

Abstract
In this study, the complete mitochondrial (mt) genome sequence of the South China deep‐sea giant isopod Bathynomus sp. was determined, and this study is the first to explore in detail the mt genome of a deep‐sea member of the order Isopoda. This species belongs to the genus Bathynomus, the members of which are saprophagous residents of the deep‐sea benthic environment; based on their large size, Bathynomus is included in the “supergiant group” of isopods. The mt genome of Bathynomus sp. is 14,965 bp in length and consists of 13 protein‐coding genes, two ribosomal RNA genes, only 18 transfer RNA genes, and a noncoding control region 362 bp in length, which is the smallest control region discovered in Isopoda to date. Although the overall genome organization is typical for metazoans, the mt genome of Bathynomus sp. shows a number of derived characters, such as an inversion of 10 genes when compared to the pancrustacean ground pattern. Rearrangements in some genes (e.g., cob, trnT, nad5, and trnF) are shared by nearly all isopod mt genomes analyzed thus far, and when compared to the putative isopod ground pattern, five rearrangements were found in Bathynomus sp. Two tRNAs exhibit modified secondary structures: The TΨC arm is absent from trnQ, and trnC lacks the DHU. Within the class Malacostraca, trnC arm loss is only found in other isopods. Phylogenetic analysis revealed that Bathynomus sp. (Cymothoida) and Sphaeroma serratum (Sphaeromatidea) form a single clade, although it is unclear whether Cymothoida is monophyletic or paraphyletic. Moreover, the evolutionary rate of Bathynomus sp. (dN/dS [nonsynonymous mutational rate/synonymous mutational rate] = 0.0705) is the slowest measured to date among Cymothoida, which may be associated with its relatively constant deep‐sea environment. Overall, our results may provide useful information for understanding the evolution of deep‐sea Isopoda species.
Keywords: Bathynomus, deep sea, gene rearrangement, Isopoda, mitochondrial genome
1. Introduction
Mitochondria, which are regarded as relicts of bacterial or alpha‐bacterial endosymbionts incorporated into an early eukaryotic cell, have their own genetic material and genetic system. Since the discovery of intraorganellar DNA in chick embryo mitochondria (Nass & Nass, 1963), determination of the structure of mitochondrial genomes (mitogenomes) has become an important aspect of genome research because of the important data it can provide for phylogenetic analyses. In general, the metazoan mitogenome is a circular, double‐stranded DNA molecule approximately 12–20 kb in length that consists of the same 37 genes: 13 protein‐coding subunits, two ribosomal RNAs, and 22 transfer RNAs (Wolstenholme, 1992). Due to the different rates of evolutionary change among different segments of a given mitogenome (Helfenbein, Fourcade, Vanjani, & Boore, 2004; Liebers, de Knijff, & Helbig, 2004), the gene order (Roehrdanz, Degrugillier, & Black, 2002), and the RNA secondary structure (Macey, Schulte, & Larson, 2000), these sequences can provide large datasets for phylogenetic analyses at different levels and can also serve as ideal models of gene rearrangement and genome evolution.
Since Clary and Wolstenholme (1985) sequenced the mitogenome of Drosophila yakuba in 1985, 1,160 Arthropoda mitogenomes have been determined. However, only 203 mitogenomes within the class Crustacea have been determined, and the number of isopod mitogenomes currently available is even lower, with only three complete mitogenomes (one each for Limnoriidae, Ligiidae, and Phreatoicidae) and another eight sequences that are almost complete (one each for Sphaeromatidae, Chaetiliidae, Cirolanidae, Asellidae, Armadillidiidae, Idoteidae, Cylisticidaeand, and Trachelipodidae; see Table 1). Among crustacean mitogenomes, most share the ancestral pancrustacean (crustacean + hexapod) gene order that shows only a trnL‐UUR translocation relative to the ancestral arthropod arrangement found in the horseshoe crab Limulus polyphemus (Lavrov, Boore, & Brown, 2000) or present only minor tRNA translocations (Yang & Yang, 2008). Nonetheless, a broad comparison of mitochondrial (mt) gene order within Crustacea has revealed that some taxa exhibit greater variability, e.g., Copepoda, Cirripedia, Brachyura, and Isopoda (Kilpert & Podsiadlowski, 2006).
Table 1.
All isopod mitogenomes sequenced to date and their nucleotide compositions
| Species | Suborder/infraorder | Accession number | Length (bp) | Entire genome | Protein‐coding gene | rrnL | rrnS | tRNAs | Control region | Reference | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AT% | GC skew | AT skew | Length (aa) | AT% (all) | AT% (3rd) | Length (bp) | AT% | Length (bp) | AT% | Length (bp)/number | AT% | Length (bp) | AT% | |||||
| Bathynomus sp.a | Cymothoida | KU057374 | 14,965 | 58.7 | +0.177 | −0.094 | 3,713 | 57.7 | 59.7 | 1,181 | 60.5 | 742 | 59.7 | 1,080/18 | 63.6 | 362 | 60.2 | This study |
| Limnoria quadripunctata | Cymothoida | KF704000 | 16,515 | 66.4 | +0.190 | −0.102 | 3,692 | 64.8 | 67.4 | 1,367 | 71.6 | 740 | 71.8 | 1,500/24 | 72.6 | 1,525 | 66.0 | Lloyd et al. (2015) |
| Ligia oceanica | Oniscidea | DQ442914 | 15,289 | 60.9 | +0.133 | −0.041 | 3,677 | 60.1 | 64.4 | 1,234 | 65.1 | 850 | 60.2 | 1,278/21 | 65.0 | 737 | 55.8 | Kilpert et al. (2006) |
| Eophreatoicus sp. ‐14 | Phreatoicidae | NC_013976 | 14,994 | 69.6 | +0.250 | −0.103 | 3,687 | 69.1 | 79.0 | 1,224 | 73.2 | 784 | 71.7 | 1,376/22 | 71.8 | 401 | 58.6 | Kilpert et al. (2010) |
| Sphaeroma serratum b | Sphaeromatidea | GU130256 | 13,467 | 54.4 | +0.022 | −0.070 | 3,567 | 53.6 | 50.4 | 1,154 | 62.3 | 651 | 43.1 | 917/15 | 62.8 | – | – | Kilpert et al. (2012) |
| Glyptonotus cf. Antarcticus b | Valvifera | GU130254 | 13,809 | 65.4 | +0.040 | −0.034 | 3,567 | 64.6 | 71.6 | 1,215 | 72.7 | 745 | 62.2 | 1,117/18 | 67.9 | – | – | Kilpert et al. (2012) |
| Eurydice pulchra b | Cymothoida | GU130253 | 13,055 | 55.9 | +0.197 | −0.052 | 3,386 | 55.8 | 61.4 | 1,137 | 59.0 | 698 | 50.5 | 964/16 | 57.3 | – | – | Kilpert et al. (2012) |
| Asellus aquaticus b | Asellota | GU130252 | 13,639 | 61.9 | −0.121 | +0.002 | 3,561 | 60.3 | 66 | 1,168 | 65.9 | 675 | 69.5 | 1,099/18 | 69.3 | – | – | Kilpert et al. (2012) |
| Armadillidium vulgare c | Oniscidea | EF643519 | 13,939 | 71.2 | +0.181 | −0.042 | 3,746 | 70.8 | 82.2 | 600 | 71.7 | 687 | 68.0 | 715/12 | 75.2 | – | – | Marcade et al. (2007) |
| Idotea baltica c | Valvifera | DQ442915 | 14,247 | 61.0 | +0.164 | −0.075 | 3,627 | 60.2 | 64.5 | 1,216 | 65.6 | 794 | 57.8 | 1,051/17 | 65.2 | – | – | Podsiadlowski et al. (2006) |
| Cylisticus convexus c | Oniscidea | KR013002 | 14,154 | 67.8 | +0.196 | −0.035 | 3,668 | 67.5 | 75.0 | 979 | 72.8 | 812 | 60.2 | 1,106/18 | 68.9 | – | – | Chandler et al. (2015) |
| Trachelipus rathkei c | Oniscidea | KR013001 | 14,129 | 67.4 | +0.184 | −0.030 | 3,625 | 66.5 | 74.0 | 975 | 71.9 | 807 | 64.1 | 846/14 | 69.4 | – | – | Chandler et al. (2015) |
Results for Bathynomus sp. from this study are given in bold.
