermore, our data entice us to speculate that the proton transfer from Lys-73 to His-166 occurs via their H-bond interaction. This speculated route is distinct from the one via Ser-70 or a water molecule as suggested by the QM/MM study of the wild-type TEM-1 enzyme (10) because the active site of E166H lacks the equivalent water molecule. Finally, in the wild-type active site, Glu-166 is unlikely to undergo the flipped-out motion like His-166 because Glu-166's protonated carboxylate side chain would be neutral and thus unlikely to be electrostatically expelled by Lys-73. Without the cumbersome movement, the wild-type active site can ensure ultrafast kinetics. In summary, the flipped-out motion of His-166 provides additional experimental evidence to confirm that the substrate-induced change in Lys-73 protonation state is a critical transition event to initiate the acylation reaction of the catalytic cycle within the wild-type active site (Fig. 7).
FIGURE 7.

Reaction scheme for wild-type class A β-lactamase after incorporating our findings regarding conformational changes for E166H. PDB codes reported in this work are 5GHX, 5GHY, and 5GHZ.
Another interesting finding from our studies concerns how Lys-73 alternates between the two subtly different conformations during the catalytic cycle, including the Ser-70-leaning conformation observed during the apo-state and upon the completion of the deacylation reaction as well as the Ser-130-leaning conformation observed in the ES*-acylation and ES*-deacylation structures. Multiple studies have reported these two alternative conformations in class A β-lactamases such as SHV-1 and Toho-1, mostly occurring within an active site that is acylation-competent but deacylation-stalled by mutation of Glu-166 or non-hydrolyzable substrates (24, 25, 30, 31). The importance of the Ser-70-leaning conformation is obvious because only such an arrangement would allow Lys-73 to abstract a proton from Ser-70 to initiate acylation. The functional significance of the Ser-130-leaning conformation is less understood and has been proposed to facilitate proton shuttling between Lys-73 and Ser-130 as part of the proton transfer path for the acylation reaction. Here, our study reveals that the Ser-130-leaning conformation is not only adopted by Lys-73 during the ES*-acylation state but also sustained through the ES*-deacylation state. Lys-73 only reverts back to the Ser-70-leaning conformation at the end of the catalytic cycle, when the ES* acyl adduct is hydrolyzed and the –OH group of Ser-70 side chains is regenerated. Thus, in addition to possibly participating in the proton shuttling pathway for acylation, the Ser-130-leaning conformation may serve to stabilize Lys-73 and suspend it from interfering with Glu-166's role of general base in the deacylation reaction (Fig. 7).
Additionally, our tracking of the in crystallo deacylation process uncovers two subtly different conformations for His-166 even when it is positioned inside the active site. In the apo structure the “in” conformation of His-166 allows it to form an H-bond with Lys-73 via the Nϵ atom, and in the ES*-deacylation structure the “revert-in” conformation of His-166 is flipped 180° so that its Nϵ atom is positioned toward the DW to serve as a general base for deacylation instead of forming an H-bond with Lys-73 (Fig. 7). This difference, reported for the first time by us, reveals that the Lys-73/His-166 interaction is suspended when His-166 serves as the general base for the deacylation reaction and is only re-established after completion of the catalytic cycle. For the wild-type active site, we propose that Glu-166 can alternate between Lys-73 and DW for H-bond or proton abstraction via adopting different resonant forms so that the titratable oxygen atom is positioned toward the correct partner (Fig. 7).
In summary, the subtle yet distinct conformational changes undergone by Lys-73 and His-166 through a full cycle of catalysis serve as “surrogate readout” to clarify the individual catalytic roles for Lys-73 and Glu-166 within the wild-type active site and uncover underappreciated coordination between these two key catalytic residues. These findings help to improve mechanistic understanding of this important family of enzymes that impact healthcare on a global scale.
