

Abstract
Introduction
High-titer Factor VIII (FVIII) inhibitors complicate peri-operative hemostasis. Recombinant porcine FVIII (r-pFVIII) may provide an alternative hemostatic agent for high-risk procedures and allow FVIII activity monitoring.
Aim
Devise an effective hemostatic plan for repair of a progressively symptomatic aortic coarctation in a 5-year-old male with immune tolerance induction (ITI) refractory high-titer FVIII inhibitors.
Methods
Pre-procedure human FVIII inhibitor titer was 58 Bethesda Units/ml (BU) and cross-reacted to neutralize porcine FVIII at 30 BU. Daily ITI with plasma-derived FVIII concentrate was supplemented with anti-B-cell and anti-plasma cell immunotherapy to reduce FVIII inhibitor titers. Potential hemostatic agents were evaluated in comparative ex vivo thrombin generation assays (TGA).
Results
Four weeks after immunosuppression human and porcine inhibitor titers declined to 16 and 2 BU, respectively. TGA with r-pFVIII was less robust than with activated Prothrombin Complex Concentrate (aPCC); however, r-pFVIII was selected for cardiac surgery to secure the ability to assay FVIII levels throughout this high bleeding risk procedure. Hemostasis with r-pFVIII was excellent; initial trough FVIII activity levels ranged from 0.81–1.17 IU/ml. On post-operative day 3, peak and trough levels markedly declined suggesting a rising porcine inhibitor titer. Post-procedure prophylaxis was transitioned to aPCC, informed by TGA.
Conclusions
R-pFVIII provided effective peri-procedural hemostasis with no adverse events. Rapid neutralization of r-pFVIII after the first 60 hours, despite intensive immune suppression, accentuates the importance of careful monitoring. Use of TGA can support bypassing agent selection for convalescence. The comparative cost of r-pFVIII may limit its use to high morbidity clinical scenarios.
Keywords: bypassing agents, r-pFVIII, rFVIIa, FEIBA, Obizur
Introduction
The presence of high-titer neutralizing alloantibodies (‘inhibitors’) complicates the peri-operative hemostasis management for patients with hemophilia. Bypassing agents (BPA), activated prothrombin complex concentrate (aPCC) and recombinant activated factor VII (rFVIIa) currently provide the mainstay of treatment [1–3]; however, inconsistent hemostatic effect and lack of validated laboratory assays for pharmacodynamic monitoring limit the advantages of these agents [4, 5]. Recombinant porcine FVIII (r-pFVIII, OBIZUR, Shire) received orphan drug designation for acquired hemophilia A (AHA) in adults and was licensed in 2014 [6, 7]. The majority of human FVIII (hFVIII) inhibitors targets the A2 and C2 domains, creating allosteric hindrance to activation. The reduced sequence homology between hFVIII and porcine FVIII (pFVIII) at these domains (84% and 76%, respectively) enables effective pFVIII replacement in humans [8].
Limited studies, focused primarily on comparative pharmacokinetic profiles with plasma derived pFVIII (pd-pFVIII, Hyate-C, Ipsen), were conducted in patients with congenital hemophilia A and inhibitors until its production was discontinued in 2004. R-pFVIII demonstrated greater recovery and AUC compared to pd-pFVIII and a mean half-life of 11 hours [9]. Even in the absence of prior exposure to pFVIII, hFVIII inhibitors can cross-neutralize pFVIII activity, impacting the potential efficacy of r-pFVIII [6, 9]. Use of global measures of hemostasis, such as thrombin generation assays, may guide selection of the optimal peri-procedure hemostatic agent for a given patient to minimize risk of bleeding [10, 11].
While use of pd-pFVIII has been well described for congenital hemophilia with inhibitors, minimal investigation of r-pFVIII has been conducted in this population [12]. Here, we present our experience using r-pFVIII for a pediatric patient with high-titer inhibitors requiring cardiac surgery for repair of progressively symptomatic hypoplastic aortic arch and coarctation.
Materials and Methods
Patient
The patient was a 5-year-old male diagnosed with severe hemophilia A (nonsense mutation in the F8 gene (FVIII c.6853C>T)) and emergence of very high titer inhibitors at age 14 months. Peak inhibitor titer was 563 BU prior to any attempt at immune tolerance induction (ITI). An incidental diagnosis of hypoplastic transverse arch and aortic coarctation was made during an echocardiogram for central venous line (CVL) evaluation at 18-months of age. Plans for an open heart surgical approach included the anticipated need for cardiopulmonary bypass and intra-operative anti-coagulation. Definitive repair was initially deferred due to lack of systemic hypertension, the acceptable ascending to descending aortic gradient, and the significant bleed risk with cardiac repair secondary to his high-titer FVIII inhibitors.
During the interval between high-titer inhibitors development and the patient’s need for aortic repair due to symptom progression, several attempts at ITI were made. Initially a high-dose daily FVIII concentrate regimen was used, then immunosuppression regimens, including mycophenolate as monotherapy and later combined B and T cell immune modulation with rituximab, mycophenolate, intravenous immunoglobulin, and dexamethasone as per Beutel et al, were added [13]. The peak inhibitor titer following start of ITI #1 was 3,967 BU. Despite multiple approaches to ITI spanning more than 2 years, high-titer inhibitors persisted (30–100 BU), [Fig.1A]. Use of rFVIIa for daily prophylaxis and peri-operatively for CVL placement yielded suboptimal clinical outcomes with breakthrough musculoskeletal hemorrhages and poor post-operative hemostasis. The patient subsequently underwent operative central venous port-a-cath replacement under coverage of aPCC without bleeding complications.
Fig 1.


Longitudinal course of patient’s hFVIII and pFVIII inhibitor titers. Star (*) denotes the date of the patient’s cardiac surgery and first exposure to porcine factor VIII. A. hFVIII inhibitor titers over the patient’s extended clinical course. ITI #1 rFVIII 100 IU/kg daily and mycophenolate; ITI #2 pd-FVIII 200 IU/kg daily and combined B- and T-cell immunosuppression per Beutel et al [13]; ITI #3 repeat Beutel regimen as per ITI #2; ITI #4 pd-FVIII 100 IU/kg daily, bortezomib, rituximab [15]. B. Trend of anti-porcine FVIII titer (open squares, □) before and after first exposure to any pFVIII product; also shown are the concurrent anti-human FVIII inhibitor titers (closed circles, ●).
Thrombin Generation Measurements
Given the anticipated need for definitive aortic repair despite persistent high-titer inhibitor, pre-operative characterization of the patient’s thrombin generation in response to bypassing agents was evaluated. Blood collection and analyses were performed after obtaining approval from the University of North Carolina Institutional Review Board and informed consent. TGA were performed using both platelet-rich (data not shown) and platelet-poor plasma to characterize the relative thrombin generation of r-pFVIII, rFVIIa and aPCC at a time when the patient’s hFVIII and pFVIII inhibitor titers were 37 BU and 3 BU, respectively. This patient had no prior exposure to porcine FVIII.
Blood collected from the patient at baseline (prior to infusion of his daily ITI dose of FVIII/VWF concentrate) was spiked in vitro with r-pFVIII, rFVIIa, or aPCC. The final assay concentrations were chosen to reproduce clinically relevant dosing. R-pFVIII was tested at 2 IU/ml and 4 IU/ml (equivalent to doses of 100 IU/kg and 200 IU/kg), rFVIIa was tested at 25 nM, 50 nM, and 75 nM (equivalent to doses of 90 µg/kg, 180 µg/kg, and 270 µg/kg), and aPCC was tested at 1 IU/ml and 2 IU/ml (equivalent to doses of 50 IU/kg and 100 IU/kg). Reactions with platelet-rich and platelet-poor plasma were performed with 1 pM tissue factor and 1 pM tissue factor per 4 µM phospholipids, respectively, [14].
Pre-operative ITI and immunosuppressive regimen
Subsequent to the thrombin generation measurements, an interval increase in both human and porcine inhibitor titers was observed, hFVIII inhibitor titer 58 BU and pFVIII inhibitor 30 BU, despite absence of exposure to pFVIII. Prophylaxis and bleed treatment with aPCC and ITI were ongoing. . Safety and efficacy of r-pFVIII has not been established in patients with a porcine factor VIII inhibitor titer of greater than 20 BU [7]. A pre-operative attempt to minimize both human and porcine inhibitor titers was begun with ITI consisting of continuing antihemophilic factor/von Willebrand factor complex with the addition a single dose of rituximab 375 mg/m2 on day 1 and bortezomib 1.3 mg/m2 on days 1, 4, 8, 11 (following the regimen of Khandelwal et al. however omitting plasmapheresis) [15]. Following administration of initial immunosuppressive agents human and porcine anti-FVIII titers were measured every two weeks.