Incomplete determination with a partial cob sequence, and a few tRNAs and the control region were not sequenced.
Incomplete determination, with a few tRNAs and the control region not sequenced.
Approximately 77% of the ocean floor and 60% of our planet's surface are covered with deep‐sea habitats that remain largely unexplored. In these deep‐sea environments, a lack of sunlight, extremely high pressures, and low oxygen levels prevent the formation of typical biological assemblages due to the lack of photosynthesis, which is responsible for biosphere primary production. Nonetheless, the deep sea is a species‐rich habitat, as has been well documented since marine biologists began to extensively sample the bathyal and abyssal depths (Grassle, 1989; Hessler & Sanders, 1967; Wolff, 1977). For example, Isopoda comprises a highly diverse and species‐rich group of crustaceans, with many members living in the abyssal benthos in all oceans (Hessler, Wilson, & Thistle, 1979; Wolff, 1962), and the existence of such biological communities has produced a profound change in our perception of deep‐sea life (Van Dover, German, Speer, Parson, & Vrijenhoek, 2002). In addition, mitochondria are the energy metabolism centers of the cell because more than 95% of cellular energy is generated by mitochondria through oxidative phosphorylation (OXPHOS). Mitochondrial‐encoded OXPHOS genes may therefore evolve under selection due to metabolic requirements and display evidence of adaptive evolution in mammals, birds, and fishes (Shen, Shi, Sun, & Zhang, 2009; Sun, Shen, Irwin, & Zhang, 2011).
Because of their large size, Bathynomus spp. (Crustacea: Isopoda: Cirolanidae) are classified into the “supergiant group” of isopods (Lowry & Dempsey, 2006). These animals are important scavengers in the deep‐sea benthic environment, from the gloomy sublittoral zone, at a depth of 170 m, to the dark of the bathypelagic zone at 2,140 m, and they are often found at depths between 365 and 730 m (Holthuis & Mikulka, 1972). Sankar et al. (2011) also mentioned these species as the first recorded deep‐sea isopods in the waters off India. However, no complete mitogenome data are available to date for any deep‐sea isopod, even though studying the early mitogenomic evolution of deep‐sea isopods using mt DNA fragments is facilitated by effective Crustacea‐specific versatile primers (Crandall & Fitzpatrick, 1996; Folmer, Black, Hoeh, Lutz, & Vrijenhoek, 1994; Merritt et al., 1998).
In this study, we report the first complete mitogenome sequence of the Bathynomus sp. mitogenome, consisting of the same 13 protein‐coding genes and two rRNAs found in most metazoans but only containing 18 tRNAs, with tRNA‐H, tRNA‐I, tRNA‐L1, and tRNA‐S1 missing from the typical structure. Moreover, the genes show surprising rearrangements compared to the ancestral pancrustacean order and the isopod ground pattern. Phylogenetic analyses indicate that Bathynomus sp. (Cymothoida) and Sphaeroma serratum (Sphaeromatidea) form one clade and that the evolutionary rate of Bathynomus sp. (dN/dS = 0.0705) is the slowest measured to date within Cymothoida. Our results may provide useful information for understanding the evolution of deep‐sea isopod species.
2. Materials and Methods
Experiments were performed in accordance with the recommendations of the Ethics Committee of the Institute of Hydrobiology, Chinese Academy of Sciences. These policies were enacted according to the Chinese Association for Laboratory Animal Sciences and the Institutional Animal Care and Use Committee protocols.
2.1. Sampling, identification, and DNA extraction
Six individuals of Bathynomus sp. were collected at a depth of 898 m in the South China sea (110°38.217′E, 17°46.845′N) using the “ROV Lander 01,” which was a remotely operated vehicle (ROV) with sampling equipment made by the Sanya Institute of Deep‐sea Science and Engineering, Chinese Academy of Sciences, in November 2014 (Fig. S1). All specimens were preserved in 99% ethanol until DNA extraction. Species‐level morphological identification was performed based on original morphological descriptions, locality data, and additional information about the giant deep‐sea scavenger genus Bathynomus (Lowry & Dempsey, 2006). Total genomic DNA was extracted from one or several pleopods using the EZNA® Tissue DNA Kit (OMEGA, Wuhan, China) following the manufacturer's instructions.
2.2. Genome determination
The first partial genetic fragments of the genes encoding cytochrome oxidase subunit 1 (cox1), cytochrome oxidase subunit 2 (cox2), large ribosomal RNA (rrnL), small ribosomal RNA (rrnS), cytochrome b (cob), and NADH dehydrogenase subunit 5 (nad5) were amplified using primers LCO1490/HCO2198 (Folmer et al., 1994), CO2f/CO2r1 (Yamauchi, Miya, & Nishida, 2004), 16sf‐cray/16s1472 (Crandall & Fitzpatrick, 1996), SR‐J14197/SR‐N14745 (Simon, Buckley, Frati, Stewart, & Beckenbach, 2006), Cytb151F/Cytb270R (Merritt et al., 1998), and DEnad5F/DEnad5R (Yang & Yang, 2008), respectively (Table S1). The reactions were carried out in a volume of 30 μl containing 21.125 μl of sterilized ultrapure water, 3.0 μl of 10× PCR buffer (including MgCl2), 1.5 μl of each primer (10 mmol/L), 1.5 μl of dNTPs (2.5 mmol/L each), 0.375 μl of Taq DNA polymerase (2.5 U/μl, Takara Bio, Shanghai, China), and 1.0 μl of DNA template (50–100 ng/μl). The cycling parameters were as follows: 94°C for 5 min; 32 cycles of 94°C for 40 s, 43–48°C for 30 s, and 72°C for 1 min; and a final elongation step at 72°C for 10 min. The six amplified fragments were sequenced and used for designing gene‐specific primers.