Experimental Procedures
Protein Expression and Purification
Wild-type PenP was subcloned into a modified pET 30a vector containing an N-terminal His6 tag and the human rhinovirus 3C protease cleavage site. Substitutions of Glu-166 with other amino acid residues were done by site-directed mutagenesis (Finnzyme). Protein expression for the wild-type and substitution constructs were all done using E. coli strain BL21(DE3) following standard procedures. Briefly, inoculated bacteria cultures were grown at 37 °C until the OD600 reached 0.6–0.8; then protein expression was induced by adding isopropyl 1-thio-β-d-galactopyranoside at the final concentration of 500 μm, and the cell was grown at 30 °C for an additional 5 h and was collected by centrifugation. The His6-tagged PenP proteins were purified by HisTrap affinity column (GE Healthcare), and then protease 3C was used to cleave the tag. The target proteins were further purified by gel filtration chromatography (Superdex 75, GE Healthcare) in a buffer of 20 mm Tris (pH 7.5) and 50 mm NaCl. The desired fractions were collected and concentrated by Amicon® Ultra-15 centrifugal filter devices (Millipore nominal molecular weight limit = 10,000).
Nitrocefin Assay
0.0015 g of nitrocefin powder was dissolved in 200 μl of N,N-dimethylformamide and then diluted by 30-fold into 50 mm potassium phosphate (pH 7.0) to get the working solution. Then 50 μl of nitrocefin solution and 50 μl of protein solution in the same buffer were mixed; the absorbance of the mixture was measured at 500 nm over a time period of 200 s, and thus a curve of absorbance (A) against time was obtained. To get a linear correlation of absorbance to time, the protein concentration usually had to be optimized by serial dilutions. The activity of the enzyme then can be calculated using Equation 1,
| (Eq. 1) |
Whereas 1 unit (U) of enzyme is defined as 1 nmol of nitrocefin hydrolyzed by 1 mg of enzyme per min at 25 °C; Δϵ (nitrocefin) = 15,900 m−1 cm−1; ΔT is set as 10/3 min or 200 s; V indicates the volume of the mixture, and m is the amount of the enzyme added (units in mg).
ESI-MS Measurements for Enzyme Kinetics
The enzyme/substrate binding interaction was initiated by mixing 35 μl of 5 μm enzyme (E166H, E166W, and E166Q) in 20 mm ammonium acetate (pH 7.0) with 35 μl of 10 μm cephaloridine in the same buffer. At the desired time intervals, the reaction was quenched by addition of 70 μl of 1% formic acid (v/v) in acetonitrile. The resulting solution was characterized by ESI-MS. Normally there are two major peaks in the mass spectrum, one for the enzyme itself (E) the other for the acyl-adduct (ES*). The relative concentration of free enzyme ([E]) and the acyl-adduct ([ES*]) can be determined by integrating the area under measuring the intensity of these two peaks, whereas the total amount of enzyme ([Etotal]) can be calculated from the sum of [E] and [ES*] ([Etotal] = [E] + [ES*]). The deacylation constant (k3) then can be determined by fitting the value of [ES*]/[Etotal] versus time (t) to Equation 2.
| (Eq. 2) |
UV Spectroscopy Measurements for Enzyme Kinetics
Two antibiotics, penicillin G and cephaloridine, were used in this study, and their corresponding wavelength and extinction coefficient was as follows: penicillin G, 232 nm, Δϵ = 1100 m−1 cm−1; cephaloridine, 260 nm, Δϵ = 10.2 m−1 cm−1. A brief procedure is as follows. The reaction was started by mixing the substrate and protein in a buffer of 50 mm potassium phosphate (pH 7.0); the mixture was quickly submitted to detection by UV-visible spectrophotometer, and the absorbance change of this mixture within a time period of 5 min was monitored at the wavelength mentioned above. Six different concentrations of the substrate ([S]) were tested to give six absorbance versus time curves, each curve was used to calculate an initial hydrolysis rate (v0) according to the equation v0 = ΔA/ΔTΔϵ, and then all the v0 and [S] values were fitted into the Michaelis-Menten equation (Equation 3) to give the maximum reaction rate (Vmax) and Michaelis constant (Km).
| (Eq. 3) |
Furthermore the turnover number (kcat) was also obtained through the equation kcat = Vmax[E0], whereas [E0] is the initial concentration of enzyme.
