

Abstract
Cervical cancer remains one of the leading causes of cancer-related deaths worldwide. Reported here is an extensive molecular characterization of 228 primary cervical cancers, the largest comprehensive genomic study of cervical cancer to date. We observed striking APOBEC mutagenesis patterns and identified SHKBP1, ERBB3, CASP8, HLA-A, and TGFBR2 as novel significantly mutated genes in cervical cancer. We also discovered novel amplifications in immune targets CD274/PD-L1 and PDCD1LG2/PD-L2, and the BCAR4 lncRNA that has been associated with response to lapatinib. HPV integration was observed in all HPV18-related cases and 76% of HPV16-related cases, and was associated with structural aberrations and increased target gene expression. We identified a unique set of endometrial-like cervical cancers, comprised predominantly of HPV-negative tumors with high frequencies of KRAS, ARID1A, and PTEN mutations. Integrative clustering of 178 samples identified Keratin-low Squamous, Keratin-high Squamous, and Adenocarcinoma-rich subgroups. These molecular analyses reveal new potential therapeutic targets for cervical cancers.
Cervical cancer accounts for 528,000 new cases and 266,000 deaths worldwide each year, more than any other gynecologic tumor1. Ninety-five percent of cases are caused by persistent infections with carcinogenic human papillomaviruses (HPV)2. Effective prophylactic vaccines against the most important carcinogenic HPV types are available, but uptake remains poor. Although early cervical cancer can be treated with surgery or radiation, metastatic cervical cancer is incurable and new therapeutic approaches are needed3.
While most HPV infections are cleared within months, some persist and express viral oncogenes that inactivate p53 and Rb, leading to increased genomic instability, accumulation of somatic mutations, and in some cases integration of HPV into the host genome4. The association with cancer risk and histological subtypes varies substantially among carcinogenic HPV types, but the reasons for these differences are poorly understood. Further, clinically relevant cervical cancer patient subgroups have yet to be identified. Presented here is a comprehensive study of invasive cervical cancer conducted as part of The Cancer Genome Atlas (TCGA) project, with a focus on identifying novel clinical and molecular associations as well as functionally altered signaling pathways that may drive tumorigenesis and serve as prognostic or therapeutic markers.
Samples and clinical data
Primary frozen tumor tissue and blood were obtained from women with cervical cancer without prior chemotherapy or radiotherapy (Supplemental Information S1 and Supplemental Tables 1 and 2). DNA, RNA, and protein were processed as previously described5 (Supplementary Information S1, S3, S5, and S8). Mutations were called for 192 samples (Extended Set), while all other platform (aside from protein) and integrated analyses were performed on a subset of 178 samples (Core Set). Protein levels were measured on 155 samples (119 samples from both the Core and Extended Sets plus 36 additional samples). The total number of non-overlapping samples in these three sets was 228 (Extended Data Fig. 1a). Of the 178 Core Set samples, surgery was the primary treatment in 121 cases, median follow-up was 17 months, and 145 patients were alive at the time of last follow-up. A committee of expert gynecologic pathologists reviewed most cases (Supplemental Information S1 and Extended Data Fig. 1b–g). The Core Set included 144 squamous cell carcinomas, 31 adenocarcinomas, and 3 adenosquamous cancers.
Somatic genomic alterations
Whole exome sequencing was performed on 192 Extended Set tumor-blood normal pairs. All samples had at least 32 Mbp of target exons covered with a median of 49× (range: 7–351×) coverage for tumor samples and 47× (range: 9–341×) coverage for normal samples. Collectively, the samples harbored 43,324 somatic mutations, including 24,551 missense, 2,470 nonsense, 9,260 silent, 5,841 non-coding, 535 splice site, 74 nonstop, 475 frameshift indels, and 118 in-frame indels. Eleven tumors with outlier mutation frequencies (>600 per sample) were classified as “hypermutant.” The aggregate mutation density was 4.04 mutations per megabase across all tumors, and 2.53 when the hypermutant tumors were excluded.
Fourteen significantly mutated genes (SMGs) with false discovery rates (FDR) < 0.1 were found using the MutSig2CV6 algorithm (Supplemental Table 4). We identified SHKBP1, ERBB3, CASP8, HLA-A, and TGFBR2 as novel SMGs in cervical cancer, and confirmed PIK3CA, EP300, FBXW7, HLA-B, PTEN, NFE2L2, ARID1A, KRAS, and MAPK1 which have been previously reported (Fig. 1, Extended Data Fig. 2a–g, and Supplemental Fig. S6)7,8. Supplemental Table 4 shows the comparison of SMGs identified in the TCGA and Ojesina et al. datasets. Mutations in 7 of the 14 SMGs in the TCGA set were present in at least one squamous cell carcinoma and one adenocarcinoma; however, mutations in HLA-A, HLA-B, NFE2L2, MAPK1, CASP8, SHKBP1, and TGFBR2 were found exclusively in squamous tumors.
Figure 1. Somatic alterations in cervical cancer and associations with molecular platform features.
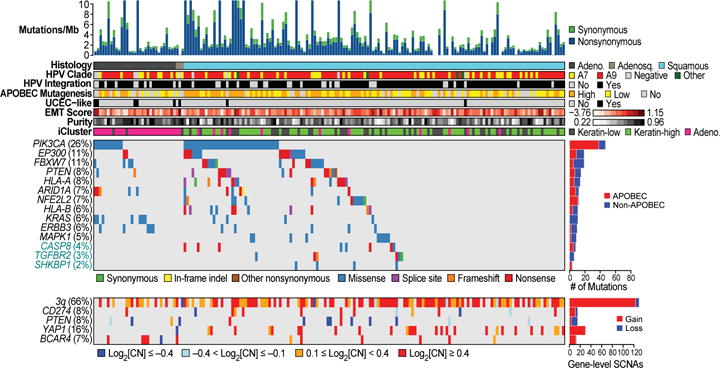
CESC samples are ordered by histology and mutation rate (top panel), clinical and molecular platform features (second panel), significantly mutated genes (SMGs; third panel), and select somatic copy number alterations (SCNAs; fourth panel) are presented. SMGs are ordered by the overall mutation frequency and color-coded by mutation type. Novel SMGs identified in squamous cell carcinomas are labeled in turquoise text. The number of APOBEC signature mutations (red) and other mutations (blue) present in every SMG is plotted to the right of the SMG panel and the number of gene level SCNAs across all genes is plotted as gain (red) and loss (blue) to the right of the SCNA panel.
PIK3CA harbored mostly activating helical domain mutations E542K and E545K, with a marked relative decrease in mutations elsewhere in the gene (Extended Data Fig. 2g). This observation resembles findings in bladder cancer9 and HPV-positive head and neck squamous cell cancers (HNSC)10, but it differs from observations in breast and most other cancers11. The underlying nucleotide substitution pattern in the E542K and E545K mutations is associated with mutagenesis by a subclass of APOBEC cytidine deaminases8,12–15, with 150 of 192 exomes displaying statistically significant (q<0.05) enrichment (up to 6-fold) for the APOBEC signature. Further, the APOBEC mutation load correlated strongly with the total number of mutations per sample (Extended Data Fig. 2h), suggesting that APOBEC mutagenesis is the predominant source of mutations in cervical cancers.
We found an average of 88 somatic copy number alterations (SCNAs) per tumor, fewer than in HNSC, ovarian, and serous endometrial carcinomas but more than in endometrioid endometrial carcinomas10,16,17. GISTIC2.0 analysis (threshold q< 0.25) revealed 26 focal amplifications and 37 focal deletions along with 23 recurrently altered whole arms (Extended Data Fig. 3c and Supplemental Table 7). Novel recurrent focal amplification events were identified (in genomic order) at 7p11.2 (EGFR; 17%), 9p24.1 (CD274, PDCD1LG2; 21%), 13q22.1 (KLF5; 18%), and 16p13.13 (BCAR4; 20%). Other previously reported amplification events occurred at 3q26.31 (TERC, MECOM; 78%), 3q28 (TP63; 77%), 8q24.21 (MYC, PVT1; 42%), 11q22.1 (YAP1, BIRC2/3; 17%), and 17q12 (ERBB2; 17%). Novel recurrent deletions were identified at 3p24.1 (TGFBR2; 36%) and 18q21.2 (SMAD4; 28%), in addition to previously identified deletions at 4q35.2 (FAT1; 36%) and 10q23.31 (PTEN; 31%). A CN high cluster largely contained squamous tumors with amplification events involving 11q22 (YAP1, BIRC2/3) and 7p11.2 (EGFR), while the CN low cluster included most adenocarcinomas and was enriched for tumors with deletions in TGFBR2 and SMAD4 and gains in ERBB2 and KLF5 (Extended Data Fig. 3a, b). Notably, both groups had amplifications involving CD274 (PD-L1) and PDCD1LG2 (PD-L2) that correlated significantly (p<0.0001) with expression of two key immune cytolytic effector genes, granzyme A and perforin18 (Extended Data Fig. 3d). This highlights the potential of immunotherapeutic strategies for a subset of cervical cancers.
Structural rearrangements were identified through analysis of RNA-seq (Core Set, n=178) and whole genome sequencing (WGS) data with low-pass (n=50) and deep (n=19) coverage. Both RNA-seq and WGS detected 22 putative structural rearrangements in 14 patients (Supplemental Table 8). In total, 26 recurrent fusions were found (Supplemental Table 9, with examples in Extended Data Fig. 4d). RNA-seq analysis revealed 4 cases harboring 16p13 ZC3H7A-BCAR4 gene fusions, with exon 1 of ZC3H7A linked to the last exon of BCAR4. Whole genome sequencing revealed tandem duplication and copy number gain of BCAR4 on chromosome 16p13.13 (Extended Data Fig. 4c). BCAR4 is a metastasis-promoting lncRNA that enhances cell proliferation in estrogen-resistant breast cancer by activating the HER2/3 pathway. Lapatinib, an EGFR/HER2 inhibitor, counteracts BCAR4-driven tumor growth in vitro, and warrants evaluation as a possible therapeutic agent in BCAR4-positive cervical cancer19.
Integrated analysis of molecular subgroups and pathways
Integration of copy number, methylation, mRNA, and miRNA data using iCluster20 highlighted the molecular heterogeneity of cervical carcinomas. Three clusters were identified that largely corresponded to mRNA clusters (Supplemental Fig. S9): a squamous cluster with high expression of keratin gene family members (Keratin-high), another squamous cluster with lower expression of keratin genes (Keratin-low), and an Adenocarcinoma-rich cluster (Adenocarcinoma). Keratin-high and Keratin-low clusters included 133 of 144 squamous cell carcinomas and the Adenocarcinoma cluster contained 29 of 31 adenocarcinomas (Fig. 2). KRAS (p=9.7e-5), ERBB3 (p=2.6e-3), and HLA-A (p=0.03) mutations were significantly associated with iClusters, with KRAS mutations absent from the Keratin-high cluster and HLA-A mutations missing in the Adenocarcinoma cluster (Fig. 2). Members of the SPRR and TMPRSS cornification gene families and the SMGs ARID1A (p=0.02), NFE2L2 (p=6.9e-6), and PIK3CA (p=0.01) were differentially expressed between Keratin-low and Keratin-high clusters (Extended Data Fig. 4b).
Figure 2. Multiplatform integrative clustering of cervical cancers.

Integrative clustering of 178 Core Set cervical cancer cases using mRNA, methylation, miRNA, and copy number (CNV) data identifies two squamous carcinoma-enriched groups (Keratin-low and Keratin-high) and one adenocarcinoma-enriched group as shown in the feature bars. Features presented include histology, HPV clade, HPV integration status, UCEC-like status, APOBEC mutagenesis level, mRNA EMT score, tumor purity, and three SMGs that are significantly associated across the three iClusters (ERBB2 is presented for comparison purposes with its family member ERBB3). The cluster of cluster panel displays subtypes defined independently by mRNA, miRNA, methylation, reverse phase protein array (RPPA), CNV, and PARADIGM data. Black indicates that the sample is not represented in the cluster, red indicates that the sample is represented in the cluster, and gray represents data not available. The bottom heatmap panel shows select mRNAs, miRNAs, proteins, and CNVs that are either significantly associated with iCluster groups or identified as markers in other analyses. The heatmap color scale bar represents the scale for the features presented in the heatmap panel with a breakpoint of zero represented by white. APOBEC Mut., APOBEC Mutagenesis; inter., intermediate.
Unsupervised hierarchical clustering of variable DNA methylation probes produced three groups (Extended Data Fig. 5a), including a small “CpG island hypermethylated” (CIMP-high) cluster, a CIMP-intermediate cluster, and a CIMP-low cluster that were associated with an epithelial-mesenchymal transition (EMT) mRNA score (Extended Data Fig. 5b)10,21. Most of the cases in the Adenocarcinoma iCluster were CIMP-high, while the other iClusters contained a mixture of CIMP-intermediate and CIMP-low samples (Fig. 2). Comparing all cervical carcinomas to 120 normal samples drawn from 12 TCGA projects, we identified 1026 epigenetically silenced genes that were methylated to a greater extent in cancers than in normal tissues, including several zinc-finger (ZNF), protease (ADAM, ADAMTS), and collagen (COL) genes (Supplemental Tables 11 and 12).
Unsupervised clustering resulted in 6 miRNA clusters associated with iClusters (p=1.7e-19) (Extended Data Fig. 6a). Samples from the Adenocarcinoma iCluster almost exclusively overlapped with miRNA cluster 5, and were characterized by high expression of miR-375 and low expression of miR-205-5p and miR-944 (Supplemental Table 31). Expression levels of tumor suppressors miR-99a-5p and miR-203a were significantly higher in Keratin-high cluster samples than Keratin-low cluster samples (Supplemental Table 31; p=0.01 and 0.008, respectively). Among miRNAs with significant and functionally validated gene and protein anti-correlations22, one large subnetwork involved miR-200-family and other miRs with expression patterns anti-correlated with those of the EMT-related transcription factors ZEB1, ZEB2, and SNAI2, the Hippo and p73 transcriptional co-factor YAP1, the receptor tyrosine kinases (RTKs) ERBB2, ERBB3, and AXL, and the hormone receptor ESR1 (Extended Data Fig. 6b, Supplemental Fig. S17, Supplemental Fig. S18, and Supplemental Table 15).
Reverse Phase Protein Array (RPPA) analysis of 155 samples with 192 antibodies (Extended Data Fig. 1a and Supplemental Table 17) identified three clusters significantly associated with iClusters (p=1.8e-4) and EMT mRNA score (Fig. 3a, c, d and Supplemental Table 16). EMT cluster samples were enriched in the Keratin-low iCluster, while PI3K/AKT and Hormone cluster samples were enriched in the Keratin-high and Adenocarcinoma iClusters, respectively, suggesting distinct pathway activation across integrated cervical cancer subtypes. Differential expression levels of Phospho-MAPK, Phospho-EGFR (Y1068), Phospho-Src (Y416), IGFBP2, and TIGAR between Keratin-high and Keratin-low iClusters suggest diverse activation patterns of RTK, MAPK, PI3K, and metabolic signaling pathways that may underlie the molecular diversity of cervical squamous cancers (Fig. 2).
Figure 3. Proteomic landscape of cervical cancer.
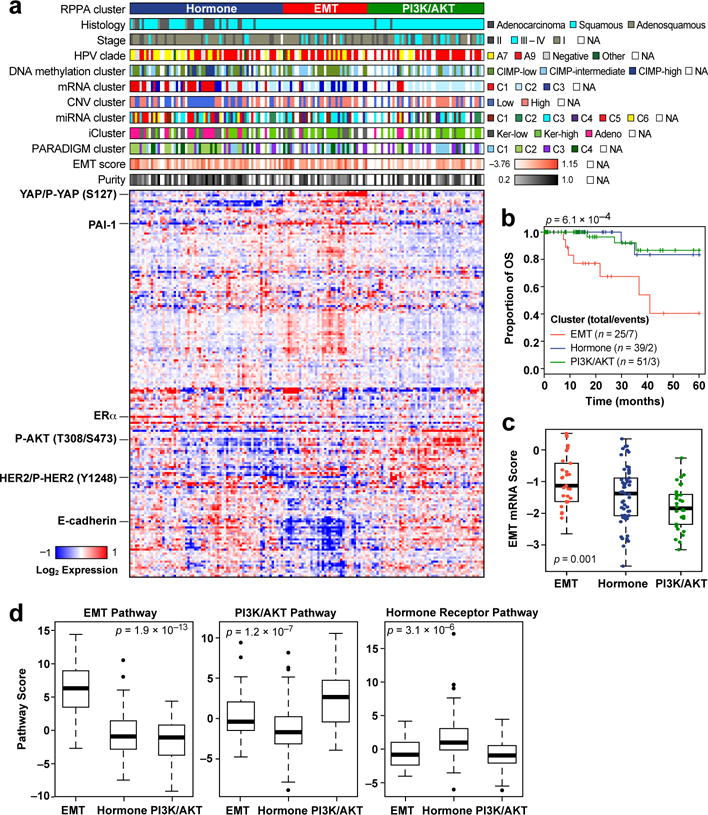
a, Clustered heatmap of samples (columns) and 192 antibodies (rows) for 155 samples (112 overlap with the Core Set of 178; see Extended Data Fig. 1a). Clusters presented from left to right include Hormone (dark blue), EMT (red), and PI3K/AKT (green). A subset of proteins differentially expressed between the clusters is highlighted. Clinical and molecular feature tracks are shown for those features which were significantly associated with RPPA clusters (p<0.05). Correlation between RPPA clusters and other categorical variables were detected by Chi-Squared test, while correlations with continuous variables were examined using the non-parametric Kruskal-Wallis test. In the heatmap blue color represents downregulated expression, red represents upregulated expression, and white represents no change in expression. NA represents data not available. b, Five-year Kaplan-Meier survival curves and log-rank test’s p-value comparing overall survival (OS) across all RPPA clusters using 115 Silhouette Width Core samples (Silhouette Core; see Supplemental Information S8). c, EMT mRNA score levels were calculated for all samples and compared across RPPA clusters. A significant p-value is presented for a one-way ANOVA analysis. d, Pathway scores for EMT, hormone receptor, and PI3K/AKT signaling pathways are presented for all RPPA clusters (x-axis), with significant pathway score differences between the clusters measured by Kruskal Wallis test.
The core members of each RPPA cluster with the highest silhouette width (>0.02, n=115) were associated with five-year survival (Fig. 3b; p=6.1e-4), with the EMT group exhibiting worse outcome. Interestingly, this was the only platform where clusters associated with outcomes (Supplemental Figs. S8, S9, S12, and S22; Supplemental Information S6). Samples in the EMT cluster exhibited high “reactive” pathway scores (Supplemental Fig. S20)11, illustrating for the first time in cervical cancer the presence of a subset of stromal “reactive” tumors with high expression of Caveolin-1, MYH11, and Rab11, which also appear in other diseases (Supplemental Table 16)23. YAP was the most significantly differentially expressed protein distinguishing EMT cluster samples from all others (Supplemental Table 18; p=1.7e-15) and YAP1 was significantly amplified in the EMT cluster samples compared with the Hormone (p=1.1e-5) and PI3K/AKT cluster (p=6.4e-4) samples. Regulation of the EMT-related molecules YAP and ZEB124–26 may also be driven by significantly lower expression levels of miR-200a-3p in the EMT cluster samples compared with other RPPA cluster samples (Extended Data Fig. 6b and Extended Data Fig. 7a; p=3.8e-3). These results highlight potential roles for YAP and reactive stroma in the context of EMT-regulated cervical cancer progression.
The Mutual Exclusivity Modules in cancer (MEMo) algorithm27 uses somatic mutation and copy number data to identify oncogenic networks with mutually exclusive genomic alterations. Since miR-200a and miR-200b (miR-200a/b) expression were negatively correlated with EMT mRNA scores (Extended Data Fig. 7b, d), we used MEMo to examine alterations in miR-200a/b and EMT networks and found a potential link between TGFβ pathway and miR-200a/b alterations in regulating EMT28,29. Deletions and mutations affecting the receptor gene TGFBR2, the modulating genes CREBBP and EP300, and the transcription factor SMAD4 all likely impinge on growth suppressive and pro-apoptotic functions driven by TGFβ (Fig. 4c) and were observed in 30% of squamous cell carcinomas (Fig. 4d). Tumors with both hypermethylation and downregulation of miR-200a/b (referred to as altered) were restricted to squamous cell carcinomas, were enriched in the Keratin-low iCluster (Fig. 4d and Extended Data Fig. 8; p=0.001 for both miR-200a and miR-200b), showed significant upregulation of both ZEB1 and ZEB2 (Extended Data Fig. 9a–d), and were mutually exclusive with TGFβ signaling pathway alterations (Fig. 4d). Importantly, samples with altered miR-200a/b exhibited higher EMT mRNA scores than unaltered samples, while there was no significant difference between samples with or without TGFβ pathway alterations (Fig. 4d and Extended Data Fig. 7c, e). These findings highlight potential treatment approaches for this subgroup of cervical cancer patients, as targeting EMT may render tumors more sensitive to small molecule inhibitors and cytotoxic chemotherapy21,30,31.
Figure 4. Mutual exclusivity of somatic alterations within the PI3K/MAPK and TGFβR2 pathways.
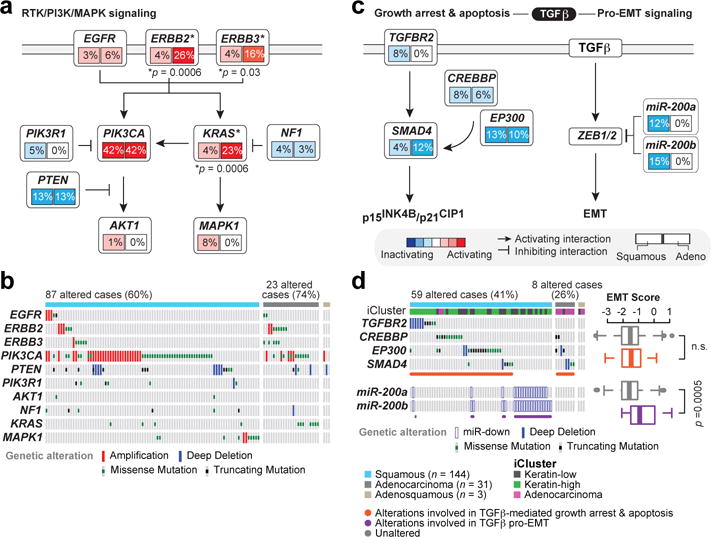
a, Multiple alterations affect receptor tyrosine kinase (RTK), AKT, and MAPK signaling in both squamous cell and adenocarcinoma cases. A schematic diagram of the pathways is shown for altered genes along with percentage of alteration in squamous cell and adenocarcinoma cases. Significant (p<0.05) Student’s t-test p-values for alteration frequency differences between squamous cell and adenocarcinomas are listed at the gene level, with genes marked with an asterisk (*). b, Distinct types of alterations (amplification, deletion, missense mutation, and truncating mutation) affect genes (rows) in these pathways in each sample (columns). c, TGFβ signaling is frequently altered in cervical tumors. Alterations in this pathway are divided between those likely impinging on TGFβ tumor suppressive functions and those affecting the TGFβ-driven EMT program. Legend also corresponds to layout in panel a. d, Samples with alterations targeting TGFβ tumor suppressive functions do not show significantly different EMT scores compared with all other samples (n.s = not significant); however, samples with low expression/high methylation of miR-200a/b have significantly higher EMT scores than all other samples. miR-down: met double-threshold of methylated and downregulated as described in Methods.
MEMo analysis also showed differences in therapeutically-relevant RTK, PI3K, and MAPK pathway alterations across cervical cancers. MEMo identified mutual exclusivity modules involving alterations within both the PI3K and MAPK pathways (Supplemental Table 27; adjusted p=0.06); however, there was a strong tendency for co-occurrence of ERBB2 and ERBB3 alterations within adenocarcinomas (p<0.001, log odds-ratio > 3), indicating that a subset of these tumors may exhibit aberrant HER3 signaling through interactions between mutant HER3 and activated HER2 and therefore could potentially benefit from HER2- and HER3-targeted therapies (Fig. 4a, b)32. Although not statistically significant, aberrations in PIK3CA also tended to co-occur with PTEN somatic mutations and deletions (p=0.078, log-odds ratio=0.71), which is similar to copy number-low endometrial tumors and suggests potential therapeutic benefit from PI3K pathway targeting agents17.
PARADIGM33,34, which integrates copy number, RNA-seq, and pathway interaction data, showed markedly different pathway activation profiles between squamous carcinomas and adenocarcinomas (Extended Data Fig. 10 and Supplemental Fig. S48). PARADIGM identified higher inferred activation of p53, p63, p73, AP-1, MYC, HIF1A, FGFR3, and MAPK signaling as key distinguishing signaling features of squamous cell carcinomas, similar to other squamous cancers35. In contrast, adenocarcinomas exhibited higher inferred activation of ERα, FOXA1, FOXA2, and FGFR1 pathways (Extended Data Fig. 10, Supplemental Fig. S25, Supplemental Fig. S48, and Supplemental Table 18). Possible underlying mechanisms for ERα upregulation may stem from the expression of miR-193b-3p, a direct regulator of ESR1 that was significantly downregulated in adenocarcinomas compared with squamous carcinomas (Fig. 2, Extended Data Fig. 6, and Supplemental Table 14; p=0.04), or from estrogen signaling in stromal cells36.
Cross-cancer analysis
To evaluate the relationship of cervical cancer subtypes with endometrial cancer, an adjacent cancer site with hormone-related carcinogenesis, and HNSC, a subset of which is caused by HPV, hierarchical clustering of cervical, uterine corpus endometrial (UCEC)17, and HNSC10 mRNA expression data was performed. Three major groups were observed, with Cluster 1 including all UCEC samples and most cervical adenocarcinomas and characterized by overexpression of hormone receptor genes ESR1 and PGR (Extended Data Fig. 4a). Cluster 2 included predominantly squamous cervical carcinomas and 23/27 HPV-positive HNSC samples. Cluster 3 included few cervical cancers and the remaining HNSC cancers, which were mostly HPV-negative. This highlights the similarity of HPV-related squamous cancers at different anatomical sites.
Since a subset of cervical cancers clustered with endometrial samples, a gene expression classifier was developed to predict whether carcinomas were cervical or endometrial (Supplemental Information S5). We classified 8 of 178 (4.5%) cervical cancer samples as endometrial-like (UCEC-like) cancers, which were confirmed to be cervical cancers by study pathologists (Extended Data Fig. 1f, g). These tumors included 7 of 9 HPV-negative cancers and 5 of the 8 were adenocarcinomas. Six UCEC-like cancers were in the Adenocarcinoma iCluster and 2 were in the Keratin-low iCluster. Despite their low number, the UCEC-like tumors accounted for 33%, 27%, and 20% of mutations in ARID1A, KRAS, and PTEN, respectively. They were associated with the RPPA Hormone and miRNA C6 clusters, and all but one sample was CIMP-low and CN low (Supplemental Table 1).
HPV genotypes, variants, and integration
Of 178 Core Set tumors, 169 (95%) were HPV-positive, 120 (67%) had alpha-9 (A9) types (103 HPV16), 45 (25%) had alpha-7 (A7) types (27 HPV18), and 9 (5%) were HPV-negative (Supplemental Table 3). HPV variants were predominantly European (137 of 169, 81% A variants), and there was a significant association of non-European HPV16 variants with cervical adenocarcinomas (Supplemental Table 3; OR 5.3, p=3e-3). All HPV-positive cancers had detectable expression of HPV E6 and E7 oncogene mRNAs, which encode proteins that inhibit p53 and Rb function, respectively37,38. Interestingly, HPV18 cancers had significantly higher levels of unspliced/spliced transcripts encoding active E6 oncoprotein than the HPV16 cancers (Extended Data Fig. 11a; p=2e-10), suggesting different functional implications of E6 and E7 in cancers associated with different HPV genotypes.
HPV A7 types were enriched in Keratin-low and Adenocarcinoma iClusters (p=5e-4). Most HPV clade A7 tumors were CIMP-low, and HPV-negative tumors formed a distinct subgroup within the CIMP-low cluster with a significantly lower mean promoter methylation level than other samples in that cluster (Extended Data Fig. 5a; p=5e-3). Samples with the highest rate of silencing were HPV-positive adenocarcinomas, particularly those related to A9 types (t-test p-values <0.001). Functional Epigenetic Module (FEM; Supplemental Information S13) analysis39, which integrates DNA methylation and gene expression data using protein-protein-interaction networks, identified inverse correlations between methylation and gene expression in HPV-positive vs. HPV-negative cervical cancers and HPV-positive (n=36) vs. HPV-negative (n=243) HNSCs. The analysis revealed 12 statistically significant subnetworks for cervical cancer and 11 for HNSCs, with one common subnetwork centered around Forkhead Box A2 (FOXA2) (Supplemental Table 19 and Supplemental Fig. S32). miR-944, miR-767-5p, and miR-105-5p were the most differentially expressed miRNAs between HPV-positive and HPV-negative samples (Supplemental Fig. S14e). miR-944 expression was also significantly higher while miR-375 expression was significantly lower in HPV16-positive squamous cancers compared with HPV18-positive squamous cancers (Supplemental Fig. S14d). Interestingly, HPV-negative cancers displayed a significantly higher EMT mRNA score and a lower frequency of the APOBEC mutagenesis signature compared with HPV-positive tumors (Extended Data Fig. 11b and Supplementary Figure S27; p=0.02 and p=0.004, respectively).
PARADIGM was used to evaluate molecular pathways differentially activated in squamous samples with A7 and A9 HPV infections. We observed higher inferred activation of p53 and p63 signaling and lower FOXA1 signaling in tumors infected with A9 types (Fig. 5a and Supplemental Fig. S23a). Higher SFN pathway activation was also observed for A9-positive tumors, which is consistent with the low methylation and high gene expression patterns of SFN revealed by FEM analysis (Fig. 5a and Supplemental Table 19). Interestingly, the SFN-encoded Stratifin/14-3-3σ adapter protein has previously been associated with epithelial immortalization and squamous cell cancers40,41, altered p53 pathway activation42, and Wnt-mediated β-catenin signaling43.
Figure 5. HPV integration and differential pathway activation between HPV subtypes.
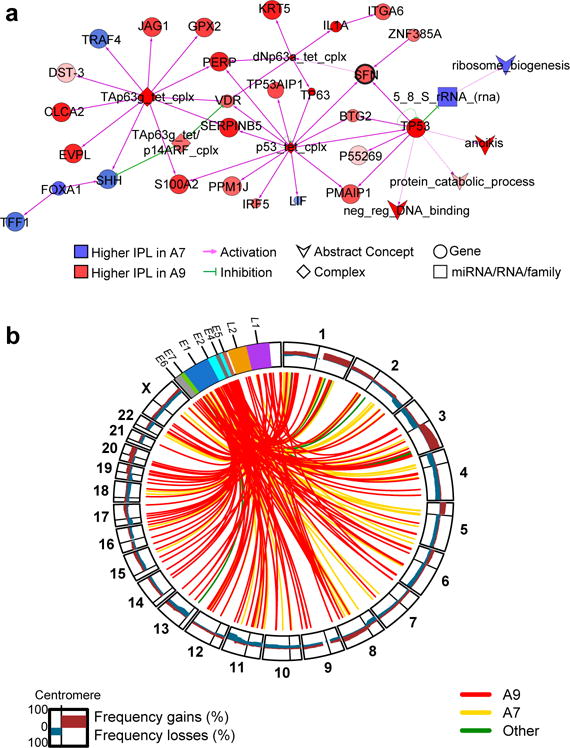
a, Cytoscape display of the largest interconnected regulatory network of PARADIGM integrated pathway level (IPL) features showing differential inferred activation between HPV A9 and A7 squamous carcinomas (n= 101 and n=35, respectively). Node color and intensity reflect the level of differential activation. Node size represents level of significance. Regulatory nodes with at least 5 downstream targets are highlighted in bold text. SFN is within a subnetwork identified by Functional Epigenetic Module (FEM) analysis (Supplemental Information S13) as disrupted between HPV A9 and A7 squamous cell carcinomas, and is highlighted using a bold black outline. b, Circos plot showing frequency (0–100%) of gains and losses for regions of each chromosome (outer circle). Lines within inner circle indicate integration breakpoints from the HPV genome to the human genome as defined in Methods, Supplemental Information S2, and Supplemental Table 3. Lines are color coded by HPV clade.
Viral-cellular fusion transcripts indicating integration of HPV into the host genome were observed in 141 of 169 (83%) HPV-positive cancers, including all HPV18-positive cancers. Of these 141 cases, 90 (64%) had a single HPV integration event, 35 had two events, and 16 had three or more events (totaling 220 unique integration events) (Supplemental Table 3). HPV integration events affected all chromosomes, including some previously described hotspots such as 3q28 and 8q24 (Fig. 5b)44. Genomic loci affected by integration were characterized by increased SCNAs (p=6.9e-13 for HPV16 and p=0.058 for HPV18) and increased gene expression (p=1.6e-11 for HPV16 and p=0.011 for HPV18) (Extended Data Fig. 11c, d). One hundred fifty-three (70%) fusion transcripts included known or predicted genes, while the remainder included intergenic regions (Fig. 5b and Supplemental Table 3).
Conclusion
Through comprehensive molecular and integrative profiling, we identified novel genomic and proteomic characteristics that subclassify cervical cancers. Integrated clustering identified Keratin-low squamous, Keratin-high squamous, and Adenocarcinoma-rich clusters defined by different HPV and molecular features (Extended Data Fig. 8). ERBB3, CASP8, HLA-A, SHKBP1, and TGFBR2 were identified as SMGs for the first time in cervical cancer, with ERBB3 (HER3) immediately applicable as a therapeutic target. Notably, we report amplifications and fusion events involving the BCAR4 gene for the first time in cancer, which can be targeted indirectly by lapatinib. Further, we identified amplifications in CD274 and PDCD1LG2, two genes that encode for well-known immunotherapy targets. A set of endometrial-like cervical cancers comprised predominantly of HPV-negative tumors and characterized by mutations in KRAS, ARID1A, and PTEN was discovered, with PTEN and potentially ARID1A proteins serving as therapeutic targets. Importantly, over 70% of cervical cancers exhibited genomic alterations in either one or both of the PI3K/MAPK and TGFβ signaling pathways (Extended Data Fig. 9e), illustrating the potential clinical significance of therapeutic agents targeting members of these pathways. For the first time, we report distinct molecular pathways activated in cervical carcinomas caused by different HPV types, highlighting the biologic diversity of HPV.
Together, these findings provide insight into the molecular subtypes of cervical cancers and rationales for developing clinical trials to treat populations of cervical cancer patients with distinct therapies.
Methods
Samples and data freeze
The Core Data Freeze (Core Set) included 178 cases from cervical carcinoma (CESC) batches 88, 114, 127, 148, 169, 179, 200, 217, 236, 256, 280, 297, 335, and 350 (Supplemental Table 1). Samples in the Core Set had mRNA-seq, whole exome DNA-seq (WES), miRNA-seq, methylation, SNP6 copy number, and clinical data available. Additional cases having multicenter mutation calls and/or RPPA data included 67 cases from CESC batches 88, 114, 127, 148, 169, 179, 200, 217, 236, 256, 280, 297, 335, 350, 361, 373, 380, 394, and 420 (Supplemental Table 2). Of these cases, 14 had mutations called and 60 had RPPA data available; however, RPPA data for 17 cases was excluded due to low protein content within samples (Supplemental Table 2). Mutations were called for 192 samples (Extended Set), while all other platform and integrated analyses (aside from protein) were performed on the subset of 178 Core Set samples. Protein levels were measured on 155 samples, which included 119 total samples from both the Core and Extended Sets as well as 36 samples outside of these sets. The total number of non-overlapping samples across Core, Extended, and RPPA datasets is 228 (Extended Data Fig. 1a).
HPV detection, variant calling, and transcript analysis
HPV status was determined using consensus results from MassArray and RNA-seq (Supplemental Information S2). MassArray uses real-time competitive polymerase chain reaction and matrix-assisted laser desorption/ionization-time of flight mass spectroscopy with separation of products on a matrix-loaded silicon chip array, similar to the work described in Tang et al45. Two approaches for pathogen detection from RNA-seq data were used. The first used the microbial detection pipeline at the British Columbia Cancer Agency’s Genome Sciences Centre (BC), which is based on BioBloom Tools (BBT, v1.2.4b1)46. The second used the PathSeq algorithm47 at the Broad Institute (BI) to perform computational subtraction of human reads followed by alignment of residual reads to a combined database of human reference genomes and microbial reference genomes including HPV. In 97% of samples, complete agreement between MassArray and both RNA-seq approaches was observed. The remaining discrepant samples were resolved by majority decision, assigning the genotype called by at least two of the methods. RNA-seq data in FASTA format was used to identify HPV variants (Supplemental Fig. S1). Unaligned reads were taken from the PathSeq analysis and aligned to HPV reference genomes using TopHat48 with default parameters49. The HPV variant lineages/sublineages were assigned based on the phylogenetic topology and confirmed visually using the SNP patterns50. HPV splice junctions from RNA-seq were determined using TopHat. Two transcript types were distinguished for HPV16 and HPV18: (a) transcripts that included evidence of an unspliced sequence of E6, and (b) a transcript spliced at the E6 splice donor site (position 226 for HPV16 and position 233 for HPV18) (Supplemental Fig. S2). Read counts for unspliced, spliced, as well as the ratio of unspliced/spliced transcripts were categorized into quartiles separately for HPV16 and HPV18.
HPV integration analysis
Using RNA-seq data, concordance of integration events based on alignments of contigs from de novo transcriptome assembly (BC) and read alignments (BI) was evaluated (Supplemental Fig. S3). We identified method-specific integration events by assigning all sites within a 500-kb sliding window to a single integration event located at the median coordinate of that event’s assigned sites. An integration event was labeled as ‘confident’ when the total read support for each of its supporting integration sites passed center-specific read evidence thresholds. To take advantage of differences between the two integration methods (i.e. contig and read), for the concordance analysis we used all method-specific integration events (both confident and non-confident events). We labeled an integration event as ‘concordant’ when both methods reported an integration event within 500 kb in the same patient. For some concordant events, both methods reported a confident event. An integration event was labeled as ‘discordant’ when only one center reported a confident integration event within 500 kb (Supplemental Figs. S4 and S5). For both intragenic and intergenic concordant events, we reported a range of coordinates that extends from the most proximal to the most distal supporting integration site. We assessed gene-level expression relative to somatic copy number and structural variant data for genes into which we had mapped viral-human junctions from RNA sequencing data and for genes that were associated with enhancers into which we had mapped RNA junctions.
DNA sequencing and mutation calling
Detailed methods for library hybrid capture, read alignments, and somatic variant calling are documented in Supplemental Information S3. MutSig2CV6 was utilized to identify significantly mutated genes (SMGs) within the cervical cancer exome sequencing data. Mutations were analyzed for the Core Set plus 14 samples to total 192 Extended Set samples. Eleven samples were identified to exhibit greater than average mutations rates and were termed “hypermutants” (somatic mutations >600). These 11 samples were excluded from the analysis for identifying SMGs. All 3 sample subsets (all samples, squamous carcinomas only, adenocarcinomas only) without “hypermutants” (Supplemental Table 4) were analyzed using an FDR cutoff of 0.1. FDR values are shown in Supplemental Table 4. SMG analysis using the entire sample cohort in Ojesina et al. was performed as described previously8.
Copy number analysis
DNA from each tumor or germline sample was hybridized to Affymetrix SNP 6.0 arrays using protocols at the Genome Analysis Platform of the Broad Institute as previously described51. Briefly, Birdseed was used to infer a preliminary copy number at each probe locus from raw. CEL files52. For each tumor, genome-wide copy number estimates were refined using tangent normalization, in which tumor signal intensities are divided by signal intensities from the linear combination of all normal samples that are most similar to the tumor16. Individual copy number estimates then underwent segmentation using Circular Binary Segmentation53, and segmented copy number profiles for tumor and matched control DNAs were analyzed using Ziggurat Deconstruction54. Significance of copy number alterations were assessed from the segmented data using GISTIC2.0 (Version 2.0.22)54. For the purpose of this analysis, an arm-level event was defined as any event spanning more than 50% of a chromosome arm. For copy number based clustering, tumors were clustered based on log2 copy number at regions revealed by GISTIC analysis. Clustering was done in R based on Euclidean distance using Ward’s method. Allelic and integer copy number, tumor purity, and tumor ploidy were calculated using the ABSOLUTE algorithm55.
Detecting structural variants from RNA-seq and WGS data
Integrative analysis was performed to identify putative driver fusions using both WGS (low-pass and high-coverage) and RNA-seq data. RNA-seq data for 178 Core Set cases were analyzed using the TopHat-Fusion and BreakFusion, PRADA, and MapSplice algorithms. To identify structural variations in WGS data, 50 low-pass WGS and 19 high-pass WGS samples were analyzed. Detection of structural variations in low-pass WGS data was performed using two algorithms, BreakDancer56 and Meerkat57, with a requirement for at least two discordant read pairs supporting each event and at least one read covering the breakpoint junction. High-pass WGS data were analyzed to detect somatic structural variations using two runs of BreakDancer and one run of SquareDancer (https://github.com/ding-lab/squaredancer). The gene fusion lists generated by all methods and platforms were integrated (See Supplemental Tables 8–10).
APOBEC mutagenesis analysis
Analysis is based on previous findings that APOBECs deaminate cytidines predominantly in a tCw motif and that the APOBEC mutagenesis signature is composed of approximately equal numbers of two kinds of changes in this motif: tCw→tTw and tCw→tGw mutations (flanking nucleotides are shown in small letters; w=A or T). Using mutation data from all 192 Extended Set samples, we calculated on a per sample basis the enrichment of the APOBEC mutation signature among all mutated cytosines in comparison to the fraction of cytosines that occur in the tCw motif among the +/- 20 nucleotides surrounding each mutated cytosine (“APOBEC_enrich” column in data files). The minimum estimate of the number of APOBEC-induced mutations in a sample (APOBEC_MutLoad_MinEstimate) was calculated using the formula: [“tCw→G+tCw→T”]x[(“APOBEC_enrich”-1)/“APOBEC_enrich”], which allows estimating the number of APOBEC signature mutations in excess of what would be expected by random mutagenesis. “APOBEC_MutLoad_MinEstimate” was calculated only for samples passing 0.05 FDR threshold for APOBEC enrichment ([“BH_Fisher_p-value_tCw”]<0.05. Samples with “BH_Fisher_p-value_tCw” value greater than 0.05 received a value of 0. The “APOBEC_MutLoad_MinEstimate” value shows high correlation (0.9–0.95) with all other parameters used to characterize the APOBEC mutagenesis pattern, such as APOBEC enrichment as well as absolute and relative APOBEC mutation loads. For some analyses and figures, the “APOBEC_MutLoad_MinEstimate” parameter was converted into categorical values as follows:
“no”: “APOBEC_MutLoad_MinEstimate”=0
“low”: 0<“APOBEC_MutLoad_MinEstimate”≤median of non-zero values
“high”: “APOBEC_MutLoad_MinEstimate”>median of non-zero values
The median of non-zero values in the Extended Set = 33.
Methylation analysis
The Illumina Infinium HM450 array58 was used to evaluate DNA methylation in the Core Set of cervical cancer samples. Unsupervised consensus clustering was performed with Euclidean distance and partitioning around medoids (PAM) using the most variable 1% of CpG island promoter probes. Epigenetically silenced genes were identified as previously described59. A total of 120 normal samples were used for this analysis by selecting 10 samples at random from the 12 TCGA projects that included normal samples.
RNA-seq analysis
RNA was extracted, converted into mRNA libraries, and paired-end sequenced (paired 50 nt reads) on Illumina HiSeq 2000 Genome Analyzers as previously described5. RNA reads were aligned to the hg19 genome assembly using Mapsplice v12_0760. Gene expression was quantified for the transcript models corresponding to the TCGA GAF2.1 (https://tcga-data.nci.nih.gov/docs/GAF/GAF.hg19.June2011.bundle/outputs/TCGA.hg19.June2011.gaf.) using RSEM461 and normalized within a sample to a fixed upper quartile. To predict whether a cancer sample was from the cervix or the uterus, the data matrix of normalized gene-level RSEM values from 170 UCEC samples was merged with the data matrix from the Core Set (n=178) of cervical cancers. This merged dataset was then randomly split into a training set (87 CESC samples; 86 UCEC samples) and a test set (91 CESC samples; 84 UCEC samples). A sample was predicted to be CESC if the t-statistic vs. UCEC was significant (p<0.05), but was not significantly different from the CESC mean (and vice versa for the UCEC prediction). A data matrix of RSEM values from 178 CESC, 170 UCEC, and 279 HNSC samples was used to identify expression patterns across the 3 cancer types. The gene expression matrix was further filtered to only include the top 25% most variable genes by mean absolute deviation (n=4,039 genes).
EMT mRNA score analysis
The EMT score was computed as previously described10,21. Briefly, the EMT score was the value resulting from the difference between the average expression of mesenchymal (M) genes minus the average expression of epithelial (E) genes. All NA values were removed from the calculation. Two-sample t-test and ANOVA were applied to each comparison accordingly.
miRNA sequencing and analysis
MicroRNA sequence (miRNA-seq) data was generated for the Core Set of tumor samples using methods described previously11. We identified miRNAs that have been associated with EMT62–66 and then calculated Spearman correlations between the EMT scores and RPMs for 5p and 3p mature strands for each of these miRNAs using MatrixEQTL and filtering by FDR<0.05. An miRNA was considered to be epigenetically controlled if BH-corrected p-values were less than 0.01 for both a) a Spearman correlation of miRNA abundance (RPM) to beta for probes in promoter regions associated with the miRNAs, and for b) a t-test of RPM between unmethylated (β<0.1) and methylated (β>0.3) samples (an “epigenetically-controlled pattern”). We assessed potential miRNA targeting for all 178 samples and then separately for the 144 squamous samples by calculating miR-mRNA and miR-protein (RPPA) Spearman correlations with MatrixEQTL v2.1.1 using gene-level normalized abundance RNA-seq (RSEM) data and normalized RPPA data. Correlations were calculated with a p-value threshold of 0.05, and then the anti-correlations were filtered at FDR<0.05. We extracted miR-gene pairs that corresponded to functional validation publications reported by miRTarBase v4.522. For miR-RPPA anti-correlations, all gene names that were associated with each antibody were used. Results were displayed with Cytoscape v2.8.3.
PARADIGM analysis
Integration of copy number, RNA-seq, and pathway interaction data was performed on the Core Set of samples using PARADIGM33,34. Briefly, PARADIGM infers integrated pathway levels (IPLs) for genes, complexes, and processes using pathway interactions, genomic, and functional genomic data from each patient sample. One was added to all expression values, which were then log2-transformed and median-centered across samples for each gene. The log2-transformed, median-centered mRNA data were rank-transformed based on the global ranking across all samples and all genes and discretized (+1 for values with ranks in the highest tertile, -1 for values with ranks in the lowest tertile, and 0 otherwise) prior to PARADIGM analysis.
Pathways were obtained in BioPax Level 3 format, and included the NCIPID and BioCarta databases from http://pid.nci.nih.gov and the Reactome database from http://reactome.org. Gene identifiers were unified by UniProt ID and then converted to Human Genome Nomenclature Committee’s HUGO symbols using mappings provided by HGNC (http://www.genenames.org/). Altogether, 1524 pathways were obtained. Interactions from all of these sources were then combined into a merged Superimposed Pathway (SuperPathway). Genes, complexes, and abstract processes (e.g. “cell cycle” and “apoptosis”) were retained and henceforth referred to collectively as pathway features. The resulting pathway structure contained a total of 19504 features, representing 7369 protein-coding genes, 9354 complexes, 2092 families, 82 RNAs, 15 miRNAs, and 592 abstract processes.
The PARADIGM algorithm infers an IPL for each pathway element that reflects the log likelihood contrasting the probability of activity against inactivity. An initial minimum variation filter (at least 1 sample with absolute activity > 0.05) was applied, resulting in 15502 concepts (5898 protein-coding genes, 7307 complexes, 1916 families, 12 RNAs, 15 miRNAs, and 354 abstract processes) with relative activities showing distinguishable variation across tumors.
iCluster analysis
Integrative clustering of RNA-seq, methylation, copy number, and miRNA data was performed using R package “iCluster20.” The Core Set of samples was used since all samples in this Set had data available across these four platforms. RNA-seq, methylation, copy number, and mature-strand miRNA datasets had 20531, 395552, 23109, and 1213 features, respectively. The 500 most variable features based on the standard deviation from each dataset were selected for the integrative clustering analyses. For analysis involving the RNA-seq and miRNA datasets, a log(x+1) transformation was used in order to deal with skewness in the data67. Methylation data was logit transformed to make it closer to normal distribution. The copy number variation data included the regions determined from GISTIC2.0, with copy number variation treated as a continuous measurement based on the segmentation mean value for the region.
MEMo analysis
High DNA methylation levels upstream of miR-200a and miR-200b corresponded to transcriptional downregulation of the miRs (Extended Data Fig. 9a). For a sample to be called altered for either miR-200a or miR-200b (or both), we required both high DNA methylation level upstream of the miR (β-value>0.3) and low miR expression (log2(RPM) < 9.3 for miR-200a and log2(RPM) < 9 for miR-200b). Binary calls were given to altered and unaltered samples based on this double threshold (1 = altered, 0 = unaltered).
The Mutual Exclusivity Modules in cancer (MEMo) algorithm27 was run on all Core Set samples. MEMo was initially run on 27 regions of recurrent copy number gain, 36 of copy number loss, and 22 recurrently mutated genes. In order to include alterations for miR-200a and miR-200b in the MEMo analysis, a custom network was designed where each miR was connected to its known and validated targets (see above). Second, this network was merged with the comprehensive pathway network used by MEMo to search for modules of altered genes that include at least one of the miRs. Extracted modules were tested for mutual exclusivity using MEMo’s statistical framework (Supplemental Table 27). Student’s t-test was performed for comparing EMT mRNA scores between groups.
Extended Data
Extended Data Figure 1. Sample sets and histologic patterns of cervical cancer.
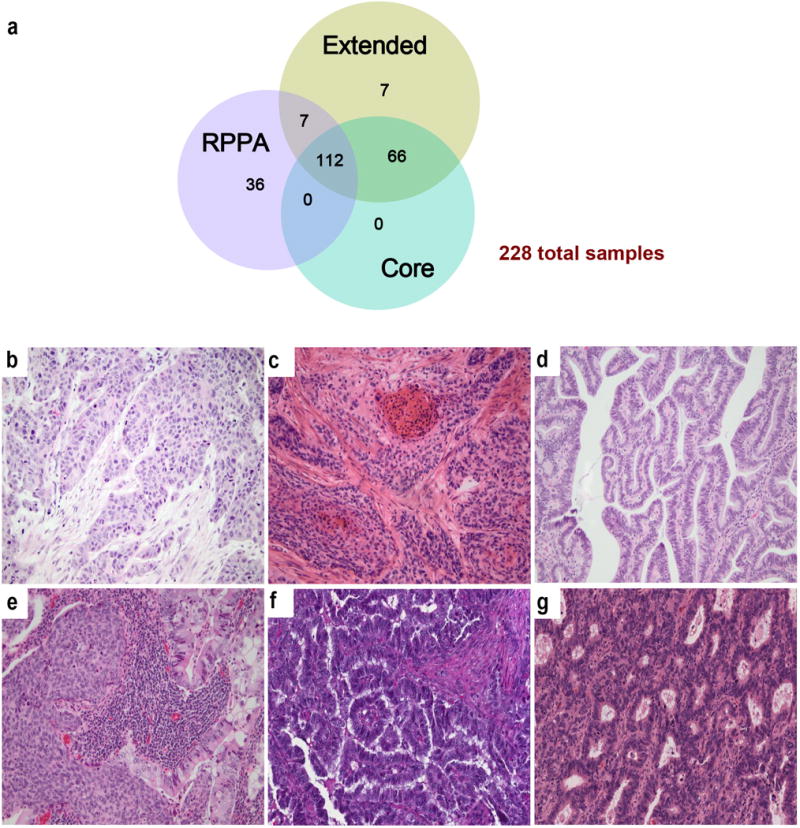
a, Summary of sample numbers and degree of overlap between the Core, Extended, and RPPA datasets. b, Squamous cell carcinoma of the large cell non-keratinizing type. Tongues of highly atypical polygonal neoplastic squamous cells infiltrate through a fibrotic stroma. The cells show abundant eosinophilic cytoplasm with pleomorphic nuclei and prominent mitotic figures. Although the tumor cells contain abundant cytokeratin filaments, this tumor has traditionally been termed “non-keratinizing” because of the absence of characteristic keratin pearls. c, Squamous cell carcinoma of the large cell keratinizing type. Nests of atypical squamous cells infiltrate through a fibrotic stroma. In addition, this tumor shows highly eosinophilic keratin pearls with small, inky dark nuclei that imperfectly mimic the normal keratinization that is found in the epidermis. This differentiation pattern is aberrant in the cervix in which the squamous epithelium is normally a non-keratinizing squamous mucosa. d, Adenocarcinoma of endocervical type (well-differentiated). Closely set, atypical glands with enlarged nuclei and scattered mitotic figures infiltrate through the connective tissue of the cervix. The tall columnar tumor cells show basally-placed, crowded, enlarged nuclei that show frequent mitotic figures. Compared with normal endocervical cells, the tumor cells show relative loss of intra-cytoplasmic mucin and are frequently called “mucin-depleted,” although most, but not all endocervical adenocarcinomas show varying amounts of intracytoplasmic mucin at least focally. e, Adenosquamous carcinoma of cervix. This tumor shows both nests of non-keratinizing squamous cell carcinoma and glands composed of tall columnar adenocarcinoma reflecting the origin of most cervical cancers in the transformation zone of the cervix in which both squamous and glandular cells normally differentiate. Despite this biphasic differentiation potential, adenosquamous carcinomas are relatively uncommon in the cervix. f, UCEC-like HPV negative adenocarcinoma of endocervical type from a radical hysterectomy specimen. The endometrium in the uterus was benign. g, UCEC-like HPV positive adenocarcinoma of endocervical type from a radical hysterectomy specimen. The endometrium in the uterus was benign. All samples were stained with hematoxylin and eosin (20×).
Extended Data Figure 2. Significantly mutated genes and the role of APOBEC in cervical cancer mutagenesis.
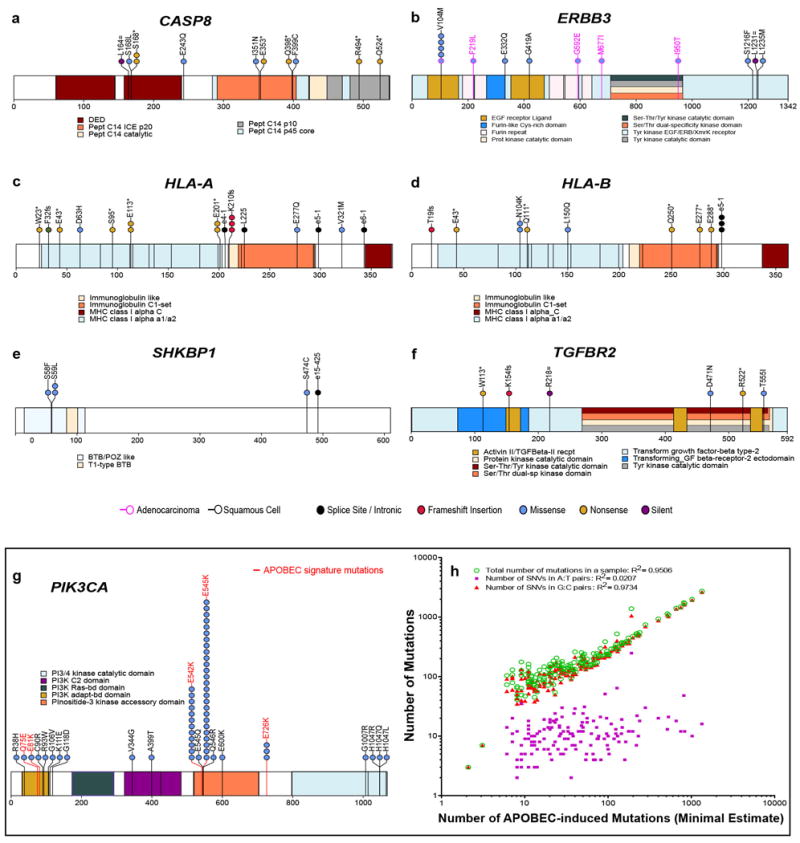
a–f, High-confidence somatic mutations in significantly mutated genes (SMGs) among 192 exome-sequenced samples in the Extended case set are shown. Domains are labeled in accordance with Gencode 19 corresponding to Ensembl 74. Mutations at canonical intronic splice acceptor (e-1 and e-2) are labeled based on proximity to the nearest coding exon. Panels display somatic mutations detected in novel cervical cancer SMGs, with HLA-B included for comparison with its family member HLA-A. Each axis is the protein-coding portion of a gene and each highlighted section represents the UniProt functional domain. Vertical lines indicate the boundaries of multiple annotation sources within common domain annotations as outlined in Supplemental Table 5. Horizontal lines distinguish overlapping domains. Circles represent a single mutation and are colored based on mutation type. Mutations present in squamous cell carcinomas are outlined in black while those present in adenocarcinomas are outlined in pink. g, PIK3CA mutations and recurrence are shown in a stacked circle plot, as above. Additionally, lolliplot sticks are colored red if the mutation type coincides with patterns of APOBEC mutagenesis. h, The minimal estimated number of APOBEC-induced mutations (“APOBEC_MutLoad_MinEstimate” column in Supplemental Table 1) strongly correlates with total number of mutations in a sample, as well as with the number of single nucleotide variants (SNVs) in G:C pairs which are the exclusive substrate for mutagenesis by APOBEC cytidine deaminases. While correlation with mutagenesis in A:T base pairs, which cannot be mutated by APOBEC enzymes is statistically significant (two-tailed P=0.047), it is very weak. Pearson correlation and R2 were calculated for all 192 exome-sequenced samples, including samples with zero values. Only samples with non-zero values of “APOBEC_MutLoad_MinEstimate” are presented.
Extended Data Figure 3. Copy number alterations in cervical cancer.
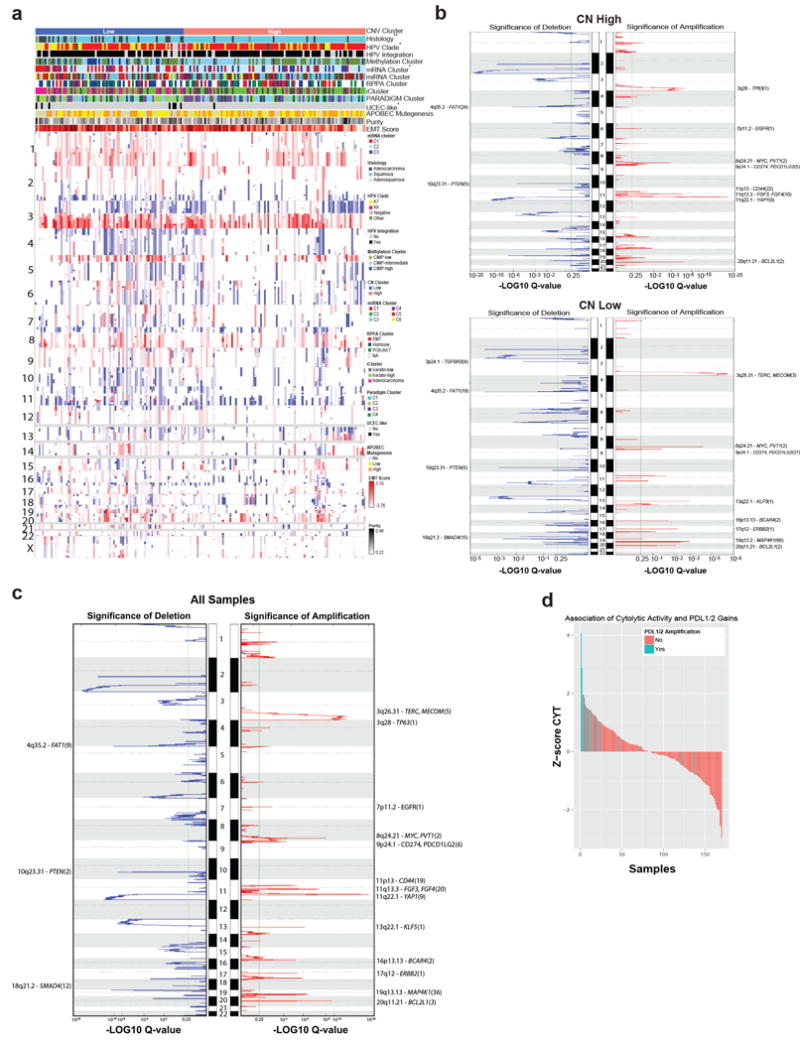
a, Log2-centered heatmap of somatic copy number alterations across 178 Core Set cervical tumors. The x-axis includes samples that have been ordered based on the cluster assignment. The y-axis is based on genomic position, from 1p to Xq. Features associated with copy number clusters are annotated with * or **. *: p<0.05; **: p<0.01. b, GISTIC2.0 amplification and deletion plots within copy number clusters. Chromosomal locations for peaks of significantly recurrent focal amplifications (red, right side) and deletions (blue, left side) are plotted by −LOG10 q-value for the CN High (top) and CN Low (bottom) copy number clusters. Peaks are annotated with cytoband and candidate driver genes. The total number of genes in the peak region is indicated in parenthesis. Peaks with more than 30 genes in the peak region are excluded. Any genes annotated have a significant positive correlation with mRNA expressions. c, Chromosomal locations for peaks of significantly recurrent focal amplifications (red, right side) and deletions (blue, left side) are plotted by −LOG10 q-value for all Core Set samples. Peaks are annotated with cytoband and candidate driver genes. The total number of genes in the peak region is indicated in parentheses. Peaks consisting of more than 30 genes in the peak region are excluded. Annotated genes have a significant positive correlation with mRNA expression. d, Cytolytic activity (CYT) associations with PDL-1/2 amplification. Each bar represents a single tumor and the height of that bar represents the z-score of that tumor’s CYT compared with the rest of the cohort. Bars are colored according to their PD-L1/2 amplification status and sorted from high z-scores to lowest.
Extended Data Figure 4. Gene expression patterns and fusion genes found in cervical cancer.
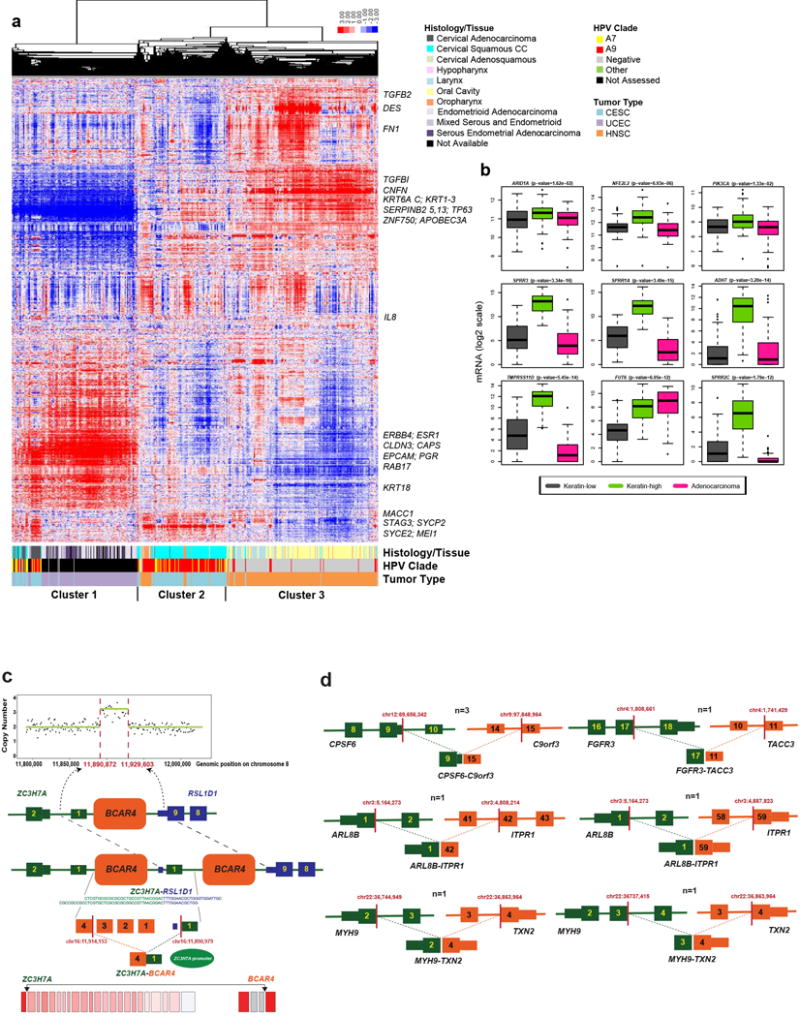
a, Hierarchical clustering (uncentered correlation with centroid linkage as the clustering method) was performed on 4,039 expressed and highly variable genes across 178 cervical, 170 endometrial, and 279 head and neck cancer samples. Normalized gene-level RSEM values were median-centered prior to clustering and relative increased expression values are indicated by red color while relative decreased expression values are indicated by blue color. Cervical, endometrial, and head and neck cancer samples are indicated by different colors as noted in the figure at the bottom of the heatmap. Also included are indications of HPV status, histology of cervical and endometrial cancers, and tissue site for head and neck cancer samples. Select genes are noted to the right of their locations on the heatmap. b, Boxplots of the three differentially expressed SMGs and top six significantly differentially expressed non-SMGs across the iCluster groups using Kruskal Wallis test. All genes are significantly different across the Keratin-low and Keratin-high clusters. Significant p-values across Keratin-low and Keratin-high clusters are presented. c, A schematic of BCAR4 tandem duplication in one case (C5-A3HF), detected by analysis of somatic copy number (top) and structural variation (middle). Split reads and genomic breakpoints indicating the tandem duplication are shown. At the RNA level (bottom) the last exon of BCAR4 forms a fusion gene with the first exon of ZC3H7A (red bars indicate location of mRNA breakpoints; NR_024049 shown as BCAR4 representative transcript). d, Schematic of recurrent fusions (CPSF6-C9orf3, ARL8B-ITPR1, and MYH9-TXN2) or fusions with known occurrences in other cancer types (FGFR3-TACC3), detected by at least two RNA-seq fusion callers in 178 samples. Red bars indicate the mRNA breakpoints.
Extended Data Figure 5. Unsupervised clusters of DNA methylation data.
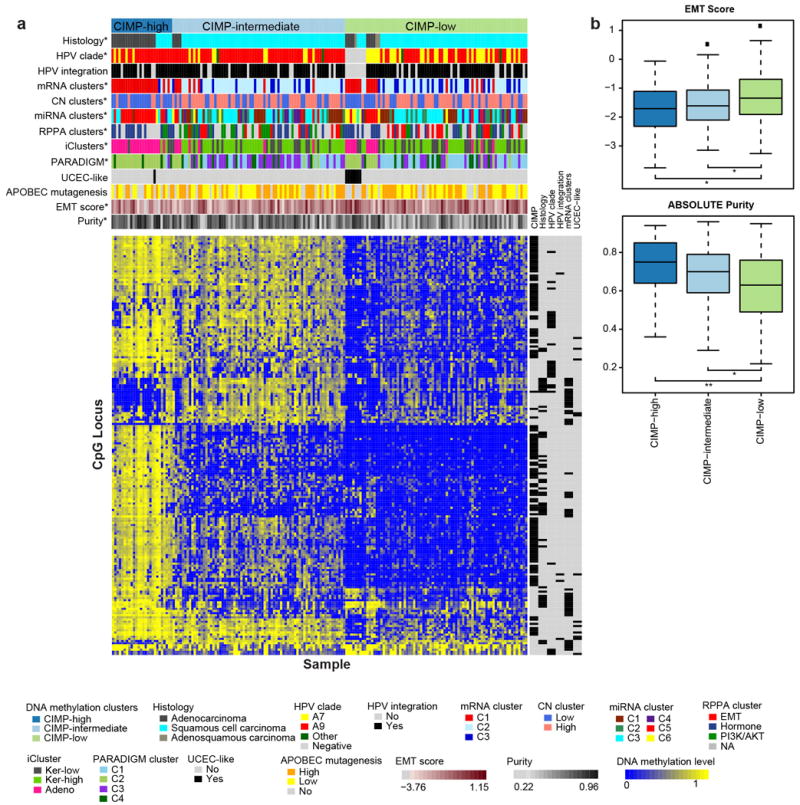
a, Heatmap showing beta values of 178 Core Set samples ordered by CIMP clusters. Samples are presented in columns and the CpG island promoter CpG loci are presented in rows. An annotation panel on the right of the heatmap indicates CpG loci that are differentially methylated within a particular feature (see Supplemental Table 13). All features (marked with *) are statistically significantly associated with DNA methylation clusters (Fisher’s Exact test p-value <0.01) except APOBEC mutagenesis level, UCEC-like status, and HPV integration status. b, Box plots of the EMT mRNA score and tumor purity by CIMP clusters. Student’s t-test p-value <0.01 (**) and <0.05 (*) are reported.
Extended Data Figure 6. miRNA clusters and miR-gene/protein anti-correlations in cervical cancer.
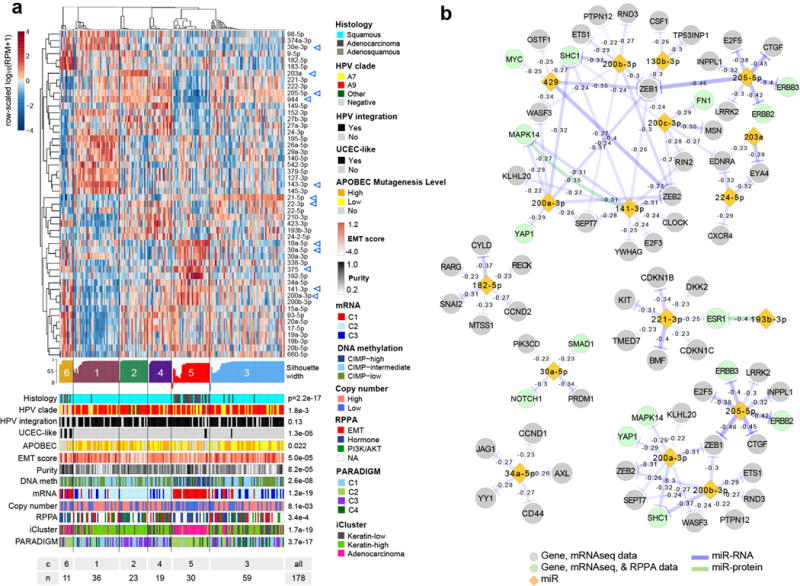
a, Unsupervised clustering for miR profiles across 178 Core Set tumor samples. Top to bottom: a normalized abundance heatmap for the fifty 5p or 3p strands that were highly ranked as differentially abundant by a SAMseq multiclass analysis, silhouette width profile calculated from the consensus membership matrix, a heatmap of tumor sample purity, covariates with association p-values, and a summary table of the number of samples in each cluster. The scale bar shows row-scaled log10(RPM+1) normalized abundances. b, Subnetworks of potential targeting relationships for a subset of miRs, as significance-thresholded (FDR<0.05) miR-mRNA and miR-RPPA anti-correlations that are supported by functional validation publications. For genes (nodes), color distinguishes those that are only present in mRNA data (grey) from those that are present in both mRNA and RPPA data (green). Edges represent anti-correlations, and color distinguishes anti-correlations between a miR and mRNA (purple) and a miR and an unphosphorylated protein (green). In the n=178 Core Set cohort, no correlations satisfying FDR<0.05 were reported between a miR and a phosphorylated protein.
Extended Data Figure 7. EMT-associated miRs and their relationship to miR clusters and TGFβR2 somatic alterations.

a, Normalized miR-200a-3p abundance (RPM) across RPPA clusters for all 112 (top) and 92 squamous (bottom) samples of the Core Set for which RPPA data is available. P-values presented are from two-sided Kolmogorov-Smirnov tests for RPPA-based EMT cluster vs non-EMT cluster samples. For n=112 samples, median miR-200a-3p RPM=296.4 within the EMT cluster (n=29) and 410.0 (n=83) in non-EMT cluster samples. For squamous samples, median miR-200a-3p RPM=296.4 (n=29) within the EMT cluster and 393.4 (n=63) in non-EMT cluster samples. EK-A2R7, which is in the Hormone RPPA cluster, has an RPM value of 4267 and is not shown. Results are not presented for adenocarcinoma samples separately due to limiting sample numbers (n=18 from the Core Set with RPPA data available). b, Negative and positive Spearman correlation coefficients (FDR<0.05) between EMT mRNA score and normalized abundance (RPM) for miRNA mature strands (n=178). miRNAs that have been reported as associated with EMT (see Methods) are highlighted by purple bars. c, Normalized abundance heatmap of miRs most strongly negatively and positively correlated with EMT mRNA scores, with samples grouped by miRNA cluster and sorted by EMT score within each cluster. Somatic mutations (MUT) and deletions (HOMDEL) are shown for TGFBR2, CREBBP, EP300, and SMAD4. Methylation and concomitant downregulated expression alterations (ALT) as defined in Methods for miR-200a/b are also shown. miRs in blue text represent those highlighted by purple bars in b. d–e, Same as b-c, but for the n=144 squamous tumor samples.
Extended Data Figure 8. Distinguishing features of cervical cancer integrated molecular subtypes.
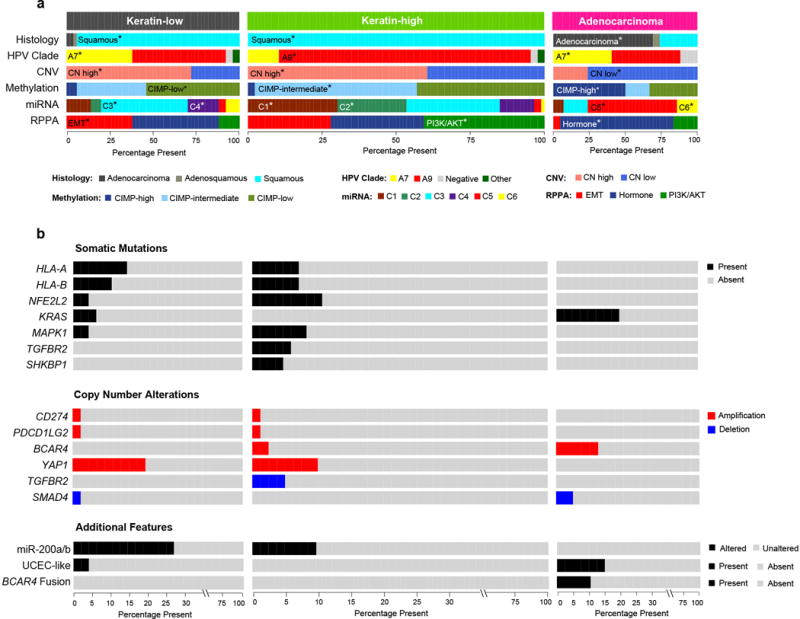
a, Integrative clustering of 178 cervical cancer Core Set cases using mRNA, methylation, miRNA, and copy number data identified three iClusters: (i) Keratin-low, (ii) Keratin-high, and (iii) Adenocarcinoma-rich (Adenocarcinoma; top feature bar). Relative frequencies of various cervical cancer classifications defined by histology, HPV clade, copy number variation (CNV), methylation, miRNA, and RPPA are plotted. The color key for each feature is presented at the bottom. For each category, the statistically significantly enriched features in each iCluster (chi-squared test; p<0.05) are highlighted with asterisks and a listing of the name of the enriched feature. The width of each plot is scaled according to the number of samples within each cluster. b, The frequencies of somatic alterations and additional novel features that distinguish the iClusters, specifically those that do not occur in all three iClusters, are plotted. The “Somatic Mutations” panel shows the presence/absence of mutations for 7 of the identified significantly mutated genes. The “Copy Number Alterations” panel shows select copy number alterations (high level amplifications and focal deletions) that are differentially present across the iClusters. The “Additional Features” panel highlights miscellaneous features that also distinguish the iClusters, including the presence of miR-200a/b alterations, UCEC-like cases, and BCAR4 fusion events. The color key for each feature is present to the right of the plots.
Extended Data Figure 9. miR-200a/b associations with EMT-regulating genes and somatic alterations within RTK, PI3K, MAPK, and TGFβR2 pathways in cervical cancer.
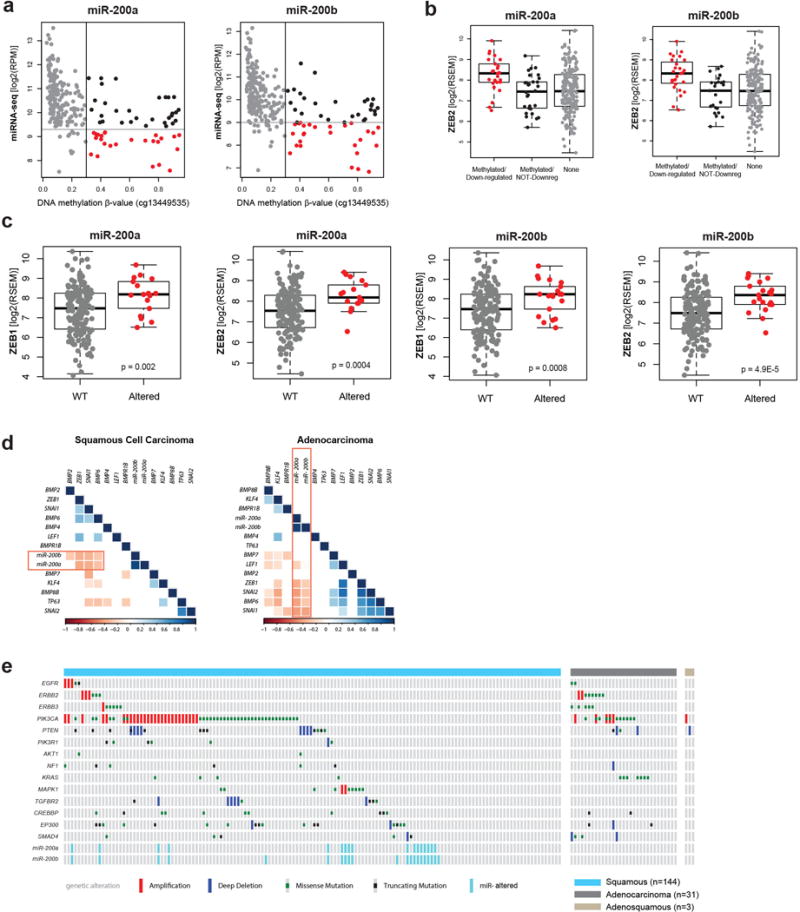
a, Expression levels for miR-200a and miR-200b compared to DNA methylation level at their promoter. Samples were called altered if the miRs were concurrently hypermethylated (β > 0.3) and downregulated (red cases). b, mRNA expression levels for ZEB2, a target of both miR-200a and miR-200b, in subsets of miR-200a/b altered samples. ZEB2 is upregulated in cases with concurrent hypermethylation and downregulation of the miRs. c, mRNA expression levels of both ZEB1 and ZEB2 in miR-200a/b hypermethylated/downregulated (Altered) and all other (WT) samples. d, Correlations of miR-200a and miR-200b expression with multiple genes involved in EMT signaling across squamous cell carcinomas and adenocarcinomas. e, Extent of genetic alterations and miR downregulation in the RTK, PI3K, MAPK, and TGFβ pathways across all cervical tumors.
Extended Data Figure 10. Pathway biomarkers differentiating squamous cell carcinomas and adenocarcinomas.
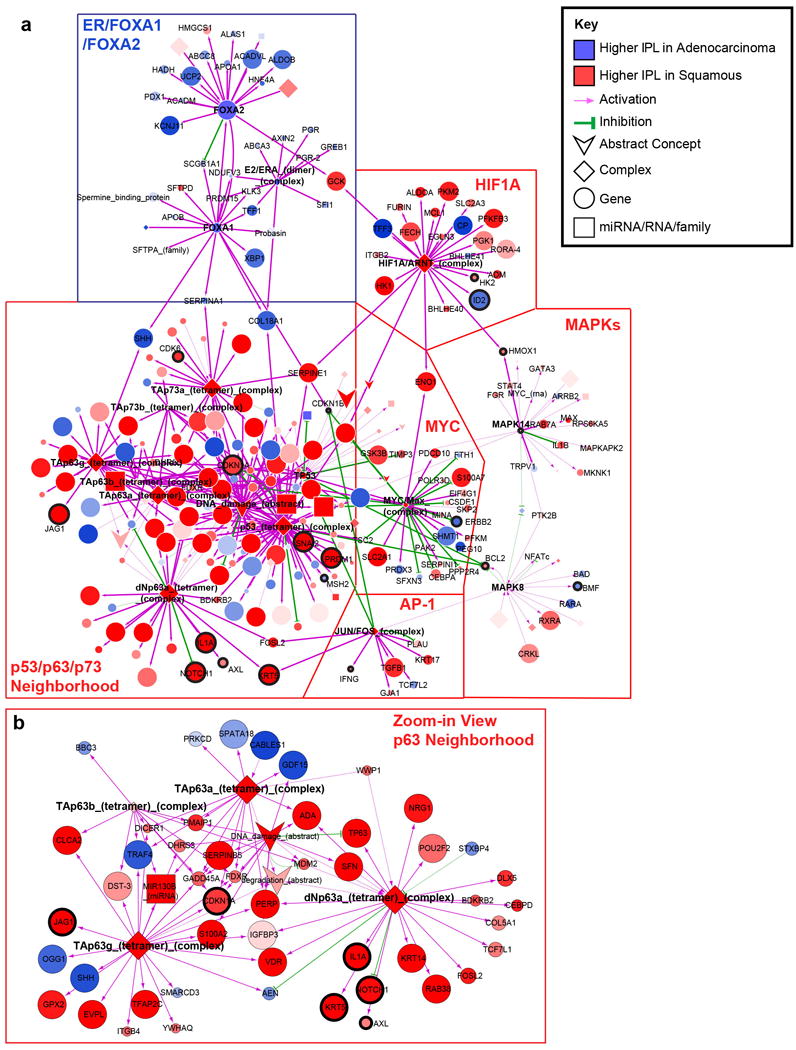
a, Cytoscape display of the largest interconnected regulatory network of PARADIGM pathway features differentially activated between squamous cell carcinomas and adenocarcinomas connected through hubs with ≥ 10 downstream targets. Hubs with ≥ 10 downstream targets are labeled. Genes showing mRNA-miRNA expression anti-correlation with strong evidence support are highlighted with thicker black outline and labeled. Top differentially expressed genes relating to immune function are also labeled. Node size is proportional to significance of differential activation. b, Zoom-in display of the p63 sub-network neighborhood. First neighbors (upstream or downstream) of four p63 complexes (bold text) are displayed in this view.
Extended Data Figure 11. HPV integration and molecular characteristics in cervical cancer.
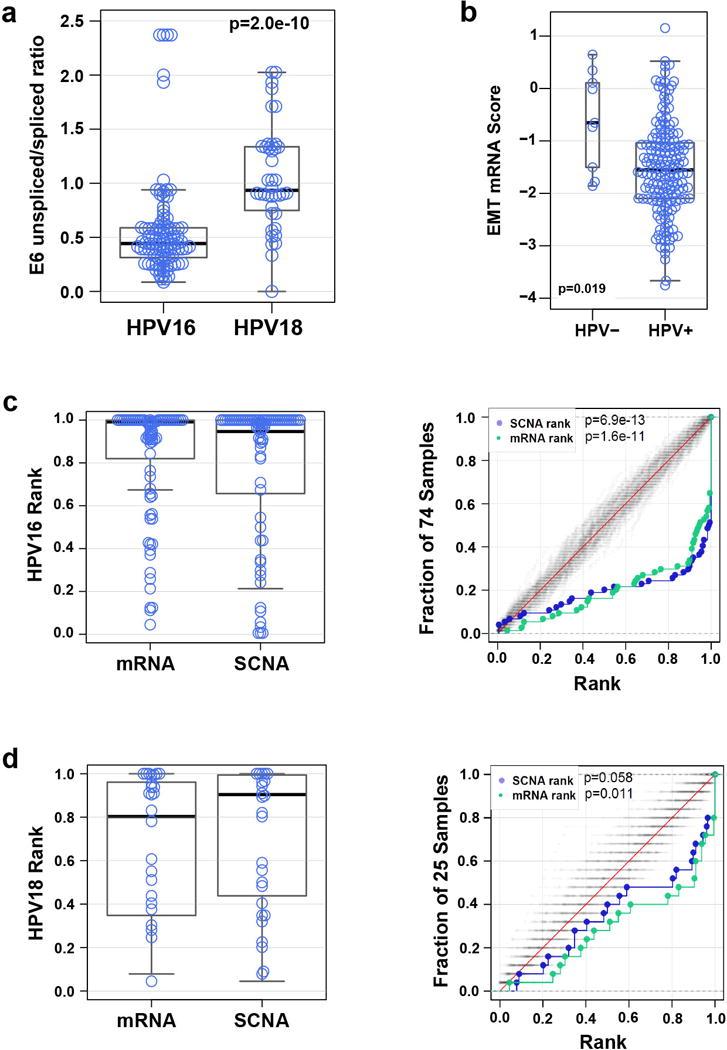
a, E6 unspliced/spliced ratio for HPV16 and HPV18 intragenic, enhancer, and intergenic sites. HPV16: median=0.44 (n=102), HPV18: median=0.93 (n=40). The p-value is from a two-sided Kolmogorov-Smirnov test. b, Distribution of RNAseq-based EMT score for HPV-negative (HPV-) and HPV-positive (HPV+) samples (n=178). c, Distributions of SCNA and mRNA abundance ranks (left panel) and distribution functions for SCNA and mRNA abundance ranks with 100 random expectation samples close to the diagonals (grey) (right panel) for genomic loci integrated with HPV16. d, Distributions described in c for genomic loci integrated with HPV18. BH-corrected p-values for the SCNA and mRNA abundance ranks (median p-values) are reported.
Supplementary Material
*The Cancer Genome Atlas Research Network (Participants are arranged by institution)
Albert Einstein College of Medicine49: Robert D. Burk, Zigui Chen
Analytical Biological Services, Inc.18: Charles Saller, Katherine Tarvin
Barretos Cancer Hospital29: Andre L. Carvalho, Cristovam Scapulatempo-Neto, Henrique C. Silveira, José H. Fregnani
Baylor College of Medicine20: Chad J. Creighton, Matthew L. Anderson, Patricia Castro
Beckman Research Institute of City of Hope19: Sophia S. Wang
Buck Institute for Research on Aging10: Christina Yau, Christopher Benz
Canada’s Michael Smith Genome Sciences Centre5: A. Gordon Robertson, Karen Mungall, Lynette Lim, Reanne Bowlby, Sara Sadeghi, Denise Brooks, Payal Sipahimalani, Richard Mar, Adrian Ally, Amanda Clarke, Andrew J. Mungall, Angela Tam, Darlene Lee, Eric Chuah, Jacqueline E. Schein, Kane Tse, Katayoon Kasaian, Yussanne Ma, Marco A. Marra, Michael Mayo, Miruna Balasundaram, Nina Thiessen, Noreen Dhalla, Rebecca Carlsen, Richard A. Moore, Robert A. Holt, Steven J. M. Jones, Tina Wong
Harvard Medical School14: Angeliki Pantazi, Michael Parfenov, Raju Kucherlapati, Angela Hadjipanayis, Jonathan Seidman, Melanie Kucherlapati, Xiaojia Ren, Andrew W. Xu, Lixing Yang, Peter J. Park, Semin Lee
Helen F. Graham Cancer Center and Research Institute at Christiana Care Health Services39: Brenda Rabeno, Lori Huelsenbeck-Dill, Mark Borowsky, Mark Cadungog, Mary Iacocca, Nicholas Petrelli, Patricia Swanson
HudsonAlpha Institute for Biotechnology56: Akinyemi I. Ojesina
ILSbio, LLC52: Xuan Le
Indiana University School of Medicine45: George Sandusky
Institute of Human Virology, Nigeria46: Sally N. Adebamowo, Teniola Akeredolu, Clement Adebamowo
Institute for Systems Biology21: Sheila M. Reynolds, Ilya Shmulevich
International Genomics Consortium41: Candace Shelton, Daniel Crain, David Mallery, Erin Curley, Johanna Gardner, Robert Penny, Scott Morris, Troy Shelton
Leidos Biomedical42: Jia Liu, Laxmi Lolla, Sudha Chudamani, Ye Wu
Massachusetts General Hospital48: Michael Birrer
McDonnell Genome Institute at Washington University6: Michael D. McLellan, Matthew H. Bailey, Christopher A. Miller, Matthew A. Wyczalkowski, Robert S. Fulton, Catrina C. Fronick, Charles Lu, Elaine R. Mardis, Elizabeth L. Appelbaum, Heather K. Schmidt, Lucinda A. Fulton, Matthew G. Cordes, Tiandao Li, Li Ding, Richard K. Wilson
Medical College of Wisconsin17: Janet S. Rader, Behnaz Behmaram, Denise Uyar, William Bradley
Medical University of South Carolina35: John Wrangle
Memorial Sloan-Kettering Cancer Center11: Alessandro Pastore, Douglas A. Levine, Fanny Dao, Jianjiong Gao, Nikolaus Schultz, Chris Sander, Marc Ladanyi
Montefiore Medical Center36: Mark Einstein, Randall Teeter
NantOmics43: Stephen Benz
National Cancer Institute1: Nicolas Wentzensen, Ina Felau, Jean C. Zenklusen, Clara Bodelon, John A. Demchok, Liming Yang, Margi Sheth, Martin L. Ferguson, Roy Tarnuzzer, Hannah Yang, Mark Schiffman, Jiashan Zhang, Zhining Wang, Tanja Davidsen
National Hospital, Abuja, Nigeria: Olayinka Olaniyan
National Human Genome Research Institute54: Carolyn M. Hutter, Heidi J. Sofia
National Institute of Environmental Health Sciences12: Dmitry A. Gordenin, Kin Chan, Steven A. Roberts, Leszek J. Klimczak
National Institute on Deafness and Other Communication Disorders22: Carter Van Waes, Zhong Chen, Anthony D. Saleh, Hui Cheng
Ontario Tumour Bank, London Health Sciences Centre24: Jeremy Parfitt
Ontario Tumour Bank, Ontario Institute for Cancer Research25: John Bartlett, Monique Albert
Ontario Tumour Bank, The Ottawa Hospital23: Angel Arnaout, Harman Sekhon, Sebastien Gilbert
Oregon Health and Science University55: Myron Peto
Penrose-St. Francis Health Services26: Jerome Myers, Jodi Harr, John Eckman, Julie Bergsten, Kelinda Tucker, Leigh Anne Zach
Samuel Oschin Comprehensive Cancer Institute, Cedars-Sinai Medical Center33: Beth Y. Karlan, Jenny Lester, Sandra Orsulic
SRA International27: Qiang Sun, Rashi Naresh, Todd Pihl, Yunhu Wan
St. Joseph’s Candler Health System28: Howard Zaren, Jennifer Sapp, Judy Miller, Paul Drwiega
The Eli and Edythe L. Broad Institute of Massachusetts Institute of Technology and Harvard University4: Akinyemi I. Ojesina, Bradley A. Murray, Hailei Zhang, Andrew D. Cherniack, Carrie Sougnez, Chandra Sekhar Pedamallu, Lee Lichtenstein, Matthew Meyerson, Michael S. Noble, David I. Heiman, Doug Voet, Gad Getz, Gordon Saksena, Jaegil Kim, Juliann Shih, Juok Cho, Michael S. Lawrence, Nils Gehlenborg, Pei Lin, Rameen Beroukhim, Scott Frazer, Stacey B. Gabriel, Steven E. Schumacher
The Research Institute at Nationwide Children’s Hospital15: Kristen M. Leraas, Tara M. Lichtenberg, Erik Zmuda, Jay Bowen, Jessica Frick, Julie M. Gastier-Foster, Lisa Wise, Mark Gerken, Nilsa C. Ramirez
The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University7: Ludmila Danilova, Leslie Cope, Stephen B. Baylin
The University of Bergen37: Helga B. Salvesen*
The University of Texas MD Anderson Cancer Center2: Christopher P. Vellano, Zhenlin Ju, Lixia Diao, Hao Zhao, Zechen Chong, Michael C. Ryan, Emmanuel Martinez-Ledesma, Roeland G. Verhaak, Lauren Averett Byers, Yuan Yuan, Ken Chen, Shiyun Ling, Gordon B. Mills, Yiling Lu, Rehan Akbani, Sahil Seth, Han Liang, Jing Wang, Leng Han, John N. Weinstein, Christopher A. Bristow, Wei Zhang, Harshad S. Mahadeshwar, Huandong Sun, Jiabin Tang, Jianhua Zhang, Xingzhi Song, Alexei Protopopov, Kenna R. Mills Shaw, Lynda Chin
University of Abuja Teaching Hospital51: Oluwole Olabode
University of Alabama at Birmingham3: Akinyemi I. Ojesina
University of California, Irvine50: Philip DiSaia
University of California Santa Cruz38: Amie Radenbaugh, David Haussler, Jingchun Zhu, Josh Stuart
University of Kansas Medical Center9: Prabhakar Chalise, Devin Koestler, Brooke L. Fridley, Andrew K. Godwin, Rashna Madan
University of Lausanne53: Giovanni Ciriello
University of New Mexico Health Sciences Center40: Cathleen Martinez, Kelly Higgins, Therese Bocklage
University of North Carolina at Chapel Hill8: J. Todd Auman, Charles M. Perou, Donghui Tan, Joel S. Parker, Katherine A. Hoadley, Matthew D. Wilkerson, Piotr A. Mieczkowski, Tara Skelly, Umadevi Veluvolu, D. Neil Hayes, W. Kimryn Rathmell, Alan P. Hoyle, Janae V. Simons, Junyuan Wu, Lisle E. Mose, Matthew G. Soloway, Saianand Balu, Shaowu Meng, Stuart R. Jefferys, Tom Bodenheimer, Yan Shi, Jeffrey Roach, Leigh B. Thorne, Lori Boice, Mei Huang, Corbin D. Jones
University of Oklahoma Health Sciences Center13: Rosemary Zuna, Joan Walker, Camille Gunderson, Carie Snowbarger, David Brown, Katherine Moxley, Kathleen Moore, Kelsi Andrade, Lisa Landrum, Robert Mannel, Scott McMeekin, Starla Johnson, Tina Nelson
University of Pittsburgh30: Esther Elishaev, Rajiv Dhir, Robert Edwards, Rohit Bhargava
University of São Paulo, Ribeirão Preto Medical School16: Daniel G. Tiezzi, Jurandyr M. Andrade, Houtan Noushmehr, Carlos Gilberto Carlotti, Jr., Daniela Pretti da Cunha Tirapelli
University of Southern California31: Daniel J. Weisenberger, David J. Van Den Berg, Dennis T. Maglinte, Moiz S. Bootwalla, Phillip H. Lai, Timothy Triche, Jr.
University of Washington32: Elizabeth M. Swisher, Kathy J. Agnew
University of Wisconsin School of Medicine and Public Health47: Carl Simon Shelley
Van Andel Research Institute34: Peter W. Laird
Washington University in St. Louis44: Julie Schwarz, Perry Grigsby, David Mutch
* Deceased
1National Cancer Institute, Bethesda, Maryland 20892; 2The University of Texas MD Anderson Cancer Center, Houston, Texas 77030; 3University of Alabama at Birmingham, Birmingham, Alabama 35294; 4The Eli and Edythe L. Broad Institute of Massachusetts Institute of Technology and Harvard University, Cambridge, Massachusetts 02142; 5Canada’s Michael Smith Genome Sciences Centre, BC Cancer Agency, Vancouver, British Columbia, Canada V5Z 4S6; 6McDonnell Genome Institute at Washington University, St. Louis, Missouri 63108; 7The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University, Baltimore, Maryland 21287; 8University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599; 9University of Kansas Medical Center, Kansas City, Kansas 66160; 10Buck Institute for Research on Aging., Novato, California 94945; 11Memorial Sloan Kettering Cancer Center, New York, New York 10065; 12National Institute of Environmental Health Sciences, Durham, North Carolina 27709; 13University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma 73104; 14Harvard Medical School, Boston, Massachusetts 02115; 15The Research Institute at Nationwide Children’s Hospital, Columbus, Ohio 43205; 16University of São Paulo, Ribeirão Preto Medical School, Ribeirão Preto - SP, Brazil, 14049-900; 17Medical College of Wisconsin, Milwaukee, Wisconsin 53226; 18Analytical Biological Services, Inc., Wilmington, Delaware 19801; 19Beckman Research Institute of City of Hope, Duarte, California 91010; 20Baylor College of Medicine, Houston, Texas 77030; 21Institute for Systems Biology, Seattle, Washington 98109; 22National Institute on Deafness and Other Communication Disorders, Bethesda, Maryland 20892; 23Ontario Tumour Bank, The Ottawa Hospital, Ottawa, Ontario, Canada K1H 8L6; 24Ontario Tumour Bank, London Health Sciences Centre, London, Ontario, Canada N6A 5A5; 25Ontario Tumour Bank, Ontario Institute for Cancer Research, Toronto, Ontario, Canada M5G 0A3; 26Penrose-St. Francis Health Services, Colorado Springs, Colorado 80906; 27SRA International, Fairfax, Virginia 22033; 28St. Joseph’s Candler Health System, Savannah, Georgia 31406; 29Barretos Cancer Hospital, Barretos, Sao Paulo, Brazil; 30University of Pittsburgh, Pittsburgh Pennsylvania 15213; 31University of Southern California, Los Angeles, California 90033; 32University of Washington, Seattle, Washington 981095; 33Samuel Oschin Comprehensive Cancer Institute, Cedars-Sinai Medical Center, Los Angeles, California 90048; 34Van Andel Research Institute, Grand Rapids, Michigan 49503; 35Medical University of South Carolina, Charleston, South Carolina 29425; 36Montefiore Medical Center, Bronx, New York 10461; 37The University of Bergen, Bergen, Norway; 38University of California Santa Cruz, Santa Cruz, California 95064; 39Helen F. Graham Cancer Center and Research Institute at Christiana Care Health Services, Inc., Newark, Delaware 19713; 40University of New Mexico Health Sciences Center, Albuquerque, New Mexico 87131; 41International Genomics Consortium, Phoenix, Arizona 85004; 42Leidos Biomedical, Rockville, Maryland 20850; 43NantOmics, Santa Cruz, California 95060; 44Washington University in St. Louis, St. Louis, Missouri 63110; 45Indiana University School of Medicine, Indianapolis, Indiana 46202; 46Institute of Human Virology, Nigeria, Abuja, Nigeria; 47University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin 53705; 48Massachusetts General Hospital, Boston, Massachusetts 02114; 49Albert Einstein College of Medicine, Bronx, New York 10461; 50University of California, Irvine, Orange, California 92668; 51University of Abuja Teaching Hospital, Gwagwalada, Abuja, Nigeria; 52ILSbio, LLC, Chestertown, Maryland 21620; 53University of Lausanne, Lausanne, Switzerland; 54National Human Genome Research Institute, Bethesda, Maryland 20892; 55Oregon Health and Science University, Portland, Oregon 97201; 56HudsonAlpha Institute for Biotechnology, Huntsville, Alabama 35806
Author Contributions
The Cancer Genome Atlas research network contributed collectively to this work. Biospecimens were collected at the Tissue Source Sites (TSSs) and processed by the Biospecimen Core Resource (BCR). Data was generated by the genome sequencing and genome data analysis centers, with analyses performed by members across the network. Data was stored and released through the data coordinating center (DCC). The NCI Project Coordinator was Ina Felau1 and the overall Analysis Coordinator and Data Coordinator was Christopher P. Vellano2. Special thanks also go out to TCGA network members who made substantial contributions to this work: Christopher P. Vellano2 (Analysis Coordinator; Data Coordinator; Co-manuscript Coordinator; RPPA analysis), Nicolas Wentzensen1 (Co-manuscript Coordinator; HPV analysis subgroup co-leader), Akinyemi I. Ojesina3,4,56 (Co-manuscript Coordinator; HPV analysis subgroup co-leader; somatic alteration analysis), A. Gordon Robertson5 (miRNA analysis; HPV analysis), Michael D. McLellan6 (mutation calling); Ludmila Danilova7 (methylation analysis); Bradley A. Murray4 (copy number and ABSOLUTE analysis); Zhenlin Ju2 (RPPA analysis); J. Todd Auman8 (mRNA sequencing analysis; fusion analysis); Prabhakar Chalise9 (iCluster analysis); Christina Yau10 (PARADIGM pathway analysis); Giovanni Ciriello53 (MEMo pathway analysis); Dmitry A. Gordenin12 (APOBEC analysis); Rosemary Zuna13 (Pathologist); Hailei Zhang4 (mutation analysis; Firehose); Angeliki Pantazi14 (structural variant analysis subgroup leader; low-pass sequencing); Matthew H. Bailey6 (mutation analysis); Lixia Diao2 (EMT analysis); Devin Koestler9 (methylation data processing; FEM analysis); Karen Mungall5 (HPV analysis); Lynette Lim5 (HPV analysis); Reanne Bowlby5 (miRNA analysis); Sara Sadeghi5 (HPV analysis); Denise Brooks5 (miRNA analysis); Chandra Sekhar Pedamallu4 (HPV analysis); Ken Chen2 (fusion analysis); Hao Zhao2 (fusion analysis); Zechen Chong2 (fusion analysis); Emmanuel Martinez-Ledesma2 (fusion analysis); Roeland G. Verhaak2 (fusion analysis); Kristen M. Leraas15 (BCR); Tara M. Lichtenberg15 (BCR); Daniel G. Tiezzi16 (immune response gene analysis); Michael C. Ryan2 (splicing analysis); Sheila M. Reynolds21 (Regulome Explorer analysis); Gordon B. Mills2 (Project Co-chair); and Janet S. Rader17 (Project Co-chair).
Footnotes
Author Information: The primary and processed data used in analyses can be downloaded by registered users from https://gdc-api.nci.nih.gov/v0/data/a0bb9765-3f03-485b-839d-7dce4a9bcfeb and the TCGA publication page (https://tcga-data.nci.nih.gov/docs/publications/cesc_2016/).
Competing Financial Interests:
There are no competing financial interests.
References
- 1.Ferlay J, et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Schiffman M, et al. Human papillomavirus testing in the prevention of cervical cancer. J Natl Cancer Inst. 2011;103:368–383. doi: 10.1093/jnci/djq562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uyar D, Rader J. Genomics of cervical cancer and the role of human papillomavirus pathobiology. Clin Chem. 2014;60:144–146. doi: 10.1373/clinchem.2013.212985. [DOI] [PubMed] [Google Scholar]
- 4.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 5.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawrence MS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung TKH, et al. Genomic aberrations in cervical adenocarcinomas in Hong Kong Chinese women. Int J Cancer. 2015;137:776–783. doi: 10.1002/ijc.29456. [DOI] [PubMed] [Google Scholar]
- 8.Ojesina AI, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371–375. doi: 10.1038/nature12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet. 2013;45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henderson S, Chakravarthy A, Su X, Boshoff C, Fenton TR. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Reports. 2014;7:1833–1841. doi: 10.1016/j.celrep.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 15.Roberts SA, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Godinho MFE, et al. BCAR4 induces antioestrogen resistance but sensitises breast cancer to lapatinib. Br J Cancer. 2012;107:947–955. doi: 10.1038/bjc.2012.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen R, Olshen AB, Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009;25:2906–2912. doi: 10.1093/bioinformatics/btp543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byers LA, et al. An epithelial–mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279–290. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu SD, et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014;42:D78–D85. doi: 10.1093/nar/gkt1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akbani R, et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun. 2014;5:3887. doi: 10.1038/ncomms4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seton-Rogers S. Oncogenes: All eyes on YAP1. Nat Rev Cancer. 2014;14:514–515. doi: 10.1038/nrc3791. [DOI] [PubMed] [Google Scholar]
- 25.Shao Diane D, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014;158:171–184. doi: 10.1016/j.cell.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vandewalle C, Van Roy F, Berx G. The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci. 2009;66:773–787. doi: 10.1007/s00018-008-8465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012;22:398–406. doi: 10.1101/gr.125567.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gregory PA, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 29.Massague J. TGF[beta] signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haslehurst A, et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer. 2012;12:91. doi: 10.1186/1471-2407-12-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taube JH, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci USA. 2010;107:15449–15454. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaiswal Bijay S, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013;23:603–617. doi: 10.1016/j.ccr.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 33.Sedgewick AJ, Benz SC, Rabizadeh S, Soon-Shiong P, Vaske CJ. Learning subgroup-specific regulatory interactions and regulator independence with PARADIGM. Bioinformatics. 2013;29:i62–i70. doi: 10.1093/bioinformatics/btt229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vaske CJ, et al. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;26:i237–i245. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoadley KA, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 158:929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.den Boon JA, et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc Natl Acad Sci USA. 2015;112:E3255–E3264. doi: 10.1073/pnas.1509322112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roman A, Munger K. The papillomavirus E7 proteins. Virology. 2013;445:138–168. doi: 10.1016/j.virol.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vande Pol SB, Klingelhutz AJ. Papillomavirus E6 oncoproteins. Virology. 2013;445:115–137. doi: 10.1016/j.virol.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiao Y, Widschwendter M, Teschendorff AE. A systems-level integrative framework for genome-wide DNA methylation and gene expression data identifies differential gene expression modules under epigenetic control. Bioinformatics. 2014;30:2360–2366. doi: 10.1093/bioinformatics/btu316. [DOI] [PubMed] [Google Scholar]
- 40.Dellambra E, et al. Downregulation of 14-3-3σ prevents clonal evolution and leads to immortalization of primary human keratinocytes. J Cell Biol. 2000;149:1117–1130. doi: 10.1083/jcb.149.5.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moreira JMA, Gromov P, Celis JE. Expression of the tumor suppressor protein 14-3-3σ is down-regulated in invasive transitional cell carcinomas of the urinary bladder undergoing epithelial-to-mesenchymal transition. Mol Cell Proteomics. 2004;3:410–419. doi: 10.1074/mcp.M300134-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Hermeking H, et al. 14-3-3σ is a p53-regulated inhibitor of G2/M progression. Mol Cell. 1997;1:3–11. doi: 10.1016/s1097-2765(00)80002-7. [DOI] [PubMed] [Google Scholar]
- 43.Chang TC, et al. 14-3-3σ regulates β-catenin-mediated mouse embryonic stem cell proliferation by sequestering GSK-3β. PLoS One. 2012;7:e40193. doi: 10.1371/journal.pone.0040193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wentzensen N, Vinokurova S, von Knebel Doeberitz M. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res. 2004;64:3878–3884. doi: 10.1158/0008-5472.CAN-04-0009. [DOI] [PubMed] [Google Scholar]
- 45.Tang AL, et al. UM-SCC-104: A New human papillomavirus-16–positive cancer stem cell–containing head and neck squamous cell carcinoma cell line. Head & Neck. 2012;34:1480–1491. doi: 10.1002/hed.21962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chu J, et al. BioBloom tools: fast, accurate and memory-efficient host species sequence screening using bloom filters. Bioinformatics. 2014;30:3402–3404. doi: 10.1093/bioinformatics/btu558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kostic AD, et al. PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat Biotechnol. 2011;29:393–396. doi: 10.1038/nbt.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25–R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schiffman M, et al. A population-based prospective study of carcinogenic human papillomavirus variant lineages, viral persistence, and cervical neoplasia. Cancer Res. 2010;70:3159–3169. doi: 10.1158/0008-5472.CAN-09-4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCarroll SA, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 52.Korn JM, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 54.Mermel CH, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41–R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carter SL, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen K, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang L, et al. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell. 2013;153:919–929. doi: 10.1016/j.cell.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bibikova M, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 59.The Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159:676–690. doi: 10.1016/j.cell.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang K, et al. MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323–323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carstens JL, Lovisa S, Kalluri R. Microenvironment-dependent cues trigger miRNA-regulated feedback loop to facilitate the EMT/MET switch. J Clin Invest. 2014;124:1458–1460. doi: 10.1172/JCI75239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ceppi P, Peter ME. MicroRNAs regulate both epithelial-to-mesenchymal transition and cancer stem cells. Oncogene. 2014;33:269–278. doi: 10.1038/onc.2013.55. [DOI] [PubMed] [Google Scholar]
- 64.Díaz-Martín J, et al. A core microRNA signature associated with inducers of the epithelial-to-mesenchymal transition. J Pathol. 2014;232:319–329. doi: 10.1002/path.4289. [DOI] [PubMed] [Google Scholar]
- 65.Kiesslich T, Pichler M, Neureiter D. Epigenetic control of epithelial-mesenchymal-transition in human cancer. Mol Clin Oncol. 2013;1:3–11. doi: 10.3892/mco.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19:1438–1449. doi: 10.1038/nm.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zwiener I, Frisch B, Binder H. Transforming RNA-seq data to improve the performance of prognostic gene signatures. PLoS One. 2014;9:e85150. doi: 10.1371/journal.pone.0085150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.