To facilitate the subsequent long PCR, we chose candidates with high melting temperatures (Table S1). Long PCRs were performed in a volume of 25 μl containing 12.75 μl of sterilized ultrapure water, 2.5 μl of 10× Takara LA PCR buffer (including MgCl2), 4 μl of Takara dNTP mixture (2.5 mmol/L each), 0.25 μl of Takara LA Taq polymerase (5 units/μl), 1 μl of primer mixture (10 mmol/L each), and 4 μl of DNA template (100 ng/μl). The corresponding thermal cycler protocol consisted of an initial denaturation step (94°C, 1 min), followed by 30 cycles of denaturation (98°C, 10 s), annealing and extension (64–68°C, 12 min), and ending with another extension (72°C, 10 min). The long PCR products were sequenced using a primer‐walking strategy. Considering the lack of data regarding genome arrangement for this species and to obtain precise genomic sequences, five gene‐specific primer pairs were designed to cover any gaps and inaccurate sequencing.
All of the PCR products were visualized on 1.2% low‐melting agarose gels stained with ethidium bromide. The products were then purified and sequenced using an ABI3730XL sequencing system, and the primers are described in Table S1.
2.3. Gene annotation and sequence analysis
Overlapping fragments obtained by sequencing were edited and aligned using BioEdit v7.0.9.0 (Hall, 1999). The MITOS webserver (Bernt et al., 2013) was used to annotate the Bathynomus sp. mitogenome, and protein‐coding and ribosomal RNA genes were rechecked by aligning them with publicly available mitogenomes of 11 isopod species (Table 1). The reliable identification of tRNA genes is generally not trivial, and the most common approach is to search for base pairings that conform to a typical tRNA clover‐leaf structure. Therefore, tRNAs were initially re‐detected using two computer programs: tRNAscan‐SE version 1.21 (Lowe & Eddy, 1997) and ARWEN 1.2.3.c (Laslett & Canback, 2008). Confirmation was performed by blast searches of all tRNAs in MitoZoa 2.0 (de Meo et al., 2012).
CGView (Stothard & Wishart, 2005) was used for a circular display of the Bathynomus sp. mitogenome, which was modified manually. The complete genome sequence has been submitted to NCBI (GenBank: KU057374). The nucleotide composition was analyzed with MEGA 5 (Tamura et al., 2011). Strand skew values were calculated according to the formulae given by Perna and Kocher (1995): AT skew = (A − T)/(A + T) and GC skew = (G − C)/(G + C), where A, T, C, G are the four bases.
2.4. Phylogenetic analysis and evolutionary rate estimation
We utilized the 11 other determined isopod mitogenomes for phylogenetic analyses (Table 1); six decapod species were used as outgroups: Alvinocaris longirostris (AB821296), Austinograea rodriguezensis (JQ035658), Geothelphusa dehaani (AB187570), Halocaridina rubra strain (KF437508), Panulirus japonicus (NC_004251), and Shinkaia crosnieri (NC_011013). Both the nucleotides and amino acids of the 13 protein‐coding genes were subjected to concatenated alignments using MEGA 5 (Tamura et al., 2011). Poorly aligned positions and divergent regions were removed using Gblocks version 0.91b (Castresana, 2000) set at the default block parameters. The final nucleotide and amino acid datasets consisted of 10,071 nt and 3,313 aa, respectively.
For the nucleotide dataset, jModelTest 2 (Darriba, Taboada, Doallo, & Posada, 2012) was used to select the model GTR + I + G for both maximum likelihood (ML) and Bayesian analyses. ProtTest version 3.4 (Darriba, Taboada, Doallo, & Posada, 2011) was used to choose the model MtArt + G + F for the amino acid dataset. As MtArt could not be implemented in the amino acid Bayesian analysis, we used the best scoring alternative MtRev + I + G + F model.
Maximum likelihood analyses of the nucleotide and amino acid alignments were assembled in PhyML 3.0 (Guindon & Gascuel, 2003), with 1,000 replicates performed under the models GTR + G + I and MtArt + G + F, respectively. Bayesian analyses of the nucleotide and amino acid alignments were carried out using MrBayes 3.1.2 (Ronquist & Huelsenbeck, 2003) under the models GTR + G + I and MtRev + I + G + F (see above), respectively, with 2,000,000 generations in two runs of eight chains each.
To estimate the evolutionary rate, standard branch models calculated with the CODEML program of PAML 4.6 (Yang, 2007) were used, and an adjusted Chi‐square test (Storey & Tibshirani, 2003) was applied for testing p values.
3. Results
3.1. Mitogenome organization
The mitogenome of the South China deep‐sea giant isopod Bathynomus sp. is a 14,965‐bp circular molecule (Figure 1) with a nucleotide composition of A% = 26.6, G% = 24.3, T% = 32.1, and C% = 17.0. The genome has an overall AT content of 58.7%, which appears to be low for isopods (typical range: 54.4%–71.2%). The composition is skewed away from G in favor of C (the GC skew is +0.177) but is almost balanced for A and T (the AT skew is −0.094). This feature is well conserved among isopods (Table 1).
Figure 1.
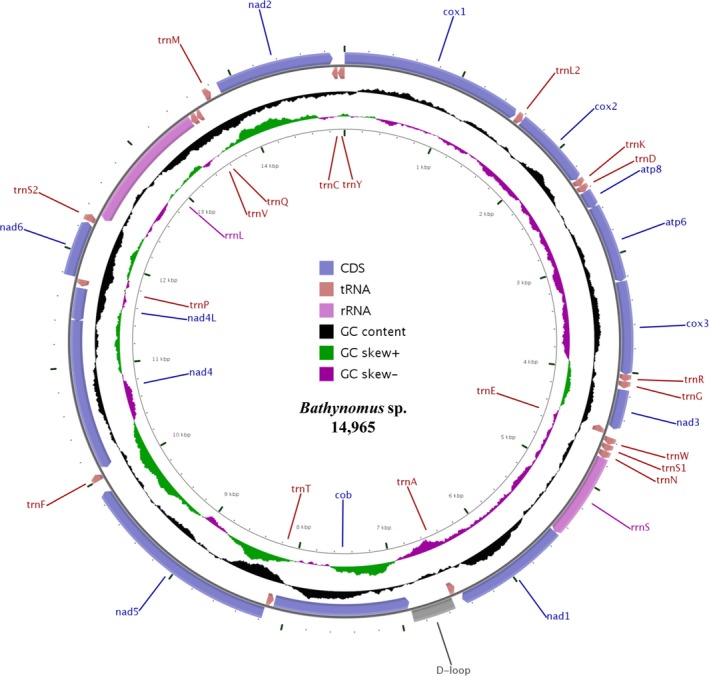
Map of the Bathynomus sp. mitogenome. Transfer RNAs are represented by their one‐letter amino acid code. Arrows pointing clockwise indicate (+) strand genes; counterclockwise arrows indicate (−) strand genes. The figure was initially generated by CGView and then modified manually
This species has the smallest complete mitogenome found in isopods (Table 1) thus far. The genome contains the same 13 protein‐coding genes and two ribosomal RNAs found in most metazoans. However, it exhibits incomplete tRNA‐encoding capacity (18 tRNAs instead of the usual 22, see Table 2). Twenty‐one mt genes are transcribed from one strand (the plus strand) and the remaining 12 from the other (the minus strand). A total of 817 noncoding intergenic nucleotides were found, with the largest continuous region (362 bp, AT% = 60.2) located between trnA and cob. Due to its location and AT richness, we predict that this part of the genome is the mt control region and that it likely contains the origins of replication and regulatory elements for transcription. However, this predicted control region is considerably smaller than that in other isopods (Table 1) and shows no sequence similarity with any previously sequenced Isopoda control region (data not shown).