Determination of pH Profile for PenP WT and E166H Mutant
The assay was performed by monitoring initial velocities of nitrocefin hydrolysis at a range of substrate concentrations and pH conditions. The buffers used for the experiment were 50 mm sodium acetate (pH 5–6), 50 mm sodium phosphate (pH 6–7), 50 mm Tris (pH 7–9), 50 mm glycine (pH 9–10.5), and 50 mm sodium bicarbonate (pH 11). Each buffer was supplemented with 150 mm NaCl. Initial velocity data were analyzed with GraphPad Prism 5 and fitted to the Michaelis-Menten equation. The pH dependence of kcat/Km was fitted to Equation 4 (27).
| (Eq. 4) |
Crystallization and Structure Determination
The crystals of PenP E166H were grown at 16 °C by the hanging drop vapor diffusion method. One μl of protein solution in a buffer of 20 mm Tris, 50 mm NaCl (pH 7.5) was mixed with 1 μl of reservoir buffer, which was composed of 0.1 m Tris (pH 8.0), 22.5% PEG3350, 0.4 m ammonium acetate. Crystals appeared in 4–5 days and were harvested after they had grown to ∼100 μm in size. Cephaloridine was soaked into the crystals by incubating E166H apo-crystals in reservoir buffer containing 0.1 m cephaloridine for 7 min. To obtain the deacylation intermediates, the crystals of E166H were soaked in 0.1 m cephaloridine-containing reservoir buffer first and then transferred to cephaloridine-free reservoir buffer for various time periods before mounting to the in-house Rigaku MicroMaxTM-007HF X-ray machine for data collection. Diffraction data were collected at 100 K, integrated by Mosflm (32), and scaled by the SCALA module (33) in CCP4. All the structures were solved by molecular replacement using the PHASER module in the CCP4i suite of programs with PenP wild-type structure (PDB code 4BLM) as search model (34). The subsequent structural refinement was conducted using REFMAC module in CCP4 (35). Manual structure rebuilding was done using WINCOOT (36). Data collection and refinement statistics are summarized in Table 2. The coordinates of all structures have been deposited to Protein Data Bank with the respective PDB code listed in Table 2. The structure figures were prepared using the CCP4mg package (37) in CCP4.
Computational Details
The initial conformation for E166H was built upon the ES*-acylation structure (PDB code 5HGY, see Fig. 3a). The protonation states of the ionizable residues were determined at pH 7 based on pKa calculations via both PROPKA (38) and H++ (39) programs. If these two programs produce inconsistent predictions, we also take into account the local hydrogen bonding network. The initial enzyme-substrate complex was constructed by docking the ligand CED into the same acyl adduct structure using Autodock 4.2 (40). The partial charges of CED were fitted with HF/6–31G(d) (32) calculations using the RESP module (32) in the AmberTools package. The whole system was solvated into explicit TIP3P water molecules (42) using a cubic box with a 12 Å buffer distance between the box wall and its nearest solute atom, and six Na+ ions were added to neutralize the charge. As a result, the whole system contains ∼54,000 atoms. We then followed the same sophisticated protocol as in a previous study (43) to conduct energy minimization and equilibration. In particular, we first performed a 2,500-step of steepest descent minimization followed by a 2,500-cycle conjugate gradient minimization by restraining the protein and ligand (with restraint force constant of 50 kcal·mol−1·Å−2). We then performed a 150-ps NVT equilibration simulation (T = 10 K) followed by another NPT (p = 1 atm) equilibration simulation, during which the restraint force constant was gradually decreased to 25 kcal·mol−1·Å−2. Next, we performed a 250-ps temperature annealing NVT simulation (T was raised from 10 K to 300 K), during which we also reduced the restraint force constant to 10 kcal·mol−1·Å−2. Finally, we performed two sequential 150-ps NPT simulations to further reduced the restraint force constant to 1 kcal·mol−1·Å−2, and then finally to zero. Three independent production NPT (T = 300 K and p = 1 atm) simulations were then carried out for 10 ns with different initial velocities. In all MD simulations, long range electrostatic interactions were treated with the particle mesh Ewald (PME) method (44), and a 12 Å cutoff was used for both PME short range and van der Waals interactions. The Velocity-rescaling thermostat (45) (with coupling constant of 0.1 ps−1), and the Parrinello-Rahman barostat (46) (with the coupling constant of 1.0 ps−1) were adopted for temperature and pressure coupling, respectively. All the MD simulations were performed using the AMBER12 (47) molecular dynamic package, and the Amber99SB-ILDN (41) force field was employed.