Peri-operative hemostasis measurements
Human and porcine Bethesda Inhibitor Assays and clinical thromboelastography (TEG) were performed at the McLendon Clinical Laboratories at the University of North Carolina, (TEG-analyzer, Haemonetcis Corp., Braintree, MA, United States), trigger kaolin. Intra-operative and post-operative one-stage FVIII activity assays were performed in the clinical coagulation laboratory at Boston Children’s Hospital.
Results
By the age of 5 years an increasing coarctation gradient indicated that aortic repair was necessary. The high bleed risk of the cardiac repair to be performed and possible need for thoracotomy and cardiac bypass made desirable the ability to monitor therapeutic FVIII levels and respond with tailored dosing as needed. Inhibitor titers were confirmed to have decreased to 16 BU hFVIII and 2 BU pFVIII at 4 weeks following bortezomib and rituximab and 2 weeks prior to the patient’s aortic repair, [Fig.1B].
Prospective hemostatic evaluation to support cardiac repair and recovery
Evaluation of thrombin generation in the patient’s platelet-poor plasma in response to r-pFVIII or bypassing agent ex vivo supplementation, collected at a baseline state of health, [Fig 2A]. The patient’s plasma demonstrated minimal thrombin generation in response to rFVIIa and no dose-response, consistent with his clinical history of recurrent and post-operative bleeding with the use of this agent. Thrombin generation following addition of r-pFVIII was modest, however, a dose-response was evident. Activated PCC, in contrast, led to substantially increased and dose-dependent thrombin generation. Plasma collected at the time of the patient’s pre-operative dose of aPCC for port-a-cath removal and replacement months before was assayed ex vivo by TGA in parallel with his baseline plasma (either untreated or spiked with aPCC 2 IU/ml), confirming good thrombin generation following in vivo infusion of aPCC, [Fig. 2B]. The observed potential to normalize clot parameters using aPCC was also confirmed by whole blood TEG performed ex vivo following aPCC infusion [Fig. 2C]. The in vitro and ex vivo TGA evaluation, the clinical TEG results, and the hemostatically benign clinical course during CVL placement and healing all suggested aPCC may provide hemostatic support during the anticipated weeks of normal wound healing following aortic repair. Nevertheless, the opportunity to monitor and manage FVIII activity levels led to the choice of r-pFVIII for hemostasis during the aortic repair.
Fig. 2.
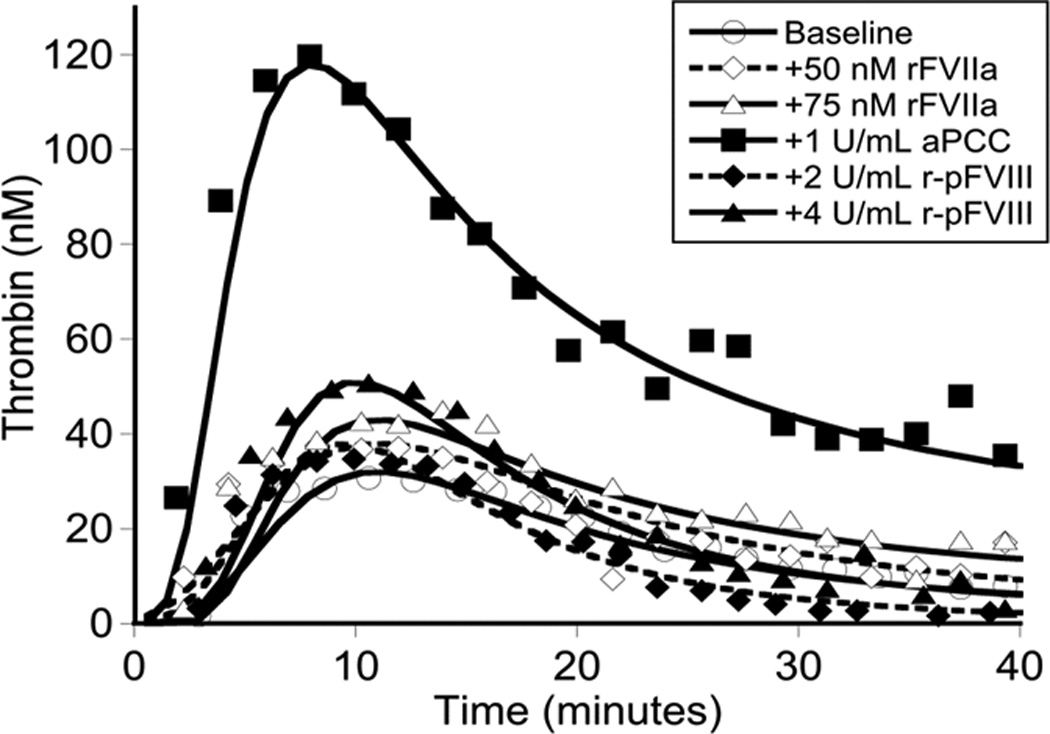
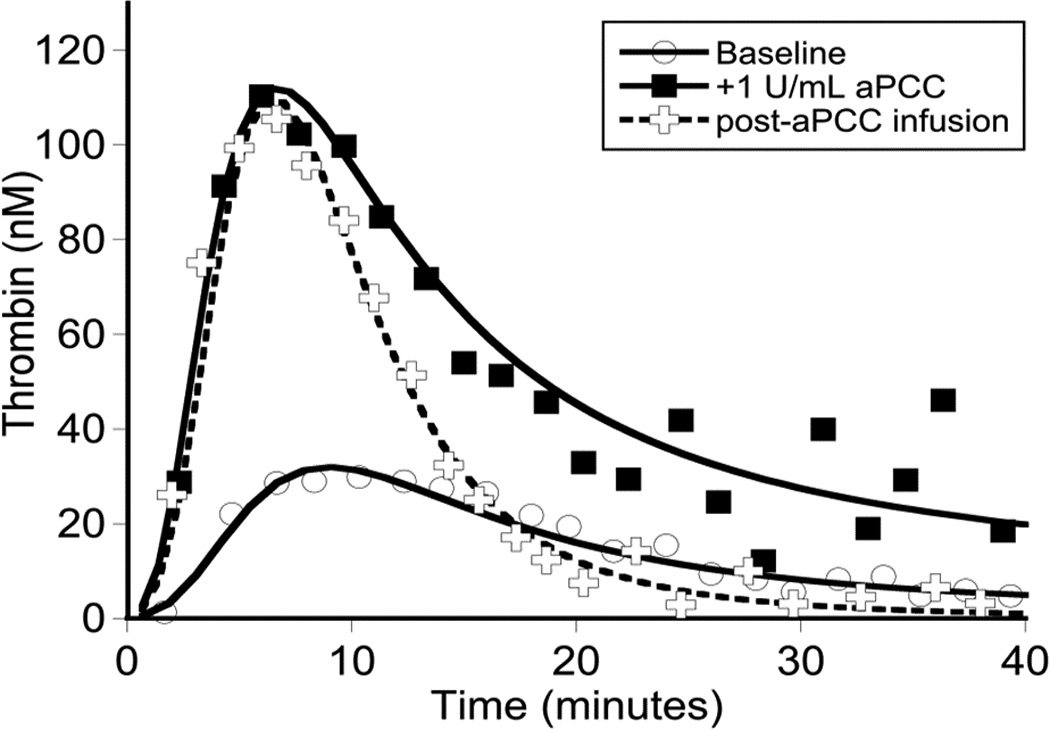
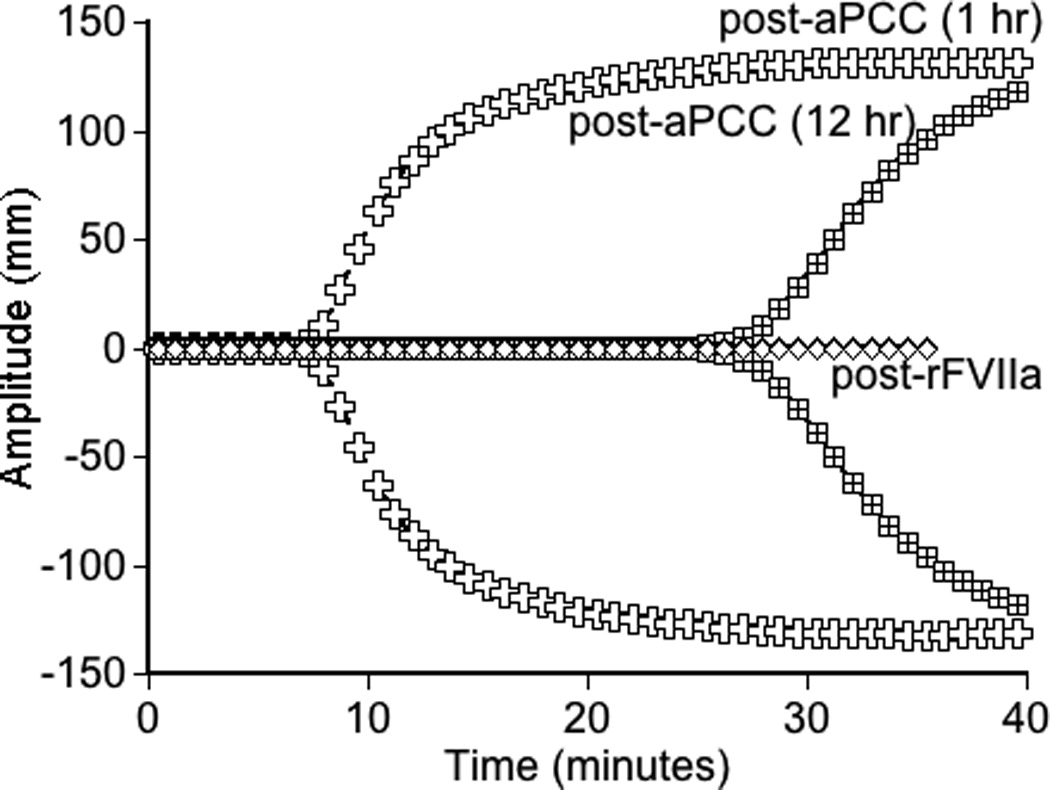
Global assays of the patient’s hemostatic potential at baseline and in response to rFVIIa, aPCC, and r-pFVIII were evaluated. At time of plasma sampling the patient’s hFVIII inhibitor titer was 37 BU and pFVIII inhibitor titer was 3 BU. A. Thrombin generation of the patient’s platelet poor plasma at baseline and following in vitro spiking with hemostatic agents. The final concentration of rFVIIa was chosen to reproduce a clinical dose of 180 µg/kg or 270 µg/kg. The final concentration of aPCC was chosen to reproduce a clinical dose of 100 IU/kg. The final concentration of r-pFVIII was chosen to reproduce a clinical dose of 100 IU/kg or 200 IU/kg. B. Thrombin generation examined in the patient’s platelet poor plasma at baseline and after ex vivo spiking with 1 IU/ml aPCC compared to the patient’s platelet poor plasma sampled after in vivo infusion of 100 IU/kg of aPCC to provide hemostatic coverage for CVL removal/replacement. C. TEG assayed in whole blood collected 1 hour and 12 hours following pre-operative infusion of 100 IU/kg of aPCC, at the same time as plasma was assayed in panel B. Also assayed in the clinical coagulation laboratory was whole blood collected at 15 minutes following in vivo infusion of 270 µg/kg of rFVIIa, demonstrating a TEG tracing that did not differ from the patient’s baseline.
Operative and post-operative hemostatic management
The pre-incision r-pFVIII dose, FVIII activity assay monitoring plan, and subsequent r-pFVIII dosing schedule for this pediatric patient were developed by considering the reported experience with r-pFVIII in acquired hemophilia A [6, 7], prior description of r-pFVIII pharmacokinetic profile in adolescent and adult patients with congenital hemophilia [9, 16, 17], and the patient’s baseline porcine inhibitor titer. The absence of available product stability testing prevented use of continuous r-pFVIII infusion for this case. A pre-procedure r-pFVIII test dose was not administered to characterize recovery or estimate half-life for this patient due to concern for prematurely stimulating an anamnestic increase in the porcine inhibitor titer [16, 18].
At incision, r-pFVIII 170 IU/kg was administered; FVIII activity was 0.85 IU/ml at 60 minutes post-infusion. A left posterolateral muscle-sparing thoracotomy and repair of the aortic arch with resection of hypoplastic segment and end-to-end aortic anastomosis were performed. Sternotomy and cardiopulmonary bypass were ultimately not required. The patient achieved excellent hemostasis throughout the 3-hour procedure. Subsequent r-pFVIII dosing at 100 IU/kg/dose started at an interval of every three hours, then the time between infusions was increased based on trough levels and clinical examination for bleeding [Fig. 3]. The target trough FVIII level for the first 24 hours post-procedure was greater than 0.8 IU/ml and thereafter greater than 0.5 IU/ml. Limited vial size availability (only nominal concentration 500 U/mL) and lag time in reporting trough levels constrained weaning the dose of r-pFVIII. Following the initial r-pFVIII dose, the patient received 3 doses at 3 hour intervals, 2 doses at 4 hour intervals, 1 dose at a 6 hour interval, then 5 doses at 8 hour intervals.
Fig. 3.

Peri-operative r-pFVIII replacement schedule and resulting peak and trough FVIII activity levels. Peri-operative dosing of r-pFVIII (closed triangles, ▼) was initiated at a dose of 170 IU/kg at the time of incision for aortic repair, hour 0. Subsequent doses of 100 IU/kg were administered at increasing intervals as indicated. Peak FVIII activity levels (open diamonds, ◊) and trough FVIII activity levels (closed squares, ▪) sampled around early post-operative doses and at later points when r-pFVIII activity was less sustained.
Initially trough FVIII activities ranged from 0.81–1.17 IU/ml; however, at the time of his tenth dose (day 3, fourth dose on an every 8-hour schedule), the trough level plummeted to 0.13 IU/ml with subsequent recovery of 1.13 IU/ml (49% of expected) following dose 11. Arterial line, chest tube and internal jugular venous catheter were removed. The subsequent trough and peak FVIII activities around dose 12 were 0.1 and 0.69 IU/ml (34% of expected), respectively. Given evidence of increasing r-pFVIII neutralization, hemostatic treatment was transitioned to aPCC 85 IU/kg/dose every 12 hours on post-procedure day 3. The patient continued to have excellent hemostasis and was discharged home by day 5 post-procedure.
Plasma for FVIII inhibitor titers was drawn at first concern for rising inhibitor and assayed at 12.5 BU human and 0.5 BU porcine. Subsequent titers measured 12 days post-procedure were 64 BU human and 13 BU porcine. These titers waned over the subsequent 4 weeks to 50 BU and 5 BU, respectively. The patient continues ITI with antihemophilic factor/von Willebrand factor complex (153 IU/kg/day) and aPCC prophylaxis (88 IU/kg every other day).
Discussion
The emergence of pd-pFVIII in the 1960s provided a therapeutic option for congenital hemophilia A patients with life-threatening bleeds at a time when concentrated human FVIII products were scarce. Despite improved fractionation procedures, which reduced common adverse events of hypersensitivity reactions and thrombocytopenia [8, 12, 18], the commercially available product, Hyate-C, discontinued production in 2004 due to concern for viral contamination (porcine parvovirus and retroviruses) [19]. The recent licensure of a recombinant porcine FVIII product for AHA has again expanded therapeutic options for patients with FVIII inhibitors; however, this product has not yet been well studied in the congenital hemophilia patients.
This case demonstrates the successful off-label use (by indication, age, and dosing) of r-pFVIII. Alloantibodies to FVIII pose the greatest challenge to the treatment of hemorrhage and perioperative hemostasis management in patients with hemophilia. The inability to anticipate and monitor the hemostatic effect of BPA makes their use in the management of high-risk bleeding challenging. R-pFVIII offers an alternate option where measurement of therapeutic effect through a one-stage clotting assay supports optimal clinical outcomes, namely effective hemostasis and protection from recurrent hemorrhage.
Comparative use of r-pFVIII in acquired hemophilia and congenital hemophilia with inhibitors
The dosing strategy used in the pivotal study for AHA included an initial dose of 200 IU/kg followed by subsequent doses of a median of 100 IU/kg approximately every 8 hours to maintain FVIII activity levels >50% (or as desired). Approximately one third of patients with AHA demonstrated positive pFVIII titers prior to exposure to r-pFVIII, with a mean cross-reactivity in this patient subset of 24.5% (porcine FVIII inhibitor titer/human FVIII inhibitor titer) [6].
Therapeutic FVIII activity levels were achieved upon repeated doses of r-pFVIII even in the patients with cross-reactivity. This trend was also observed in our patient. While the presence of cross-reacting FVIII inhibitors is generally not established at initial hemorrhagic presentation of AHA, preemptive testing for porcine inhibitor titers may be completed for congenital hemophilia patients with inhibitors prior to r-pFVIII use. The current label recommended dosing guidance is tailored toward the acquired hemophilia population. Care must be taken in the congenital hemophilia population to investigate an optimal approach to dosing and infusion schedule. Historical experience with pd-pFVIII provides some guidance; however, the pharmacokinetic profiles of pd-pFVIII and r-pFVIII are not identical.
Comparative costs of therapeutic options
Accessibility and cost of care are necessary considerations. The inpatient cost per unit for r-pFVIII at Boston Children’s Hospital is approximately $4.67 per IU. Measurable hemostasis for 62 hours (12 doses) with r-pFVIII led to an estimated cost of $103,300 prior to transition to aPCC for ongoing bleed prevention and wound healing. Using our typical BPA dosing strategies cost of hemostasis for the first 62 hours would be approximately $63,141 for rFVIIa and $16,269 for aPCC, [Table I]. Given the high risk nature of this procedure the “value-added” of being able to monitor hemostasis in near-real time without having to respond only to insufficient hemostasis (frank bleeding symptoms) made the r-pFVIII worthwhile for the intra-operative and early post-operative period. Judicious use of r-pFVIII during critical high-risk surgical procedures or emergent bleeding may be appropriate. Frequent re-assessment of when bypassing agents can be introduced for ongoing hemostasis is needed to ensure optimal balance of patient care and cost.
Table I.
Comparative regimen pricing of r-pFVIII immediately post-procedure to bypassing agents rFVIIa and aPCC.
| Product | Regimen Price | Regimen* | Total Doses | Total Usage |
|---|---|---|---|---|
| r-pFVIII | $ 103,300 | 170 IU/kg, subsequently administered doses were 100 IU/kg |
12 | 22,121 IU |
| rFVIIa | $ 63,141 | 270 mcg/kg initial dose followed by 90 mcg/kg q2 hours with subsequent spacing |
22 | 37,585 mcg |
| aPCC | $ 16,269 | 100 IU/kg initial dose followed by 50 IU/kg q6 hours |
10 | 9,571 IU |
Estimated rFVIIa and aPCC regimens based on typical site practice for a patient post-procedure with good hemostasis for the duration (62 hours) that r-pFVIII was used.
Acknowledgments
The authors would like to acknowledge helpful discussion and consultation with P Khandelwal, P Roehrs and C Kessler. SEC is the recipient of a 2014–2016 National Hemophilia Foundation (NHF)-Baxter Clinical Fellowship, supported by Baxalta. YLA was supported by the National Institutes of Health National Heart, Lung, and Blood Institute grant T32 HL007149.
SEC and EJN have acted as paid consultants for Baxalta and Novo Nordisk. Their institution receives research funding from Baxalta. BN has served on a speakers' bureau for Novo Nordisk. PEM, during the conduct of these studies, received research support through the University of North Carolina from Asklepios BioPharmaceutical and Novo Nordisk. He has received research support in the past from Baxter Healthcare, Novo Nordisk, Pfizer, and Prolor. He holds patents licensed to Asklepios, for which he receives royalties. He has received payment for consultation, services, and for speaking for Asklepios, Chatham LLC, Baxter Healthcare and Pfizer and has additionally consulted for Bayer, Novo Nordisk, and Biogen. Following the completion of these studies but during a portion of the time of manuscript preparation he has been an employee of Baxalta (now a part of Shire).
Footnotes
Author contributions
SEC performed the research, prepared figures and wrote the manuscript.
YLA performed the research, prepared figures, and wrote the manuscript.
ASW performed the research, prepared figures and reviewed the manuscript.
BIN performed the research and reviewed the manuscript.
GRM performed the research and reviewed the manuscript.
CWB performed the research and reviewed the manuscript.
EJN performed the research and reviewed the manuscript.
PEM performed the research and wrote the manuscript.
Disclosures
All other authors declared that they have no interests which might be perceived as posing a conflict or bias.
References
- 1.Valentino LA, Cooper DL, Goldstein B. Surgical experience with rFVIIa (NovoSeven) in congenital haemophilia A and B patients with inhibitors to factors VIII or IX. Haemophilia. 2011;17:579–589. doi: 10.1111/j.1365-2516.2010.02460.x. [DOI] [PubMed] [Google Scholar]
- 2.Astermark J, Donfield SM, DiMichele DM, Gringeri A, Gilbert SA, Waters J, et al. A randomized comparison of bypassing agents in hemophilia complicated by an inhibitor: the FEIBA NovoSeven Comparative (FENOC) Study. Blood. 2007;109:546–551. doi: 10.1182/blood-2006-04-017988. [DOI] [PubMed] [Google Scholar]
- 3.Croteau SE, Nakar C, Neufeld EJ, Shapiro A, Cooper DL. Safety and efficacy of recombinant factor VIIa by pediatric age cohort: reassessment of compassionate use and trial data supporting US label. Pediatr Blood Cancer. 2016;63:1822–1828. doi: 10.1002/pbc.26082. [DOI] [PubMed] [Google Scholar]
- 4.Astermark J, Santagostino E, Keith Hoots W. Clinical issues in inhibitors. Haemophilia. 2010;16(Suppl 5):54–60. doi: 10.1111/j.1365-2516.2010.02294.x. [DOI] [PubMed] [Google Scholar]
- 5.Hoffman M, Dargaud Y. Mechanisms and monitoring of bypassing agent therapy. J Thromb Haemost. 2012;10:1478–1485. doi: 10.1111/j.1538-7836.2012.04793.x. [DOI] [PubMed] [Google Scholar]
- 6.Kruse-Jarres R, St-Louis J, Greist A, Shapiro A, Smith H, Chowdary P, et al. Efficacy and safety of OBI-1, an antihaemophilic factor VIII (recombinant), porcine sequence, in subjects with acquired haemophilia A. Haemophilia. 2015;21:162–170. doi: 10.1111/hae.12627. [DOI] [PubMed] [Google Scholar]
- 7.OBIZUR [Antihemophilic Factor (Recombinant), Porcine Sequence] [package insert] Westlake Village, CA: Baxter Healthcare Corporation; 2014. [Google Scholar]
- 8.Lillicrap D, Schiviz A, Apostol C, Wojciechowski P, Horling F, Lai CK, et al. Porcine recombinant factor VIII (Obizur; OBI-1; BAX801): product characteristics and preclinical profile. Haemophilia. 2016;22:308–317. doi: 10.1111/hae.12784. [DOI] [PubMed] [Google Scholar]
- 9.Kempton CL, Abshire TC, Deveras RA, Hoots WK, Gill JC, Kessler CM, et al. Pharmacokinetics and safety of OBI-1, a recombinant B domain-deleted porcine factor VIII, in subjects with haemophilia A. Haemophilia. 2012;18:798–804. doi: 10.1111/j.1365-2516.2012.02789.x. [DOI] [PubMed] [Google Scholar]
- 10.Dargaud Y, Lienhart A, Negrier C. Prospective assessment of thrombin generation test for dose monitoring of bypassing therapy in hemophilia patients with inhibitors undergoing elective surgery. Blood. 2010;116:5734–5737. doi: 10.1182/blood-2010-06-291906. [DOI] [PubMed] [Google Scholar]
- 11.Chitlur M, Young G. Global assays in hemophilia. Semin Hematol. 2016;53:40–45. doi: 10.1053/j.seminhematol.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Hay CR. Porcine factor VIII: current status and future developments. Haemophilia. 2002;8(Suppl 1):24–27. doi: 10.1046/j.1365-2516.2002.00125.x. [DOI] [PubMed] [Google Scholar]
- 13.Beutel K, Hauch H, Rischewski J, Kordes U, Schneppenheim J, Schneppenheim R. ITI with high-dose FIX and combined immunosuppressive therapy in a patient with severe haemophilia B and inhibitor. Hamostaseologie. 2009;29:155–157. [PubMed] [Google Scholar]
- 14.Machlus KR, Colby EA, Wu JR, Koch GG, Key NS, Wolberg AS. Effects of tissue factor, thrombomodulin and elevated clotting factor levels on thrombin generation in the calibrated automated thrombogram. Thrombosis and Haemostasis. 2009;102:936–944. doi: 10.1160/TH09-03-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khandelwal P, Davies SM, Grimley MS, Jordan MB, Curtis BR, Jodele S, et al. Bortezomib for refractory autoimmunity in pediatrics. Biol Blood Marrow Transplant. 2014;20:1654–1659. doi: 10.1016/j.bbmt.2014.06.032. [DOI] [PubMed] [Google Scholar]
- 16.Lozier JN, Santagostino E, Kasper CK, Teitel JM, Hay CR. Use of porcine factor VIII for surgical procedures in hemophilia A patients with inhibitors. Semin Hematol. 1993;30:10–21. [PubMed] [Google Scholar]
- 17.Toschi V. OBI-1, porcine recombinant Factor VIII for the potential treatment of patients with congenital hemophilia A and alloantibodies against human Factor VIII. Curr Opin Mol Ther. 2010;12:617–625. [PubMed] [Google Scholar]
- 18.Brettler DB, Forsberg AD, Levine PH, Aledort LM, Hilgartner MW, Kasper CK, et al. The use of porcine factor VIII concentrate (Hyate:C) in the treatment of patients with inhibitor antibodies to factor VIII. A multicenter US experience. Arch Intern Med. 1989;149:1381–1385. [PubMed] [Google Scholar]
- 19.Gomperts E. Recombinant B domain deleted porcine factor VIII for the treatment of bleeding episodes in adults with acquired hemophilia A. Expert Rev Hematol. 2015;8:427–432. doi: 10.1586/17474086.2015.1040758. [DOI] [PubMed] [Google Scholar]