Table 2.
Gene content of the Bathynomus sp. mitogenome
| Gene | Strandb | GenBank position no. | Size (nts) | Start codon | Stop codon | Anticodon | Intergenic nucleotides |
|---|---|---|---|---|---|---|---|
| cox1 | + | 1–1,539 | 1,539 | ATG | TAG | 14 | |
| trnL2‐UUR | + | 1,554–1,614 | 61 | TAA | 0 | ||
| cox2 | + | 1,615–2,292 | 678 | ATT | TAA | 3 | |
| trnK | + | 2,296–2,355 | 60 | TTT | 0 | ||
| trnD | + | 2,356–2,415 | 60 | GTC | 0 | ||
| atp8 | + | 2,416–2,568 | 153 | ATT | TGA | −4c | |
| atp6 | + | 2,565–3,233 | 669 | ATG | TAA | 0 | |
| cox3 | + | 3,234–4,022 | 789 | ATG | TAA | −2 | |
| trnR | + | 4,021–4,078 | 58 | TCG | 0 | ||
| trnG | + | 4,079–4,138 | 60 | TCC | 12 | ||
| nad3 | + | 4,151–4,492 | 342 | ATT | TAG | 0 | |
| trnE | − | 4,493–4,551 | 59 | TTC | 16 | ||
| trnW | + | 4,568–4,628 | 61 | TCA | 59 | ||
| trnN | + | 4,688–4,751 | 64 | GTT | a | ||
| rrnS | + | 4,752–5,493 | 742 | a | |||
| nad1 | + | 5,494–6,462 | 969 | ATG | TAA | 17 | |
| trnA | − | 6,480–6,542 | 63 | TGC | a | ||
| d‐loop | 6,543–6,904 | 362 | a | ||||
| cob | − | 6,905–8,110 | 1,206 | ATG | TAA | 10 | |
| trnT | − | 8,121–8,179 | 59 | TGT | 1 | ||
| nad5 | + | 8,181–9,893 | 1,713 | ATA | TAA | 103 | |
| trnF | + | 9,997–10,056 | 60 | GAA | 9 | ||
| nad4 | − | 10,066–11,412 | 1,347 | ATG | TAA | −7 | |
| nad4L | − | 11,406–11,699 | 294 | ATT | TAA | 15 | |
| trnP | − | 11,715–11,775 | 61 | TGG | 11 | ||
| nad6 | + | 11,787–12,261 | 475 | ATT | T | 9 | |
| trnS2‐UCN | + | 12,271–12,334 | 64 | TGA | a | ||
| rrnL | − | 12,335–13,515 | 1,181 | a | |||
| trnV | − | 13,516–13,576 | 61 | TAC | 0 | ||
| trnQ | − | 13,577–13,628 | 52 | TTG | 101 | ||
| trnM | + | 13,730–13,791 | 62 | CAT | 75 | ||
| nad2 | + | 13,867–14,865 | 999 | ATA | TAG | −13 | |
| trnC | − | 14,851–14,902 | 52 | GCA | 0 | ||
| trnY | − | 14,903–14,965 | 63 | GTA |
Gene borders are defined based on borders with adjacent genes.
Plus strand (+)/minus strand (−).
Negative values represent overlapping nucleotides.
Furthermore, as found in many mitogenomes, some genes overlap; that is, atp8/atp6, nad4L/nad4, and nad2/trnC share 4, 7, and 13 nucleotides, respectively, although they are located within different reading frames. Table 2 presents a summary of the organization of the Bathynomus sp. mitogenome.
3.2. Protein‐coding genes and ribosomal RNAs
With regard to protein‐coding genes, ten (cox1–3, atp8, atp6, nad1–3, and nad5–6) are encoded by the plus strand and the remaining three (cob, nad4, and nad4L) the minus strand (Figure 1, Table 2). This orientation is shared by all isopod mitogenomes sequenced to date, except for nad1. The 13 protein‐coding genes appear to start with the codon ATN, which is typical for metazoan mitogenomes (Wolstenholme, 1992), and TAN is the termination codon for 11. The atp8 gene terminates with TGA, which has not been observed in other isopods. Truncated termination codons (T) are observed in nad6 (Table 2). Posttranscriptional polyadenylation may subsequently generate functional TAA codons (Ojala, Montoya, & Attardi, 1981).
The rrnL and rrnS genes of Bathynomus sp. are 1,181 bp (AT% = 60.5) and 742 bp (AT% = 59.7) in length, respectively. These lengths are typical for crustaceans, whereas the AT contents are slightly lower than those of other isopods (Table 1).
3.3. Transfer RNAs
We analyzed the entire genome sequence of Bathynomus sp. and successfully identified 18 tRNA genes based on their potential secondary structures (Figure 2) using the MITOS webserver. Moreover, tRNAscan‐SE was used to recheck five genes and ARWEN for 17 genes. The 18 tRNA genes are spread throughout the entire genome and are located on both strands (Figure 2, Table 2). Sixteen of these genes display a common clover‐leaf secondary structure, and the remaining two exhibit a t‐shaped secondary structure.
Figure 2.
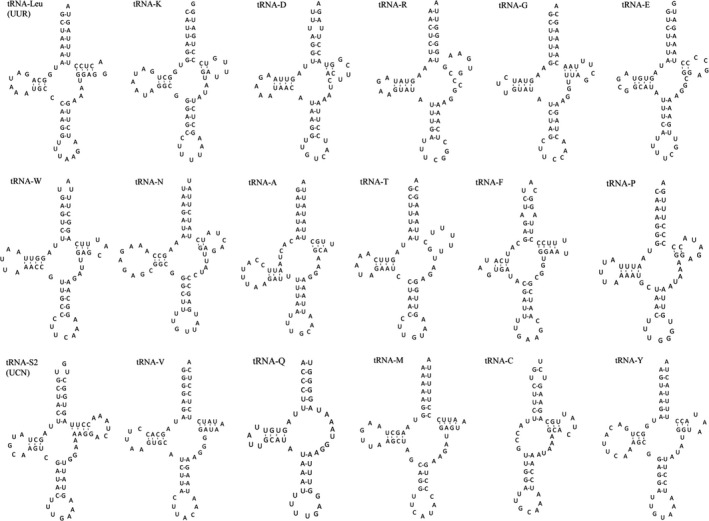
Putative secondary structures for the 18 transfer RNAs of the Bathynomus sp. mitogenome. All 18 structures were generated using the MITOS webserver and verified using tRNAscan‐SE and ARWEN. Most tRNAs feature a standard clover‐leaf structure. Exceptions: The TΨC arm is absent from trnQ, and the DHU arm is absent from trnC
3.4. Genome rearrangement
A comparison of all 12 currently available isopod mt genomes, including four complete genomes (Kilpert & Podsiadlowski, 2006, 2010; Lloyd et al., 2015) and eight partial genomes (Chandler, Badawi, Moumen, Greve, & Cordaux, 2015; Kilpert, Held, & Podsiadlowski, 2012; Marcade et al., 2007; Podsiadlowski & Bartolomaeus, 2006), is shown in Figure 3. The putative ancestral state of the pancrustacean ground pattern is also provided (Boore, Lavrov, & Brown, 1998). Between Bathynomus sp. and the pancrustacean ground pattern, six tRNA genes rearrangement, one ribosomal RNA gene rearrangement, and three protein‐coding genes rearrangement are shown (Figure 3). However, only four tRNA genes rearrangement and one protein‐coding gene rearrangement are detected between Bathynomus sp. and the isopod ground pattern (Figure 4).