Author Contributions
X. P. cloned and purified the protein, solved the structures, and carried out the in vitro biophysical and biochemical assays. Y. H. assisted with X-ray crystallography. J. L. and X. H. conducted MD simulations. Y. Z. designed the experiments, analyzed the data, supervised the project, and wrote the manuscript. All authors have given approval to the final version of the manuscript. The authors declare no competing financial interests.
Supplementary Material
Acknowledgment
We thank Shanghai Synchrotron Radiation Facility (SSRF) beam line BL17U for help with data collection.
This work was supported by Research Grants Council GRF Grants PolyU 5640/11M and PolyU 151043/14M, Research Grants Council CRF Grants C5030-14E and C6009-15G, Research Grants Council AoE Grant AoE/M-09/12, Health and Medical Research Fund Grant 14130502, and the Research Committee of Hong Kong Polytechnic University. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Fig. S1.
The atomic coordinates and structure factors (codes 5GHX, 5GHY, and 5GHZ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- QM/MM
- quantum mechanics/molecular mechanics
- r.m.s.d.
- root mean square deviation
- DW
- deacylation water
- PDB
- Protein Data Bank
- MD
- molecular dynamics.
References
- 1. Drawz S. M., and Bonomo R. A. (2010) Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 23, 160–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Papp-Wallace K. M., Endimiani A., Taracila M. A., and Bonomo R. A. (2011) Carbapenems: past, present, and future. Antimicrob. Agents Chemother. 55, 4943–4960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Molton J. S., Tambyah P. A., Ang B. S., Ling M. L., and Fisher D. A. (2013) The global spread of healthcare-associated multidrug-resistant bacteria: a perspective from Asia. Clin. Infect. Dis. 56, 1310–1318 [DOI] [PubMed] [Google Scholar]
- 4. Majiduddin F. K., Materon I. C., and Palzkill T. G. (2002) Molecular analysis of β-lactamase structure and function. Int. J. Med. Microbiol. 292, 127–137 [DOI] [PubMed] [Google Scholar]
- 5. Massova I., and Mobashery S. (1998) Kinship and diversification of bacterial penicillin-binding proteins and β-lactamases. Antimicrob. Agents Chemother. 42, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fisher J. F., and Mobashery S. (2009) Three decades of the class A β-lactamase acyl-enzyme. Curr. Protein Pept. Sci. 10, 401–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nukaga M., Mayama K., Hujer A. M., Bonomo R. A., and Knox J. R. (2003) Ultrahigh resolution structure of a class A β-lactamase: on the mechanism and specificity of the extended-spectrum SHV-2 enzyme. J. Mol. Biol. 328, 289–301 [DOI] [PubMed] [Google Scholar]
- 8. Minasov G., Wang X., and Shoichet B. K. (2002) An ultrahigh resolution structure of TEM-1 β-lactamase suggests a role for Glu-166 as the general base in acylation. J. Am. Chem. Soc. 124, 5333–5340 [DOI] [PubMed] [Google Scholar]
- 9. Golemi-Kotra D., Meroueh S. O., Kim C., Vakulenko S. B., Bulychev A., Stemmler A. J., Stemmler T. L., and Mobashery S. (2004) The importance of a critical protonation state and the fate of the catalytic steps in class A β-lactamases and penicillin-binding proteins. J. Biol. Chem. 279, 34665–34673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meroueh S. O., Fisher J. F., Schlegel H. B., and Mobashery S. (2005) Ab initio QM/MM study of class A β-lactamase acylation: dual participation of Glu-166 and Lys-73 in a concerted base promotion of Ser-70. J. Am. Chem. Soc. 127, 15397–15407 [DOI] [PubMed] [Google Scholar]
- 11. Pan X., Wong W. T., He Y., Jiang Y., and Zhao Y. (2014) Perturbing the general base residue Glu-166 in the active site of class A β-lactamase leads to enhanced carbapenem binding and acylation. Biochemistry 53, 5414–5423 [DOI] [PubMed] [Google Scholar]
- 12. Wong W. T., Au H. W., Yap H. K., Leung Y. C., Wong K. Y., and Zhao Y. (2011) Structural studies of the mechanism for biosensing antibiotics in a fluorescein-labeled β-lactamase. BMC Struct. Biol. 11, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wong W. T., Chan K. C., So P. K., Yap H. K., Chung W. H., Leung Y. C., Wong K. Y., and Zhao Y. (2011) Increased structural flexibility at the active site of a fluorophore-conjugated β-lactamase distinctively impacts its binding toward diverse cephalosporin antibiotics. J. Biol. Chem. 286, 31771–31780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao Y., and Yang W. (2016) Capture of a third Mg2+ is essential for catalyzing DNA synthesis. Science 352, 1334–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakamura T., Zhao Y., Yamagata Y., Hua Y. J., and Yang W. (2012) Watching DNA polymerase η make a phosphodiester bond. Nature 487, 196–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barends T. R., Foucar L., Ardevol A., Nass K., Aquila A., Botha S., Doak R. B., Falahati K., Hartmann E., Hilpert M., Heinz M., Hoffmann M. C., Köfinger J., Koglin J. E., Kovacsova G., et al. (2015) Direct observation of ultrafast collective motions in CO myoglobin upon ligand dissociation. Science 350, 445–450 [DOI] [PubMed] [Google Scholar]
- 17. Stojanoski V., Chow D. C., Hu L., Sankaran B., Gilbert H. F., Prasad B. V., and Palzkill T. (2015) A triple mutant in the Ω-loop of TEM-1 β-lactamase changes the substrate profile via a large conformational change and an altered general base for catalysis. J. Biol. Chem. 290, 10382–10394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chan P. H., So P. K., Ma D. L., Zhao Y., Lai T. S., Chung W. H., Chan K. C., Yiu K. F., Chan H. W., Siu F. M., Tsang C. W., Leung Y. C., and Wong K. Y. (2008) Fluorophore-labeled β-lactamase as a biosensor for β-lactam antibiotics: a study of the biosensing process. J. Am. Chem. Soc. 130, 6351–6361 [DOI] [PubMed] [Google Scholar]
- 19. Guillaume G., Vanhove M., Lamotte-Brasseur J., Ledent P., Jamin M., Joris B., and Frère J. M. (1997) Site-directed mutagenesis of glutamate 166 in two β-lactamases. Kinetic and molecular modeling studies. J. Biol. Chem. 272, 5438–5444 [DOI] [PubMed] [Google Scholar]
- 20. Chen C. C., and Herzberg O. (2001) Structures of the acyl-enzyme complexes of the Staphylococcus aureus β-lactamase mutant Glu166Asp:Asn170Gln with benzylpenicillin and cephaloridine. Biochemistry 40, 2351–2358 [DOI] [PubMed] [Google Scholar]
- 21. Chen C. C., Smith T. J., Kapadia G., Wäsch S., Zawadzke L. E., Coulson A., and Herzberg O. (1996) Structure and kinetics of the β-lactamase mutants S70A and K73H from Staphylococcus aureus PC1. Biochemistry 35, 12251–12258 [DOI] [PubMed] [Google Scholar]
- 22. Adamski C. J., Cardenas A. M., Brown N. G., Horton L. B., Sankaran B., Prasad B. V., Gilbert H. F., and Palzkill T. (2015) Molecular basis for the catalytic specificity of the CTX-M extended-spectrum β-lactamases. Biochemistry 54, 447–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tremblay L. W., Xu H., and Blanchard J. S. (2010) Structures of the Michaelis complex (1.2 A) and the covalent acyl intermediate (2.0 A) of cefamandole bound in the active sites of the Mycobacterium tuberculosis β-lactamase K73A and E166A mutants. Biochemistry 49, 9685–9687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vandavasi V. G., Weiss K. L., Cooper J. B., Erskine P. T., Tomanicek S. J., Ostermann A., Schrader T. E., Ginell S. L., and Coates L. (2016) Exploring the mechanism of β-lactam ring protonation in the class A β-lactamase acylation mechanism using neutron and X-ray crystallography. J. Med. Chem. 59, 474–479 [DOI] [PubMed] [Google Scholar]
- 25. Ibuka A. S., Ishii Y., Galleni M., Ishiguro M., Yamaguchi K., Frère J. M., Matsuzawa H., and Sakai H. (2003) Crystal structure of extended-spectrum β-lactamase Toho-1: insights into the molecular mechanism for catalytic reaction and substrate specificity expansion. Biochemistry 42, 10634–10643 [DOI] [PubMed] [Google Scholar]
- 26. Strynadka N. C., Martin R., Jensen S. E., Gold M., and Jones J. B. (1996) Structure-based design of a potent transition state analogue for TEM-1 β-lactamase. Nat. Struct. Biol. 3, 688–695 [DOI] [PubMed] [Google Scholar]
- 27. Ness S., Martin R., Kindler A. M., Paetzel M., Gold M., Jensen S. E., Jones J. B., and Strynadka N. C. (2000) Structure-based design guides the improved efficacy of deacylation transition state analogue inhibitors of TEM-1 β-lactamase. Biochemistry 39, 5312–5321 [DOI] [PubMed] [Google Scholar]
- 28. Nichols D. A., Hargis J. C., Sanishvili R., Jaishankar P., Defrees K., Smith E. W., Wang K. K., Prati F., Renslo A. R., Woodcock H. L., and Chen Y. (2015) Ligand-induced proton transfer and low-barrier hydrogen bond revealed by X-ray crystallography. J. Am. Chem. Soc. 137, 8086–8095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vandavasi V. G., Langan P. S., Weiss K. L., Parks J. M., Cooper J. B., Ginell S. L., and Coates L. (2017) Active site protonation states in an acyl-enzyme intermediate of a class A β-lactamase with a monobactam substrate. Antimicrob. Agents Chemother. 61, e01636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tomanicek S. J., Blakeley M. P., Cooper J., Chen Y., Afonine P. V., and Coates L. (2010) Neutron diffraction studies of a class A β-lactamase Toho-1 E166A/R274N/R276N triple mutant. J. Mol. Biol. 396, 1070–1080 [DOI] [PubMed] [Google Scholar]
- 31. Nukaga M., Bethel C. R., Thomson J. M., Hujer A. M., Distler A., Anderson V. E., Knox J. R., and Bonomo R. A. (2008) Inhibition of class A β-lactamases by carbapenems: crystallographic observation of two conformations of meropenem in SHV-1. J. Am. Chem. Soc. 130, 12656–12662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J. M., Cieplak P., and Kollman P. A. (2000) How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 21, 1049–1074 [Google Scholar]
- 33. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 34. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. Sect. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murshudov G. N., Skubak P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 37. Potterton L., McNicholas S., Krissinel E., Gruber J., Cowtan K., Emsley P., Murshudov G. N., Cohen S., Perrakis A., and Noble M. (2004) Developments in the CCP4 molecular-graphics project. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2288–2294 [DOI] [PubMed] [Google Scholar]
- 38. Rostkowski M., Olsson M. H., Søndergaard C. R., and Jensen J. H. (2011) Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct. Biol. 11, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gordon J. C., Myers J. B., Folta T., Shoja V., Heath L. S., and Onufriev A. (2005) H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 33, W368–W371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., and Olson A. J. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J. L., Dror R. O., and Shaw D. E. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jorgensen W. L. C. J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 43. Lei J., Zhou Y., Xie D., and Zhang Y. (2015) Mechanistic insights into a classic wonder drug–aspirin. J. Am. Chem. Soc. 137, 70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 45. Bussi G., Donadio D., and Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 [DOI] [PubMed] [Google Scholar]
- 46. Parrinello M., and Rahman A. (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 [Google Scholar]
- 47. Case D. A., Darden T. A., Cheatham T. E. III, Simmerling C. L., Wang J., Duke R. E., Luo R. C., Walker W., Zhang K. M., Merz B., Roberts S., Hayik S., Roitberg A., Seabra G., Swails J., et al. (2012) AMBER12, University of California, San Francisco [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.