Figure 3.

Comparison of mitochondrial gene arrangements in 12 species in the order Isopoda. In addition, the pancrustacean ground pattern (putative ancestral state) is provided. The mt complete genomes of Bathynomus sp., Limnoria quadripunctata, Ligia oceanica, and Eophreatoicus sp.‐14 are available; the other mt genomes are incomplete and are missing some tRNAs and the control region (CR). All tRNAs are designated by single letters (except L1, L2, S1, and S2 for trnL‐CUN, trnL‐UUR, trnS‐AGN, and trnS‐UCN, respectively). Colored genes denote translocated genes in comparison with the pancrustacean ground pattern. Red indicates genes that share a derived position in isopods. Uniquely derived gene positions of individual species are depicted in yellow
Figure 4.
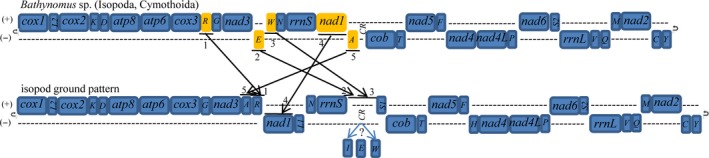
Gene rearrangements of the Bathynomus sp. mitogenome. At least five rearrangements have occurred between the Bathynomus sp. mitogenome and the putative isopod ground pattern, including (1) trnR, (2) trnE, (3) trnW, (4) nad1, and (5) trnA. All translocated genes are shown in yellow. All tRNAs are designated by single letters (except L1, L2, S1, and S2 for trnL‐CUN, trnL‐UUR, trnS‐AGN, and trnS‐UCN, respectively)
3.5. Phylogenetic analysis and evolutionary rate estimation
We examined both the nucleotide and amino acid sequences of protein‐coding genes in intraorder phylogenetic analyses. For each dataset, ML and Bayesian analyses resulted in the same tree topology (Figure 5). The evolutionary rate was estimated using the nucleotide sequences of 13 protein‐coding genes and shown in Table 3. When compared to other suborders, Cymothoida demonstrated the highest evolutionary rate values. However, Bathynomus sp. showed the lowest evolutionary rate value in Cymothoida.
Figure 5.
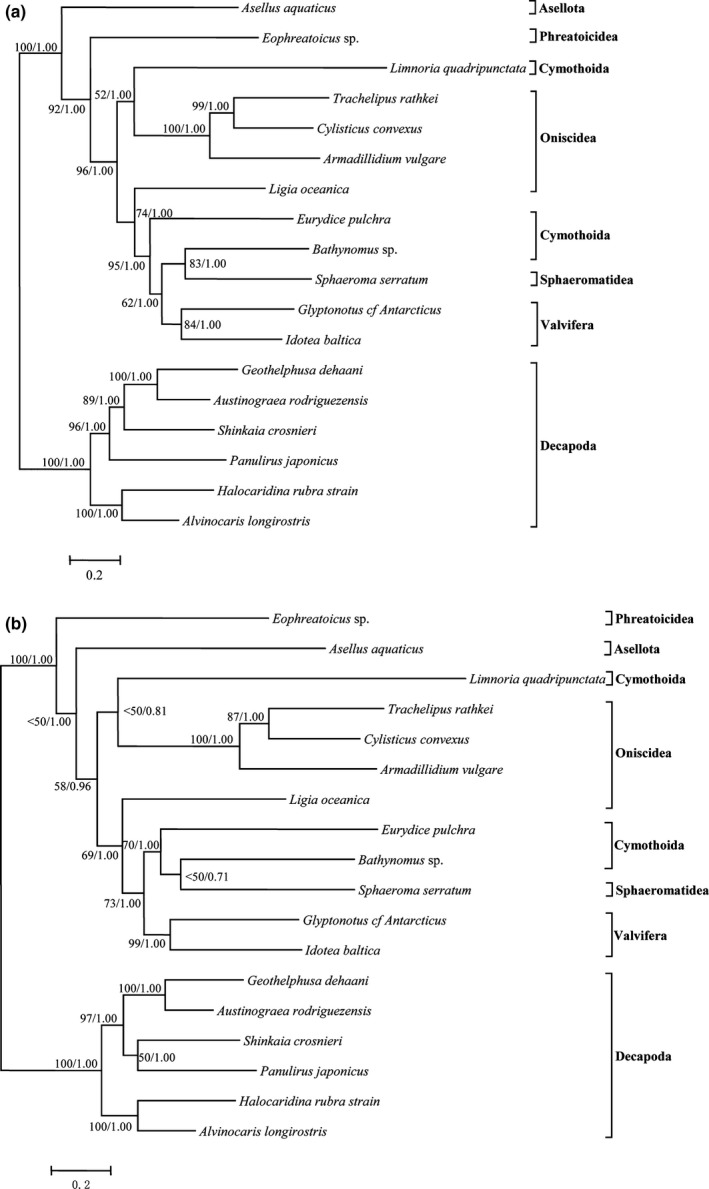
Phylogenetic trees showing relationships among isopods based on nucleotide (a) and amino acid datasets (b). Six decapod species served as outgroups. Branch lengths and topologies are derived from Bayesian analyses. Numbers next to nodes specify bootstrap percentages from the maximum likelihood (ML) analysis plus Bayesian posterior probabilities (BPP). Specifically, <50 indicates bootstrap percentages from ML values below 50%
Table 3.
Values for (non)synonymous mutation rates and dN/dS ratios of 13 tandem genes for 18 species. Ancestral branches are not included
| Suborder/infraorder | Species | Habitat type | dNc | dSc | dN/dSc |
|---|---|---|---|---|---|
| Cymothoida | Bathynomus sp.a | Deep sea | 0.1612 | 2.2875 | 0.0705 |
| Eurydice pulchra a | Intertidal zone | 0.2002 | 2.2749 | 0.0880 | |
| Limnoria quadripunctata a | Shallow sea | 0.2779 | 3.2244 | 0.0862 | |
| Oniscidea | Ligia oceanica a | Littoral zone | 0.1640 | 2.4777 | 0.0662 |
| Armadillidium vulgare a | Terrestrial | 0.1309 | 2.9097 | 0.0450 | |
| Cylisticus convexus a | Terrestrial | 0.0981 | 2.5122 | 0.0390 | |
| Trachelipus rathkei a | Terrestrial | 0.1265 | 2.3812 | 0.0531 | |
| Valvifera | Idotea baltica a | Shallow sea | 0.1335 | 1.8952 | 0.0704 |
| Glyptonotus cf. Antarcticus a | Antarctic | 0.1344 | 2.4986 | 0.0538 | |
| Phreatoicidea | Eophreatoicus sp. ‐14a | Fresh water | 0.1910 | 3.4175 | 0.0559 |
| Sphaeromatidea | Sphaeroma serratum a | Shallow sea | 0.1677 | 2.2522 | 0.0744 |
| Asellota | Asellus aquaticus a | Fresh water | 0.2183 | 3.1446 | 0.0694 |
| Decapoda | Shinkaia crosnieri b | Deep sea vent | 0.1128 | 2.5780 | 0.0438 |
| Panulirus japonicus b | Shallow sea | 0.1375 | 2.6311 | 0.0523 | |
| Halocaridina rubra strain b | Near the sea shore | 0.1212 | 1.6655 | 0.0728 | |
| Geothelphusa dehaani b | Rivers and streams | 0.0876 | 3.0085 | 0.0291 | |
| Austinograea rodriguezensis b | Deep sea | 0.0654 | 2.4248 | 0.0270 | |
| Alvinocaris longirostris b | Deep sea vent | 0.0755 | 1.7666 | 0.0427 |
12 species of isopods.
Six outgroup decapod species.
p < .001.
4. Discussion
4.1. Transfer RNAs
The DHU arm of trnC and the TΨC arm of trnQ are completely missing (Figure 2). Loss of the DHU arm from tRNA‐C has also been observed in other isopods, such as Ligia oceanica (Kilpert & Podsiadlowski, 2006) and Eophreatoicus sp.‐14 (Kilpert & Podsiadlowski, 2010). This feature might be a putative autapomorphy of isopoda, as it is absent in other malacostracan crustaceans. In general, tRNAs with a U at the wobble position (the first position) of the anticodon recognize either fourfold degenerate or NNR codons, whereas those with a G at this position only recognize NNY codons (Yang & Yang, 2008). All tRNAs of Bathynomus sp. mitogenome obey this rule.
Despite extensive efforts to identify secondary structure in noncoding regions, the genes trnH, trnI, trnL1, and trnS1 were not found in the mitogenomic sequence. Considering the absence of trnQ in the L. oceanica mitogenome (Kilpert & Podsiadlowski, 2006), this is not an exception among isopods. Indeed, a lack of these genes is also common in arthropods, such as the absence of trnQ in Aleurodicus dugesii, trnS in Schizaphis graminum (Thao, Baumann, & Baumann, 2004), and trnD in Centruroides limpidus (Davila, Pinero, Bustos, Cevallos, & Davila, 2005). tRNA gene deficiencies have often been observed in protozoans, fungi, algae, plants, and lower metazoans (Schneider & Marechal‐Drouard, 2000); in these cases, imported nuclear DNA‐encoded tRNAs compensate for the lack of mt tRNA. Furthermore, marsupial mt tRNAs exhibit interesting patterns. Janke and Paabo identified in Didelphis virginiana a pseudogene similar to trnD with an anticodon of GCC instead of the usual GUC (Janke & Paabo, 1993); the authors found that the cytosine is changed to uridine during posttranscriptional RNA editing. Based on its nonfunctional secondary structure, Dorner, Altmann, Paabo, and Morl (2001) discovered a trnK pseudogene in the same marsupial species, with compensation due to import of cytoplasmic tRNA rather than RNA editing. As there appears to be no candidate template for RNA editing in the case of Bathynomus sp., import of tRNA is a plausible explanation. In addition, some other explanations also may be plausible explanation: (1) specific bias in codon usage. Indeed, a recent work has succeeded in replacing seven codons in Escherichia coli (at least part of it), so that the genome can be decoded on 57 triplets rather than 64 (Thiele et al., 2012). It could be that some codons are missing, so that some tRNAs would not be necessary; (2) a hypothesis could be that some of the present tRNA would be edited, so that they would recognize both different amino acids and have different anticodons. This is not entirely impossible, as it is known, in some cases, that the specification of the amino acid loaded on the tRNA uses the anticodon as a signature. In a way, this would require much less genes from the host (just editing) than create a whole RNA translocation across the double membrane of mitochondria.
4.2. Genome rearrangement
Compared to the gene orders of other isopod species, the Bathynomus sp. mitogenome has undergone significant changes in gene arrangement. First, all 11 isopod mt genomes differ in gene order, but, except for Limnoria quadripunctata, most of the differences are limited to the position of one or a few tRNA genes. Second, the gene order of the isopod mt genomes can be clearly distinguished from the pancrustacean ground pattern. Inferred changes in the Bathynomus sp. genome involve tRNA genes, protein‐coding genes, rRNA genes, and the mt control region (CR).
Kilpert et al. showed that isopods share a derived order of genes, with individual species experiencing modifications. Using the similarities common to most isopods, a most parsimonious hypothesis for the mt gene order of the isopod ground pattern was inferred, which includes a unique arrangement of nad1, trnL1, rrnS, CR, trnS1, cob, trnT, nad5, and trnF (Kilpert et al., 2012). For Bathynomus sp., cob, trnT, nad5, and trnF fit the isopod ground pattern, whereas nad1, trnL1, and rrnS strongly diverge from this pattern.
We also found that the mitogenome of Bathynomus sp. harbors significant alterations in gene arrangement compared to the gene order of the putative isopod ground pattern described by Kilpert et al. (2012). In total, we identified rearrangements in this species (Figure 4): One rearrangement involves protein‐coding genes, and the remaining are tRNA translocations. Relative to its ancestral position, trnR is at a position upstream of nad3. Glyptonotus cf. Antarcticus (Kilpert et al., 2012), Cylisticus convexus, and Trachelipus rathkei (Chandler et al., 2015) share this rearrangement. The positions of trnE and trnW are changed to positions downstream of rrnS, which has not been found in other isopods to date. Finally, the nad1 gene is moved to a position downstream of nad3 and from the minus strand to the plus strand and this translocation is novel feature found only in isopods (Kilpert & Podsiadlowski, 2010; Kilpert et al., 2012; Marcade et al., 2007).
4.3. Phylogenetic analysis and evolutionary rate estimation
Indeed, the nucleotide and amino acid trees are nearly the same, except for the position of the Asellota clade (Asellus aquaticus) and the Phreatoicidea clade (Eophreatoicus sp.‐14). In the case of the nucleotide dataset, the Asellota clade is placed at the base of Isopoda (Figure 5a) and is well supported (ML/BPP = 100/1.00). However, in the amino acid tree, the Phreatoicidea clade is placed at the base of Isopoda (Figure 5b), which is in agreement with the findings of previous research (Kilpert et al., 2012) and is also well supported (ML/BPP = 100/1.00). This discrepancy may be due to the fact that the two clades are represented by only one species each. Neither tree groups L. quadripunctata the same clade with the other three Cymothoida species, and Bathynomus sp. (Cymothoida) and S. serratum (Sphaeromatidea) form one clade in both trees. Kilpert et al. (2012) also found that Eurydice pulchra (Cymothoida) and S. serratum form a single clade. According to these results, it remains unclear whether Cymothoida is monophyletic or paraphyletic. The other intraorder clade was well reconstructed according to ML and Bayesian analyses of the two dataset (Figure 5). However, as research on the phylogenetic relationships within this infraorder is rare (Kilpert et al., 2012), intensive sampling and analysis of a greater number of species are necessary to determine the phylogenetic relationships among members of Isopoda.
We found that the evolutionary rate of Bathynomus sp. is the slowest measured to date within Cymothoida (Table 3), which means that this species may have experienced nonsynonymous mutations harmful to its survival and that its mt genes have been under strong purifying selection (Yang, 2007). This slow evolutionary rate may be associated with the relatively constant deep‐sea environment of this species. For example, Bathynomus giganteus was already present as early as 160 million years ago, and a previous study showed that B. giganteus specimens from Australia, Mexico, and India are almost exactly the same, possibly because of their nearly identical environments (Parker, 2003). This phenomenon has been found in many high‐altitude habitats species (Shen et al., 2009; Sun et al., 2011).
This study is the first determination of the mitogenome of a deep‐sea member of the order Isopoda, an effort that not only increases sampling of Isopoda but also more importantly provides useful information for understanding the evolution of deep‐sea isopods.
Conflict of Interest
None declared.
Supporting information
Acknowledgments
The specimens used in the study were collected by the “ROV Lander 01” of Sanya Institute of Deep‐sea Science and Engineering, CAS, in the South China deep‐sea. The authors are grateful for their help in collecting specimens. We also sincerely thank the crews of Institute of Oceanology, Chinese Academy of Science, for their help with the preparation and photography of the specimens. This work was supported by the Strategic Priority Research Program of the Chinese Academic of Sciences (No. XDB060101).
Shen Y, Kou Q, Zhong Z, Li X, He L. The first complete mitogenome of the South China deep‐sea giant isopod Bathynomus sp. (Crustacea: Isopoda: Cirolanidae) allows insights into the early mitogenomic evolution of isopods. Ecol Evol. 2017;7:1869–1881. https://doi.org/10.1002/ece3.2737
References
- Bernt, M. , Donath, A. , Juhling, F. , Externbrink, F. , Florentz, C. , Fritzsch, G. , … Stadler, P. F. (2013). MITOS: Improved de novo metazoan mitochondrial genome annotation. Molecular Phylogenetics and Evolution, 69(2), 313–319. [DOI] [PubMed] [Google Scholar]
- Boore, J. L. , Lavrov, D. V. , & Brown, W. M. (1998). Gene translocation links insects and crustaceans. Nature, 392(6677), 667–668. [DOI] [PubMed] [Google Scholar]
- Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution, 17(4), 540–552. [DOI] [PubMed] [Google Scholar]
- Chandler, C. H. , Badawi, M. , Moumen, B. , Greve, P. , & Cordaux, R. (2015). Multiple conserved heteroplasmic sites in tRNA genes in the mitochondrial genomes of terrestrial isopods (Oniscidea). G3: Genes Genomes Genetics, 5(7), 1317–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clary, D. O. , & Wolstenholme, D. R. (1985). The mitochondrial‐DNA molecule of Drosophila yakuba—Nucleotide‐sequence, gene organization, and genetic‐code. Journal of Molecular Evolution, 22(3), 252–271. [DOI] [PubMed] [Google Scholar]
- Crandall, K. A. , & Fitzpatrick, J. F. (1996). Crayfish molecular systematics: Using a combination of procedures to estimate phylogeny. Systematic Biology, 45(1), 1–26. [Google Scholar]
- Darriba, D. , Taboada, G. L. , Doallo, R. , & Posada, D. (2011). ProtTest 3: Fast selection of best‐fit models of protein evolution. Bioinformatics, 27(8), 1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba, D. , Taboada, G. L. , Doallo, R. , & Posada, D. (2012). jModelTest 2: More models, new heuristics and parallel computing. Nature Methods, 9(8), 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila, S. , Pinero, D. , Bustos, P. , Cevallos, M. A. , & Davila, G. (2005). The mitochondrial genome sequence of the scorpion Centruroides limpidus (Karsch 1879) (Chelicerata; Arachnida). Gene, 360(2), 92–102. [DOI] [PubMed] [Google Scholar]
- de Meo, P. D. , D'Antonio, M. , Griggio, F. , Lupi, R. , Borsani, M. , Pavesi, G. , … Gissi, C. (2012). MitoZoa 2.0: A database resource and search tools for comparative and evolutionary analyses of mitochondrial genomes in Metazoa. Nucleic Acids Research, 40(D1), D1168–D1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner, M. , Altmann, M. , Paabo, S. , & Morl, M. (2001). Evidence for import of a lysyl‐tRNA into marsupial mitochondria. Molecular Biology of the Cell, 12(9), 2688–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmer, O. , Black, M. , Hoeh, W. , Lutz, R. , & Vrijenhoek, R. (1994). DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology, 3(5), 294–299. [PubMed] [Google Scholar]
- Grassle, J. F. (1989). Species‐diversity in deep‐sea communities. Trends in Ecology & Evolution, 4(1), 12–15. [DOI] [PubMed] [Google Scholar]
- Guindon, S. , & Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology, 52(5), 696–704. [DOI] [PubMed] [Google Scholar]
- Hall, T. A. (1999). BioEdit: A user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98. [Google Scholar]
- Helfenbein, K. G. , Fourcade, H. M. , Vanjani, R. G. , & Boore, J. L. (2004). The mitochondrial genome of Paraspadella gotoi is highly reduced and reveals that chaetognaths are a sister group to protostomes. Proceedings of the National Academy of Sciences of the United States of America, 101(29), 10639–10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessler, R. R. , & Sanders, H. L. (1967). Faunal diversity in deep‐sea. Deep‐Sea Research, 14(1), 65–70. [Google Scholar]
- Hessler, R. R. , Wilson, G. D. , & Thistle, D. (1979). Deep‐sea isopods—Biogeographic and phylogenetic overview. Sarsia, 64(1–2), 67–75. [Google Scholar]
- Holthuis, L. B. , & Mikulka, W. R. (1972). Biological results of University of Miami deep‐sea expeditions. 91. Notes on deep‐sea isopods of genus Bathynomus a Milne Edwards, 1879. Bulletin of Marine Science, 22(3), 575–591. [Google Scholar]
- Janke, A. , & Paabo, S. (1993). Editing of a transfer‐RNA anticodon in marsupial mitochondria changes its codon recognition. Nucleic Acids Research, 21(7), 1523–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpert, F. , Held, C. , & Podsiadlowski, L. (2012). Multiple rearrangements in mitochondrial genomes of Isopoda and phylogenetic implications. Molecular Phylogenetics and Evolution, 64(1), 106–117. [DOI] [PubMed] [Google Scholar]
- Kilpert, F. , & Podsiadlowski, L. (2006). The complete mitochondrial genome of the common sea slater, Ligia oceanica (Crustacea, Isopoda) bears a novel gene order and unusual control region features. BMC Genomics, 7, 1–18. doi:Artn 241; 10.1186/1471‐2164‐7‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpert, F. , & Podsiadlowski, L. (2010). The Australian fresh water isopod (Phreatoicidea: Isopoda) allows insights into the early mitogenomic evolution of isopods. Comparative Biochemistry and Physiology Part D, 5, 36–44. doi:10.1016/j.cbd.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Laslett, D. , & Canback, B. (2008). ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics, 24(2), 172–175. [DOI] [PubMed] [Google Scholar]
- Lavrov, D. V. , Boore, J. L. , & Brown, W. M. (2000). The complete mitochondrial DNA sequence of the horseshoe crab Limulus polyphemus . Molecular Biology and Evolution, 17(5), 813–824. [DOI] [PubMed] [Google Scholar]
- Liebers, D. , de Knijff, P. , & Helbig, A. J. (2004). The herring gull complex is not a ring species. Proceedings of the Royal Society of London B: Biological Sciences, 271(1542), 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd, R. E. , Streeter, S. D. , Foster, P. G. , Littlewood, D. T. , Huntley, J. , Beckham, G. T. , … Cragg, S. M . (2015). The complete mitochondrial genome of Limnoria quadripunctata Holthuis (Isopoda: Limnoriidae). Mitochondrial DNA, 26, 825–828. doi:10.3109/19401736.2013.855912. [DOI] [PubMed] [Google Scholar]
- Lowe, T. M. , & Eddy, S. R. (1997). tRNAscan‐SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research, 25(5), 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry, J. K. , & Dempsey, K. (2006). The giant deep‐sea scavenger genus Bathynomus (Crustacea, Isopoda, Cirolanidae) in the Indo‐West Pacific In Richer de Forges B. & Justone J. L. (Eds.), Tropical deep‐sea benthos (Vol. 24, pp. 163–192). [Google Scholar]
- Macey, J. R. , Schulte, J. A. , & Larson, A. (2000). Evolution and phylogenetic information content of mitochondrial genomic structural features illustrated with acrodont lizards. Systematic Biology, 49(2), 257–277. [PubMed] [Google Scholar]
- Marcade, I. , Cordaux, R. , Doublet, V. , Debenest, C. , Bouchon, D. , & Raimond, R. (2007). Structure and evolution of the atypical mitochondrial genome of Armadillidium vulgare (Isopoda, crustacea). Journal of Molecular Evolution, 65(6), 651–659. [DOI] [PubMed] [Google Scholar]
- Merritt, T. J. S. , Shi, L. , Chase, M. C. , Rex, M. A. , Etter, R. J. , & Quattro, J. M. (1998). Universal cytochrome b primers facilitate intraspecific studies in molluscan taxa. Molecular Marine Biology and Biotechnology, 7(1), 7–11. [PubMed] [Google Scholar]
- Nass, M. M. K. , & Nass, S. (1963). Intramitochondrial Fibers with DNA. Journal of Cell Biology, 19(3), 593–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala, D. , Montoya, J. , & Attardi, G. (1981). Transfer‐RNA punctuation model of RNA processing in human mitochondria. Nature, 290(5806), 470–474. [DOI] [PubMed] [Google Scholar]
- Parker, A. (2003). In the blink of an eye: How vision kick‐started the big bang of evolution. Cambridge, UK: Perseus Publishing. [Google Scholar]
- Perna, N. T. , & Kocher, T. D. (1995). Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. Journal of Molecular Evolution, 41(3), 353–358. [DOI] [PubMed] [Google Scholar]
- Podsiadlowski, L. , & Bartolomaeus, T. (2006). Major rearrangements characterize the mitochondrial genome of the isopod Idotea baltica (Crustacea: Peracarida). Molecular Phylogenetics and Evolution, 40, 893–899. doi:10.1016/j.ympev.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Roehrdanz, R. L. , Degrugillier, M. E. , & Black, W. C. (2002). Novel rearrangements of arthropod mitochondrial DNA detected with Long‐PCR: Applications to arthropod phylogeny and evolution. Molecular Biology and Evolution, 19(6), 841–849. [DOI] [PubMed] [Google Scholar]
- Ronquist, F. , & Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics, 19(12), 1572–1574. [DOI] [PubMed] [Google Scholar]
- Sankar, R. , Rajkumar, M. , Sun, J. , Gopalakrishnan, A. , Vasanthan, T. M. , Ananthan, G. , & Trilles, J. P. (2011). First record of three giant marine Bathynomids (Crustacea, Isopoda, Cirolanidae) from India. Acta Oceanologica Sinica, 30(1), 113–117. [Google Scholar]
- Schneider, A. , & Marechal‐Drouard, L. (2000). Mitochondrial tRNA import: Are there distinct mechanisms? Trends in Cell Biology, 10(12), 509–513. [DOI] [PubMed] [Google Scholar]
- Shen, Y. Y. , Shi, P. , Sun, Y. B. , & Zhang, Y. P. (2009). Relaxation of selective constraints on avian mitochondrial DNA following the degeneration of flight ability. Genome Research, 19(10), 1760–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, C. , Buckley, T. R. , Frati, F. , Stewart, J. B. , & Beckenbach, A. T. (2006). Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annual Review of Ecology Evolution and Systematics, 37, 545–579. [Google Scholar]
- Storey, J. D. , & Tibshirani, R. (2003). Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences of the United States of America, 100(16), 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stothard, P. , & Wishart, D. S. (2005). Circular genome visualization and exploration using CGView. Bioinformatics, 21(4), 537–539. [DOI] [PubMed] [Google Scholar]
- Sun, Y. B. , Shen, Y. Y. , Irwin, D. M. , & Zhang, Y. P. (2011). Evaluating the roles of energetic functional constraints on teleost mitochondrial‐encoded protein evolution. Molecular Biology and Evolution, 28(1), 39–44. [DOI] [PubMed] [Google Scholar]
- Tamura, K. , Peterson, D. , Peterson, N. , Stecher, G. , Nei, M. , & Kumar, S. (2011). MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28(10), 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thao, M. L. , Baumann, L. , & Baumann, P. (2004). Organization of the mitochondrial genomes of whiteflies, aphids, and psyllids (Hemiptera, Sternorrhyncha). Bmc Evolutionary Biology, 4, 439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele, I. , Fleming, R. M. T. , Que, R. , Bordbar, A. , Diep, D. , & Palsson, B. O. (2012). Multiscale modeling of metabolism and macromolecular synthesis in E. coli and its application to the evolution of codon usage. PLoS One, 7(9): e45635–e45635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dover, C. L. , German, C. R. , Speer, K. G. , Parson, L. M. , & Vrijenhoek, R. C. (2002). Evolution and biogeography of deep‐sea vent and seep invertebrates. Science, 295(5558), 1253–1257. [DOI] [PubMed] [Google Scholar]
- Wolff, T. (1962). The systematics and biology of bathyal and abyssal Isopoda Asellota Scientific Results of the Danish Deep‐Sea Expedition Round the World, Galathea Report (Vol. 6, pp. 1950–1952). Copenhagen, Denmark: Danish Science Press. [Google Scholar]
- Wolff, T. (1977). Diversity and faunal composition of deep‐sea benthos. Nature, 267(5614), 780–785. [Google Scholar]
- Wolstenholme, D. R. (1992). Animal mitochondrial‐DNA—structure and evolution. International Review of Cytology, 141, 173–216. [DOI] [PubMed] [Google Scholar]
- Yamauchi, M. M. , Miya, M. U. , & Nishida, M. (2004). Use of a PCR‐based approach for sequencing whole mitochondrial genomes of insects: Two examples (cockroach and dragonfly) based on the method developed for decapod crustaceans. Insect Molecular Biology, 13(4), 435–442. [DOI] [PubMed] [Google Scholar]
- Yang, Z. H. (2007). PAML 4: Phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution, 24(8), 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang, J. S. , & Yang, W. J. (2008). The complete mitochondrial genome sequence of the hydrothermal vent galatheid crab Shinkaia crosnieri (Crustacea: Decapoda: Anomura): A novel arrangement and incomplete tRNA suite. Bmc Genomics, 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials