

Abstract
The hypothesis of the local recurrence of preneoplastic lesions was first put forward in the 1950s. Disease recurrence may result from an inherent imbalance in cell proliferation that promotes carcinogenesis in apparently normal mucosa. Our review sheds light on how early preneoplastic lesions could be used to diagnose relapsed preneoplastic and, developing neoplastic lesions. We focus in detail on the clinical-pathological and molecular features of adenoma subtypes and their role in relapsed adenoma and their development into colorectal carcinoma. Moreover, we include the data available on microbiota and its metabolites and their role in recurrence. We strongly believe that a significant improvement could be achieved in colorectal screening by introducing personalized endoscopic surveillance for polyp-bearing patients on the basis of the presence of molecular markers that are predictive of recurrence.
Keywords: recurrence, preneoplastic lesions, suppressor and serrated pathway, microbiota
INTRODUCTION
Colorectal cancer (CRC) is the major worldwide cause of death and the majority of the cases occur in developed regions. Europe has one of the highest incidences of CRC in the world. Notwithstanding, mortality is lower (9% of the total worldwide) than in less developed regions of the world (52% of the total worldwide) [1]. The low mortality rate has been attributed to the impact of routine screening programs [2], which allow the early identification and surgical removal of preneoplastic lesions in asymptomatic patients. Indeed, the risk of developing CRC is not homogenously distributed, and a small subgroup of patients have a higher incidence of CRC that persists after baseline polypectomy [3, 4]. According to the European Society of Gastrointestinal Endoscopy (ESGE) and American Gastroenterological Association (AGA) guidelines, patients with preneoplastic lesions such as adenomatous polyps are assigned to a risk subgroup on the basis of lesion histology [3]. Specifically, patients with high-grade dysplasia adenomas, >20% villous histology, ≥ 10 mm in size, 3 or more adenomas and serrated polyps ≥ 10 mm in size are considered at high risk and advised to undergo close endoscopic surveillance for 1- 3 years. Conversely, patients with up to 2 tubular adenomas, with < 10 mm, low-grade dysplasia, or serrated polyps < 10 mm and no dysplasia are considered to be at low risk of developing CRC. Both the ESGE and AGA recommend a follow up ranging from 5 to 10 years [3]. Follow-up colonoscopy every 10 years is considered adequate for individuals with hyperplastic polyps (HPs) (Table 1) [5].
Table 1. Risk of developing colorectal cancer in adenoma patients.
| Recommended surveil lance interval (yr) | Serrated adenomas | Characteristics | Conventional adenomas | |
|---|---|---|---|---|
| No grade | 10 | MVHPs GCHPs MPHPs |
||
| Low grade | 5 | SSAs/Ps | < 1 cm < 3 adenomas Low dysplasia < 20% villous component |
TAs |
| High grade | 3 | TSAs | ≥ 1 cm | TSAs |
| ≥ 3 adenomas | TAs | |||
| SSAs/Ps | High dysplasia | TVAs | ||
| ≥ 20% villous component | TVs |
The idea of field cancerization to clarify the incidence of multiple primary tumors, local recurrence, abnormal tissue near the cancer and multifocal areas of precancerous change emerged in the 1950s [6]. Initially, CRC field cancerization was founded on histopathological parameters including size, number, localization, histology and grade of dysplasia of lesions detected during the baseline polypectomy. Molecular diagnostic procedures have shown field cancerization is caused by the accumulation of early changes in normal-appearing tissue [7–12]. Despite this, there are still no definitive molecular factors capable of identifying which early colorectal lesions are most likely to relapse. Disease recurrence may, in fact, be due to lesions that have been missed or not radically removed. Moreover, an inherent imbalance in cell proliferation may also promote carcinogenesis in apparently normal mucosa [3].
Sporadic CRC develops gradually over the years as a result of genetic and epigenetic modifications. As it is a heterogeneous disease, the analysis of molecular alterations in cancer precursors could help to discriminate between different types of pathogenesis and clinical behavior. This subtype has been classified on the basis of combinations of genetic markers, e.g. microsatellite instability (MSI), CpG island methylator phenotype (CIMP), BRAF mutation, and/or somatic KRAS mutation [13, 14] and two pathways have been identified. The progression of conventional adenoma to carcinoma is known as the suppressor pathway, while the progression of serrated adenoma to carcinoma sequence is described as the serrated pathway. Although few molecular markers are known to be related to adenoma recurrence, some authors have recently discovered that also microbiota influences adenoma and cancer etiology. However, the way in which the gut microbiota influences adenoma recurrence remains to be clarified [15].
We present an overview of emerging strategies that could help us to understand whether early preneoplastic colorectal lesions are capable of predicting the recurrence of preneoplastic and developing CRC. We shed light on current histopathological and molecular features of preneoplastic colorectal lesions and put forward hypotheses about their involvement in recurrence. Although we are not ready to change our surveillance program based on molecular and microbiota characterization, we believe that the identification of biomarkers and bacteria involved into relapse process has potential for improving the personalized endoscopic surveillance of polyp-bearing patients in a screening program.
The suppressor pathway
The suppressor pathway, also called the canonical or conventional pathway, is found in 80%-85% of CRCs and is known to follow the Fearon and Vogelstein model [16]. Early lesions detected are conventional adenomas: tubular (TAs) or villous (VAs) or tubular-villous adenomas (TVAs) [16, 17]. Conventional adenomas are usually sited in the ascending colon and rectosigmoid [18]. Villous structures constitute ≥ 80% of VAs and > 20% of TVAs and show different morphologies e.g. classic villi (long, slender, finger-like projections), palmate villi (leaf-like, broad, branched projections) and foreshortened villi (isolated, slender projections) [19]. TA is the most common subtype constituting 65-80% of all preneoplastic lesions removed. Histologically, TAs have branched tubular glands and are often pedunculated with fewer atypia than VAs. VAs have long, finger-like projections and represent 5-10% of neoplastic polyps. They are frequently sessile and are more likely to show severe atypia or dysplasia than TAs. About 10 to 25% of polyps are TVAs with cellular features of TAs and VAs [20].
These adenomas are characterized by allelic losses in the adenomatous polyposis coli (APC) gene which alters the Wnt–β-catenin pathway, activation of prostaglandin signaling induced by inflammation or mitogen-associated upregulation of cyclooxygenase-2 (COX-2), TP53 mutation and loss of heterozygosity at 18q chromosome [17, 21]. Conventional adenomas with high MSI are linked to Lynch syndrome, whereas APC germline mutations are correlated with familial adenomatous polyposis [22].
On the basis of the Fearon and Vogelstein model, the bi-allelic inactivation of APC followed by oncogenic KRAS mutation, which culminates in the inactivation of TP53, are the main molecular alterations involved in the progression from conventional adenoma to cancer [16, 17]. Moreover, cancers that develop through this conventional pathway are generally CIMP-negative and show microsatellite stability (MSS) but chromosomal instability (CIN) [23]. Jass et al. detected KRAS mutation in 18% of TAs and 50% of VAs. Moreover, mutations in KRAS and BRAF appeared to be mutually exclusive in the mitogen-activated protein kinase (MAPK) pathway. In fact, KRAS was mutated in 27% of adenomas while only 5% showed a BRAF mutation [17]. In agreement with this, Kim et al. reported that about half of the conventional adenomas harbored KRAS mutations but none showed BRAF mutations [24]. Leggett et al. reported similar results, observing that only a small proportion of conventional adenomas contained BRAF mutations [25]. It has also been seen that the loss of O6-methylguanine DNA methyltransferase (MGMT) gene expression correlates with KRAS mutation in small TAs [17]. Furthermore, Whitehall et al. reported that phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) gene mutations were exclusively observed in TVAs [26].
In studies on relapsed colorectal lesions, the promoters of the tumor suppressor genes mutL homolog 1 (MLH1), ataxia telangiectasia mutated (ATM) and fragile histidine triad (FHIT) were found to be significantly hypermethylated in recurring conventional adenomas [27], while the absence of phosphatase and tensin homolog (PTEN) expression correlated positively with local recurrence [28]. It has also been seen that conventional adenomas recur more frequently in patients with conventional adenomas at baseline polypectomy [29] (Figure 1).
Figure 1. Distribution of lesions according to molecular alterations involved in the etiology and/or recurrence of preneoplastic lesions.
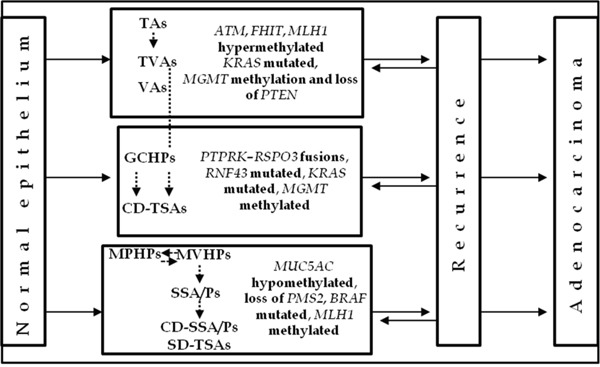
Schematic diagram of CRC progression and recurrence. Two pathways have been recognized, the suppressor and the serrated pathway. Both sequences involve the progression of normal colonocytes into early and advanced adenomas, with subsequent transformation into early and advanced cancer. Relapsed suppressor and serrated preneoplastic lesions retain the same histological subtype. The boxes show the typical molecular biomarkers for each lesion. The dotted arrows represent the potential connections between adenomas and polyps during their development.
The serrated pathway
Jass and Smith first described the serrated pathway in colorectal carcinogenesis more than twenty years ago [30]. In the literature, two terms are synonymous for serrated lesions, i.e. serrated polyps or serrated adenomas [31]. The “serrated pathway” is present in about 15%-20% of sporadic CRCs, the morphological features being the serrated or “sawtooth-like” appearance of the crypts. This feature is considered to be a consequence of cell growth in combination with the delayed migration or failure of cell apoptosis, which leads to an accumulation of epithelial cells. It is now acknowledged that several types of serrated polyps exist and can develop into a subset of invasive tumors via the serrated pathway [32]. According to 2010 WHO guidelines, this pathway has been recognized in sessile serrated lesions i.e. HPs, traditional serrated adenomas (TSAs) and sessile serrated adenomas/polyps (SSAs/Ps) [33]. However, a definitive classification for these polyps is still lacking and histological interpretation often differs among pathologists [34–36].
HPs, which represent more than 75% of serrated polyps, are flat or sessile, pale in color, generally < 5 mm, and are usually located in the distal colon. They are the only subtype of polyps with no dysplastic progression. HPs are often larger than other serrated lesions and difficult to visualize endoscopically. Although they are found more frequently than conventional adenomas in younger individuals, their incidence does not substantially increase after the age of 50 [37]. HPs can be divided into microvesicular (MVHPs), goblet cell (GCHPs) and mucin-poor polyps (MPHPs). MVHPs are frequently CIMP-high with BRAF mutation. Although they are mainly detected in the descending colon, 10-15% occur in the transverse and ascending colon. Multiple MVHPs are frequent in the rectum. GCHPs often have KRAS mutation, are more commonly found in the descending colon, and are usually small [38]. MPHPs have little to no cytoplasmic mucin and show a luminal serration pattern with increased nuclear atypia comprising large, round, hyperchromatic nuclei without pseudostratification. Observational studies report that HPs are not connected with advanced adenomas [39, 40]. Indeed, the co-existence of HPs with adenomas at index colonoscopy does not increase the risk of further adenomas or advanced adenomas at surveillance [41, 42].
In Western populations only 5% of serrated polyps are TSAs. Generally found in the descending colon and with a higher incidence in the elderly [23, 43], they are protuberant or pedunculated lesions similar to conventional adenomas, with an architecture characterized by ectopic crypts at their base. This morphological feature is used to distinguish them from SSAs. TSAs show copious eosinophilic cytoplasm and elongated, outlined nuclei. They can develop conventional or serrated dysplasia, both types of which are believed to be markers of progression to carcinoma [23]. A subset of TSAs with conventional dysplasia (CD-TSAs) is characterized by KRAS mutations and silencing of MGMT by promoter hypermethylation, CIMP-low phenotype and MSS [44]. Conversely, TSAs with serrated dysplasia (SD-TSAs) are characterized by CIMP-high, BRAF mutation and MSS [23].
Numerous studies have shown that individuals with sessile serrated lesions at baseline tend to develop additional sessile serrated lesions over time [29, 45, 46]. Of note, DNA mismatch repair (MMR) proteins are intact in all cases of TSAs, including those with dysplasia [47].
SSAs/Ps without dysplasia are the most clinically important serrated polyps due to their high incidence. These flat or slightly elevated lesions are often located in the proximal colon and generally measure > 5 mm [23]. Histologically, SSAs differ from HPs in their manifestation of atypical structural characteristics due to abnormal proliferation. The HP proliferation zone is situated at the bottom of the crypts. However, in SSAs/Ps, crypt proliferation leads to an increase in asymmetry, e.g. T-shaped or inverted L-shaped structures. SSAs/Ps may also have a yellow mucous cap, which makes them easier to identify endoscopically [23, 37]. From a molecular point of view, SSAs/Ps show a high frequency of BRAF mutation, CIMP phenotype and MSS [48, 49].
It has been reported that 19% to 27% of CRCs occur at the same site of the primary polypectomy, 18% of patients with large sessile polyps showing residual adenomatous tissue when re-examined [49–53].
Cytological dysplasia in SSAs/Ps (CD-SSAs/Ps) resembles that of conventional adenomas characterized by elongated pencillate nuclei with hyperchromasia, nuclear pseudostratification and amphophilic cytoplasm [23]. The transition between cytological dysplastic epithelium and non-dysplastic SSA/P epithelium may be abrupt, appearing as a ‘collision’ between two lesions and leading to their classification as “mixed hyperplastic adenomatous polyps” or “mixed SSA/P-TA” [54, 55]. CD-SSAs/Ps are characterized by wild type KRAS, mutated BRAF, CIMP-high status, methylated MLH1 and MSI [56].
Lazarus et al. reported that recurrence rates of serrated adenoma were higher than those of HPs or conventional adenomas [29]. In a retrospective study, Lu et al. reported that 15% of SSP patients developed cancer or adenomatous polyps with high-grade dysplasia compared to 6% of control patients with HPs or adenomatous polyps at baseline [40]. However, patients with non-dysplastic SSAs/Ps did not develop advanced neoplasia, even though the size and location of the lesions indicated that there was an increased risk of CRC [61, 62]. Moreover, the detection of serrated polyps was indicative of the subsequent development of mature serrated polyps rather than conventional adenomas [29, 57]. Indeed, 94% of patients with serrated adenomas at the first check-up later developed serrated polyps. Furthermore, Teriaky et al. observed new SSPs within the first 3 years of follow-up in patients with SSPs at baseline [45]. Renaud et al. observed that mucin 5AC (MUC5AC) gene hypomethylation was an early event in the serrated neoplasia pathway and specifically detected MVHP and SSA/P lesions [58]. Individuals with co-existing SSAs/Ps and conventional adenomas appear to be a high-risk group [59]. Sekine et al. showed that TSAs commonly harbor genetic alterations leading to the activation of the WNT pathway [60]. In particular, they found that TSA genetic features were PTPRK-RSPO3 fusions and RNF43 mutations (Figure 1) [60].
Interactions between preneoplastic lesions
As previously mentioned, various models of CRC development, starting from preneoplastic lesions, have been hypothesized, each of which is based on different molecular mechanisms. The suppressor and serrated pathways are the best characterized and most clearly distinguishable. However, to date relatively few genetic or epigenetic factors have been correlated with recurrent lesions (Figure 1). TSAs are a heterogeneous group that can be divided into CD-TSAs or SD-TSAs on the basis of specific molecular markers [47, 63]. SD-TSAs have been described as exhibiting areas with features of HPs and/or SSAs/Ps, and the hypothesis that HPs and/or SSAs/Ps may be precursor lesions to TSAs has been put forward [23]. Tsai et al. reported that TSAs with this type of dysplasia develop into in smaller, less aggressive neoplastic lesions than other preneoplastic subtypes [47]. Conversely, CD-TSAs characterized by KRAS mutations, MGMT hypermethylation, CIMP-low phenotype and MSS, may originate from TVAs and are associated with a poorer prognosis. This pathway is known as the alternative pathway [23, 64]. It has been suggested that TAs may also evolve along this alternative pathway, bypassing TVAs [23, 65]. It is also likely that MVHPs evolve into SSAs/Ps, especially when they are located in the proximal colon [66]. There is ample evidence that MVHPs and SSAs/Ps without dysplasia show overlapping molecular patterns when tested for CIMP, MLH1, KRAS and BRAF status, which is consistent with the notion that MVHPs are immediate precursors of SSAs/Ps [23]. The methylation silencing of MLH1, which leads to the MSI phenotype, is detected as variably decreased MLH1 expression in dysplastic areas of CD-SSAs/Ps. Of note, although MLH1 methylation is detectable early in SSAs/P growth, only a reduction or loss of gene expression by extensive methylation is associated with dysplasia and heralds the committed progression of the serrated cancer pathway to malignancy. In lesions with loss of MLH1, the PMS2 gene, one partner in the stabilizing of MutL heterodimer MMR, is also lost [23]. It has also been suggested that MPHPs are MVHPs that have been modified by inflammation and reactive epithelial changes [38]. However, the only molecular marker associated with MPHPs to date is the CIMP-high phenotype. Successor lesions of GCHPs are seldom observed and it is debatable whether they are self-limiting or progress to advanced KRAS-mutated serrated polyps, i.e. CD-TSAs (Table 2) [66–68].
Table 2. Main molecular alterations of all preneoplastic colorectal lesions.
| Preneoplastic lesions | APC | KRAS | BRAF | CIMP | Microsatellite | CIN | MGMT | |
|---|---|---|---|---|---|---|---|---|
| Serrated adenomas | MVHPs | Mutated | High | Stable | ||||
| GCHPs | Mutated | Low | Stable | |||||
| MPHPs | High | |||||||
| SD-TSAs | Mutated | High | Stable | |||||
| CD-TSAs | Mutated | Low | Stable | Hypermethylated | ||||
| SSAs/Ps | Mutated | High | Instable | |||||
| CD-SSAs/Ps | Mutated | High | Instable | |||||
| Conventional adenomas | TAs | Mutated | Mutated | Low | Stable | + | ||
| TVAs | Mutated | Mutated | Low | Stable | + | Hypermethylated | ||
| VAs | Mutated | Mutated | Low | Stable | + |
Gut microbiota
The microbial communities of the gastrointestinal tract can be modified by diet and environmental factors [69]. Several studies have analyzed the promoting or protecting role of microbial dysbiosis in the etiology of colorectal adenoma-to-carcinoma sequence [70–73], but few have focused on the correlation between the microbiota and colorectal adenoma recurrence. Bacterial species implicated in the carcinogenesis process are Streptococcus gallolyticus, Enterococcus faecalis, Bacteroides fragilis, Escherichia coli, Fusobacterium nucleatum and Faecalibacterium prausnitzii [74, 75]. Bacteria express their pathogenicity through chronic inflammation, DNA damage, and the production of bioactive carcinogenic metabolites. A higher proportion of the phylum Proteobacteria, of the genera Pseudomonas, Helicobacter and Acinetobacter and a lower abundance of the phylum Bacteroidetes have been reported in a number of studies focusing on adenoma [76, 77]. An overabundance of the genera Fusobacterium and Prevotella in tumor samples has been observed by authors assessing differences in the microbiota between tumors and matching normal colon tissue [78–82]. These results indicate that adenoma development is potentially related to gut microbiota modifications [15]. In particular, inflammatory processes in the gut microbiota appear to be involved in CRC progression [15–83]. In the study by Dulal et al., the transfer of fecal microbiota from tumor-bearing mice to germ-free mice promoted tumorigenesis in the recipient animals [15]. The microbiota of the latter showed an abundance of the genera Akkermansia, Odoribacter and Bacteroides similar to that of the donor mice. The authors also reported that gut microbiota can be manipulated with antibiotics or probiotics to inhibit adenomas and CRC progression [15].
It is known that colonic microbiota affects a broad range of metabolic processes leading to favorable or detrimental effects. Although metabolites produced by microbiota are thought to play a role in CRC progression, little is known about the function of the majority of gut bacteria and their metabolites. Zackular et al. reported that subjects with adenomas and CRC showed a loss of the genera Clostridium and Bacteroides and of the family Lachnospiraceae in their colonic microbiota [83]. Each of these protective bacteria is a producer of short chain fatty acids (SCFAs) in the colon. SCFAs are metabolites that supply nutrients to colonocytes, thus maintaining the epithelial homeostasis. Specifically, the SCFA butyrate has been shown to have anti-tumorigenic features, including inhibition of tumor cell proliferation, initiation of apoptosis in tumor cells and mediation of T-regulatory cell homeostasis. The depletion of these protective bacterial populations, associated with an improvement in pathogenic populations, probably acts as a tumorigenic stimulator [83].
Nugent et al. demonstrated that 23 metabolites contribute to the development of adenoma, mainly as a result of an increase in prostaglandin E2, an inflammatory metabolite, and a decrease in 5-oxoproline and diketogulonic acid, two antioxidant-related metabolites [84]. Pathway studies have shown that numerous metabolites are significantly related to inflammatory response, carbohydrate metabolism and gastrointestinal diseases. An abundance of the genus Bifidobacterium has also been found in patients with adenomas. These metabolites were differently correlated with the bacteria of healthy and sick individuals, suggesting that bacteria metabolic products may be responsible for adenoma and CRC development.
Ito et al. detected the species Fusobacterium nucleatum bacteria in HPs, SSAs, TSAs and non-serrated adenomas but found that the presence of this bacterium was not significantly associated with lesion histology [85]. Positivity was, however, significantly correlated with CIMP-high status and larger tumor size, suggesting that Fusobacterium nucleatum plays a role in the initial phase of CRC tumorigenesis [85–86].
CRC bacterial dysbiosis is associated with a decreased abundance of obligate anaerobes, an increase in potentially pathogenic bacteria, and a reduction in the proportion of beneficial butyrate-producing bacteria, implying that microbial metabolites are involved in CRC etiology [87–89]. Furthermore, some dietary elements are metabolized by symbiotic microbiota into bioactive food components believed to prevent cancer [69]. The way in which gut microbiota and specific bacteria influence adenoma and CRC development has yet to be clarified. Further research is warranted to determine a correlation between the microbiota, metabolome and preneoplastic/neoplastic lesions.
CONCLUSIONS
The current review provides an overview of the state-of-art of research into field cancerization and recurrent lesions in colorectal tissue. By examining the histopathological and molecular features of colorectal preneoplastic lesion subtypes, we aimed to show that early preneoplastic colorectal lesions can be used to identify individuals at risk of recurrent preneoplastic lesions and of developing CRC. The active surveillance of subjects with preneoplastic lesions represents an important step forward in preventing CRC and in reducing mortality [90]. It is believed that genetic and environmental factors determine the predominant type of recurrent polyps after baseline polypectomy [29]. In fact, similarities have been found between the type of lesion found at baseline colonoscopy and that detected during follow-up [29, 45]. However, the majority of studies conducted to date focus on the difference between CRC and healthy tissue, only a few evaluating the different precursors of CRC and even fewer assessing the role of recurrent preneoplastic lesions in the development of CRC.
According to the top-down model, genetic modifications occur in healthy tissue of the upper crypt compartment of early conventional lesions [91]. Conversely, genetic and/or epigenetic modifications of serrated pathway early lesions originate from the lower crypt compartment but have not yet been fully characterized. There is evidence to support that an epigenetic program regulated by polycomb repressive complexes maintains the specific functions of the lower compartment [64]. An “epigenetic memory” in lower crypt cells may be present in early preneoplastic lesions and may predispose to the development of neoplastic disease. This may explain why precursor lesions proliferate downwards or laterally, are age-related, rapidly progress, and are more likely to have a CIMP phenotype.
Generally, it has been seen that conventional adenomas follow the traditional pathway of developing cancer with CIN, CIMP-negative, MSS, BRAF wild type and KRAS mutated. Conversely, principals precursors lesions of the serrated pathway are characterized by BRAF mutation, CIMP-high and MSS phenotype.
In recent years, the microRNA signatures of conventional, serrated preneoplastic, and neoplastic lesions have been extensively studied [92, 93, 94, 95]. Some miRNA shed new light into the pathogenic mechanisms underlying adenoma-to-carcinoma progression [96]. However, no clear molecular signature exists for all of the preneoplastic colorectal lesions identified to date, a number of markers would seem to play a role in more than one lesion.
For this reason, a molecular characterization of preneoplastic lesions would help to better classify preneoplastic lesion subtypes, in particular those with a higher risk of recurrence and with worse histopathological features. It would also facilitate planning of surveillance measures for polyp-bearing patients. Furthermore, a better understanding of microbiota found in relapsed lesions could shed light on the bacteria present in preneoplastic and neoplastic colorectal lesions. Such knowledge could be implemented in CRC screening programs to improve the effectiveness of personalized endoscopic surveillance programs for polyp-bearing patients.
Acknowledgments
The authors would like to thank Dr. Sara Pignatta for revising the paper for important intellectual content.
Footnotes
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
Author contributions
All authors contributed to this review equally and approved the final version of the manuscript for submission.
REFERENCES
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Saito H. Colorectal cancer screening using immunochemical faecal occult blood testing in Japan. J Med Screen. 2006;13(Suppl 1):S6–7. [PubMed] [Google Scholar]
- 3.Hassan C, Quintero E, Dumonceau JM, Regula J, Brandão C, Chaussade S, Dekker E, Dinis-Ribeiro M, Ferlitsch M, Gimeno-García A, Hazewinkel Y, Jover R, Kalager M, et al. European Society of Gastrointestinal Endoscopy. Post-polypectomy colonoscopy surveillance. European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy. 2013;45:842–51. doi: 10.1055/s-0033-1344548. [DOI] [PubMed] [Google Scholar]
- 4.Martinez ME, Baron JA, Lieberman DA, Schatzkin A, Lanza E, Winawer SJ, Zauber AG, Jiang R, Ahnen DJ, Bond JH, Church TR, Robertson DJ, Smith-Warner SA, et al. A pooled analysis of advanced colorectal neoplasia diagnoses after colonoscopic polypectomy. Gastroenterology. 2009;136:832–41. doi: 10.1053/j.gastro.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winawer SJ, NPS investigators The achievements, impact, and future of the National Polyp Study. Gastrointest Endosc. 2006;64:975–8. doi: 10.1016/j.gie.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 6.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–68. doi: 10.1002/1097-0142(195309)6. 5<963:: AID-CNCR2820060515>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 7.Dakubo GD, Jakupciak JP, Birch-Machin MA, Parr RL. Clinical implications and utility of field cancerization. Cancer Cell Int. 2007:7–12. doi: 10.1186/1475-2867-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heaphy CM, Griffith JK, Bisoffi M. Mammary field cancerization: molecular evidence and clinical importance. Breast Cancer Res. Treat. 2009;118:229–39. doi: 10.1007/s10549-009-0504-0. [DOI] [PubMed] [Google Scholar]
- 9.Utsunomiya T, Shimada M, Morine Y, Tajima A, Imoto I. Specific molecular signatures of non-tumor liver tissue may predict a risk of hepatocarcinogenesis. Cancer Sci. 2014;105:749–54. doi: 10.1111/cas.12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee YC, Wang HP, Wang CP, Ko JY, Lee JM, Chiu HM, Lin JT, Yamashita S, Oka D, Watanabe N, Matsuda Y, Ushijima T, Wu MS. Revisit of field cancerization in squamous cell carcinoma of upper aerodigestive tract: better risk assessment with epigenetic markers. Cancer Prev Res. 2011;4:1982–92. doi: 10.1158/1940-6207.CAPR-11-0096. [DOI] [PubMed] [Google Scholar]
- 11.Di Domenico M, Santoro A, Ricciardi C, Iaccarino M, Iaccarino S, Freda M, Feola A, Sanguedolce F, Losito S, Pasquali D, A Di Spiezio Sardo, Bifulco G, Nappi C, et al. Epigenetic fingerprint in endometrial carcinogenesis: the hypothesis of a uterine field cancerization. Cancer Biol Ther. 2011;12:447–57. doi: 10.4161/cbt.12.5.15963. [DOI] [PubMed] [Google Scholar]
- 12.Baba Y, Ishimoto T, Kurashige J, Iwatsuki M, Sakamoto Y, Yoshida N, Watanabe M, Baba H. Epigenetic field cancerization in gastrointestinal cancers. Cancer Lett. 2016;375:60–66. doi: 10.1016/j.canlet.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 13.Setaffy L, Langner C. Microsatellite instability in colorectal cancer: clinicopathological significance. Pol J Pathol. 2015;66:203–18. doi: 10.5114/pjp.2015.54953. [DOI] [PubMed] [Google Scholar]
- 14.Hokazono K, Ueki T, Nagayoshi K, Nishioka Y, Hatae T, Koga Y, Hirahashi M, Oda Y, Tanaka M. A CpG island methylator phenotype of colorectal cancer that is contiguous with conventional adenomas, but not serrated polyps. Oncol Lett. 2014;8:1937–44. doi: 10.3892/ol.2014.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dulal S, Keku T. Gut microbiome and colorectal adenomas. Cancer J. 2014;20:225–31. doi: 10.1097/PPO.0000000000000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–32. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 17.Jass JR, Baker K, Zlobec I, Higuchi T, Barker M, Buchanan D, Young J. Advanced colorectal polyps with the molecular and morphological features of serrated polyps and adenomas: concept of a “fusion” pathway to colorectal cancer. Histopathology. 2006;49:121–31. doi: 10.1111/j.1365-2559.2006.02466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stulc JP, Petrelli NJ, Herrera L, Mittelman A. Colorectal villous and tubulovillous adenomas equal to or greater than four centimeters. Ann Surg. 1988;207:65–71. doi: 10.1097/00000658-198801000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lanza G, Messerini L, Gafà R, GIPAD; Risio M;, IAP. SIAPEC/ Colorectal tumors: the histology report. Dig Liver Dis. 2011;43(Suppl 4):S344–55. doi: 10.1016/S1590-8658(11)60590-2. [DOI] [PubMed] [Google Scholar]
- 20.Shussman N, Wexner SD. Colorectal polyps and polyposis syndromes. Gastroenterol Rep (Oxf) 2014;2:1–15. doi: 10.1093/gastro/got041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med. 2009;361:2449–60. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langner C. Serrated and non-serrated precursor lesions of colorectal cancer. Dig Dis. 2015;33:28–37. doi: 10.1159/000366032. [DOI] [PubMed] [Google Scholar]
- 23.Yang HM, Mitchell JM, Sepulveda JL, Sepulveda AR. Molecular and histologic considerations in the assessment of serrated polyps. Arch Pathol Lab Med. 2015;139:730–41. doi: 10.5858/arpa.2014-0424-RA. [DOI] [PubMed] [Google Scholar]
- 24.Kim MJ, Lee EJ, Suh JP, Chun SM, Jang SJ, S Kim do, Lee DH, Lee SH, Youk EG. Traditional serrated adenoma of the colorectum: clinicopathologic implications and endoscopic findings of the precursor lesions. Am J Clin Pathol. 2013;140:898–911. doi: 10.1309/AJCPDJC9VC5KTYUS. [DOI] [PubMed] [Google Scholar]
- 25.Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010;138:2088–100. doi: 10.1053/j.gastro.2009.12.066. [DOI] [PubMed] [Google Scholar]
- 26.Whitehall VL, Rickman C, Bond CE, Ramsnes I, Greco SA, Umapathy A, McKeone D, Faleiro RJ, Buttenshaw RL, Worthley DL, Nayler S, Zhao ZZ, Montgomery GW, et al. Oncogenic PIK3CA mutations in colorectal cancers and polyps. Int J Cancer. 2012;131:813–820. doi: 10.1002/ijc.26440. [DOI] [PubMed] [Google Scholar]
- 27.Rengucci C, De Maio G, Gardini A, Zucca M, Scarpi E, Zingaretti C, Foschi G, Tumedei MM, Molinari C, Saragoni L, Puccetti M, Amadori D, Zoli W, et al. Promoter methylation of tumor suppressor genes in pre-neoplastic lesions; potential marker of disease recurrence. J Exp Clin Cancer Res. 2014;33:65. doi: 10.1186/s13046-014-0065-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colakoglu T, Yildirim S, Kayaselcuk F, Nursal TZ, Ezer A, Noyan T, Karakayali H, Haberal M. Clinicopathological significance of PTEN loss and the phosphoinositide 3-kinase/Akt pathway in sporadic colorectal neoplasms: is PTEN loss predictor of local recurrence? Am J Surg. 2008;195:719–725. doi: 10.1016/j.amjsurg.2007.05.061. [DOI] [PubMed] [Google Scholar]
- 29.Lazarus R, Junttila OE, Karttunen TJ, Mäkinen MJ. The risk of metachronous neoplasia in patients with serrated adenoma. Am J Clin Pathol. 2005;123:349–59. doi: 10.1309/VBAG-V3BR-96N2-EQTR. [DOI] [PubMed] [Google Scholar]
- 30.Jass JR, Smith M. Sialic acid and epithelial differentiation in colorectal polyps and cancer-a morphological, mucin and lectin histochemical study. Pathology. 1992;24:233–42. doi: 10.3109/00313029209068874. [DOI] [PubMed] [Google Scholar]
- 31.Torlakovic EE, Gomez JD, Driman DK, Parfitt JR, Wang C, Benerjee T, Snover DC. Sessile serrated adenoma (SSA) vs. traditional serrated adenoma (TSA) Am J Surg Pathol. 2008;32:21–9. doi: 10.1097/PAS.0b013e318157f002. [DOI] [PubMed] [Google Scholar]
- 32.Ensari A, Bosman FT, Offerhaus GJ. The serrated polyp: getting it right! J Clin Pathol. 2010;63:665–68. doi: 10.1136/jcp.2010.077222. [DOI] [PubMed] [Google Scholar]
- 33.Bosman FT. WHO classification of tumours of the digestive system. Lyon: IARC Press,; 2010. World Health Organization, International Agency for Research on Cancer. NLM ID: 101553728 [Book] [Google Scholar]
- 34.Khalid O, Radaideh S, Cummings OW, O'Brien MJ, Goldblum JR, Rex DK. Reinterpretation of histology of proximal colon polyps called hyperplastic in 2001. World J Gastroenterol. 2009;15:3767–70. doi: 10.3748/WJG.15.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snover DC, Jass JR, Fenoglio-Preiser C, Batts KP. Serrated polyps of the large intestine: a morphologic and molecular review of an evolving concept. Am J Clin Pathol. 2005;124:380–91. doi: 10.1309/V2EP-TPLJ-RB3F-GHJL. [DOI] [PubMed] [Google Scholar]
- 36.Glatz K, Pritt B, Glatz D, Hartmann A, O'Brien MJ, Blaszyk H. A multinational, internet-based assessment of observer variability in the diagnosis of serrated colorectal polyps. Am J Clin Pathol. 2007;127:938–45. doi: 10.1309/NXDB6FMTE9×5CD6Y. [DOI] [PubMed] [Google Scholar]
- 37.Ł Szylberg, Janiczek M, Popiel A, Marszałek A. Serrated polyps and their alternative pathway to the colorectal cancer: a systematic review. Gastroenterol Res Pract. 2015;2015:573814. doi: 10.1155/2015/573814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rex DK, Ahnen DJ, Baron JA, Batts KP, Burke CA, Burt RW, Goldblum JR, Guillem JG, Kahi CJ, Kalady MF, O'Brien MJ, Odze RD, Ogino S, et al. Serrated Lesions of the Colorectum: Review and Recommendations From an Expert Panel. Am J Gastroenterol. 2012;107:1315–29. doi: 10.1038/ajg.2012.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lieberman DA, Prindiville S, Weiss DG, Willett W, VA Cooperative Study Group 380 Risk factors for advanced colonic neoplasia and hyperplastic polyps in asymptomatic individuals. JAMA. 2003;290:2959–67. doi: 10.1001/jama.290.22.2959. [DOI] [PubMed] [Google Scholar]
- 40.Lu FI, de W van Niekerk, Owen D, Tha SP, Turbin DA, Webber DL. Longitudinal outcome study of sessile serrated adenomas of the colorectum: an increased risk for subsequent right-sided colorectal carcinoma. Am J Surg Pathol. 2010;34:927–34. doi: 10.1097/PAS.0b013e3181e4f256. [DOI] [PubMed] [Google Scholar]
- 41.Imperiale TF, Glowinski EA, Lin-Cooper C, Larkin GN, Rogge JD, Ransohoff DF. Five-year risk of colorectal neoplasia after negative screening colonoscopy. N Engl J Med. 2008;359:1218–24. doi: 10.1056/NEJMoa0803597. [DOI] [PubMed] [Google Scholar]
- 42.Laiyemo AO, Murphy G, Sansbury LB, Wang Z, Albert PS, Marcus PM, Schoen RE, Cross AJ, Schatzkin A, Lanza E. Hyperplastic polyps and the risk of adenoma recurrence in the polyp prevention trial. Clin Gastroenterol Hepatol. 2009;7:192–97. doi: 10.1016/j.cgh.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pai RK, Hartman DJ, Gonzalo DH, Lai KK, Downs-Kelly E, Goldblum JR, Liu X, Patil DT, Bennett AE, Plesec TP, Kuan SF, Nikiforova MN, Shadrach B, et al. Serrated lesions of the appendix frequently harbor KRAS mutations and not BRAF mutations indicating a distinctly different serrated neoplastic pathway in the appendix. Hum Pathol. 2014;45:227–35. doi: 10.1016/j.humpath.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 44.Ogino S, Kawasaki T, Kirkner GJ, Suemoto Y, Meyerhardt JA, Fuchs CS. Molecular correlates with MGMT promoter methylation and silencing support CpG island methylator phenotype-low (CIMP-low) in colorectal cancer. Gut. 2007;56:1564–71. doi: 10.1136/gut.2007.119750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teriaky A, Driman DK, Chande N. Outcomes of a 5-year follow-up of patients with sessile serrated adenomas. Scand J Gastroenterol. 2012;47:178–183. doi: 10.3109/00365521.2011.645499. [DOI] [PubMed] [Google Scholar]
- 46.Salaria SN, Streppe MM, Lee LA, Iacobuzio-Donahue CA, Montgomery EA. Sessile serrated adenomas: high risk lesions? Hum Pathol. 2012;43:1808–14. doi: 10.1016/j.humpath.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsai JH, Liau JY, Lin YL, Lin LI, Cheng YC, Cheng ML, Jeng YM. Traditional serrated adenoma has two pathways of neoplastic progression that are distinct from the sessile serrated pathway of colorectal carcinogenesis. Mod Pathol. 2014;27:1375–85. doi: 10.1038/modpathol.2014.35. [DOI] [PubMed] [Google Scholar]
- 48.Kim JH, Bae JM, Cho NY, Kang GH. Distinct features between MLH1-methylated and unmethylated colorectal carcinomas with the CpG island methylator phenotype: implications in the serrated neoplasia pathway. Oncotarget. 2016;7:14095–111. doi: 10.18632/oncotarget.7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fernando WC, Miranda MS, Worthley DL, Togashi K, Watters DJ, Leggett BA, Spring KJ. The CIMP Phenotype in BRAF Mutant Serrated Polyps from a Prospective Colonoscopy Patient Cohort. Gastroenterol Res Pract. 2014;2014:374926. doi: 10.1155/2014/374926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robertson DJ, Lieberman DA, Winawer SJ, Ahnen DJ, Baron JA, Schatzkin A, Cross AJ, Zauber AG, Church TR, Lance P, Greenberg ER, Martínez ME. Colorectal cancers soon after colonoscopy: a pooled multicohort analysis. Gut. 2014;63:949–56. doi: 10.1136/gutjnl-2012-303796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pabby A, Schoen RE, Weissfeld JL, Burt R, Kikendall JW, Lance P, Shike M, Lanza E, Schatzkin A. Analysis of colorectal cancer occurrence during surveillance colonoscopy in the dietary polyp prevention trial. Gastrointest Endosc. 2005;61:385–91. doi: 10.1016/S0016-5107(04)02765-8. [DOI] [PubMed] [Google Scholar]
- 52.Robertson DJ, Greenberg ER, Beach M, Sandler RS, Ahnen D, Haile RW, Burke CA, Snover DC, Bresalier RS, McKeown-Eyssen G, Mandel JS, Bond JH, Van Stolk RU, et al. Colorectal cancer in patients under close colonoscopic surveillance. Gastroenterology. 2005;129:34–41. doi: 10.1053/j.gastro.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 53.Farrar WD, Sawhney MS, Nelson DB, Lederle FA, Bond JH. Colorectal cancers found after a complete colonoscopy. Clin Gastroenterol Hepatol. 2006;4:1259–64. doi: 10.1016/j.cgh.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 54.Khashab M, Eid E, Rusche M, Rex DK. Incidence and predictors of “late” recurrences after endoscopic piecemeal resection of large sessile adenomas. Gastrointest Endosc. 2009;70:344–349. doi: 10.1016/j.gie.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 55.Snover DC, Jass JR, Fenoglio-Preiser C, Batts KP. Serrated polyps of the large intestine: a morphologic and molecular review of an evolving concept. Am J Clin Pathol. 2005;124:380–91. doi: 10.1309/V2EP-TPLJ-RB3F-GHJL. [DOI] [PubMed] [Google Scholar]
- 56.Nosho K, Igarashi H, Ito M, Mitsuhashi K, Kurihara H, Kanno S, Yoshii S, Mikami M, Takahashi H, Kusumi T, Hosokawa M, Sukawa Y, Adachi Y, et al. Clinicopathological and molecular characteristics of serrated lesions in Japanese elderly patients. Digestion. 2015;91:57–63. doi: 10.1159/000368820. [DOI] [PubMed] [Google Scholar]
- 57.Zhu H, Zhang G, Yi X, Zhu X, Wu Y, Liang J, Zhang S, Zeng Y, Fan D, Yu X, He J, He G, Chen Z, et al. Histology subtypes and polyp size are associated with synchronous colorectal carcinoma of colorectal serrated polyps: a study of 499 serrated polyps. Am J Cancer Res. 2015;5:363–74. [PMC free article] [PubMed] [Google Scholar]
- 58.Renaud F, Mariette C, Vincent A, Wacrenier A, Maunoury V, Leclerc J, Coppin L, Crépin M, Van Seuningen I, Leteurtre E, Buisine MP. The serrated neoplasia pathway of colorectal tumors: Identification of MUC5AC hypomethylation as an early marker of polyps with malignant potential. Int J Cancer. 2016;138:1472–81. doi: 10.1002/ijc.29891. [DOI] [PubMed] [Google Scholar]
- 59.Urbanski SJ, Kossakowska AE, Marcon N, Bruce WR. Mixed hyperplastic adenomatous polyps-an underdiagnosed entity: report of a case of adenocarcinoma arising within a mixed hyperplastic adenomatous polyp. Am J Surg Pathol. 1984;8:551–6. [PubMed] [Google Scholar]
- 60.Sekine S, Yamashita S, Tanabe T, Hashimoto T, Yoshida H, Taniguchi H, Kojima M, Shinmura K, Saito Y, Hiraoka N, Ushijima T, Ochiai A. Frequent PTPRK-RSPO3 fusions and RNF43 mutations in colorectal traditional serrated adenoma. J Pathol. 2016;239:133–8. doi: 10.1002/path.4709. [DOI] [PubMed] [Google Scholar]
- 61.Vu HT, Lopez R, Bennett A, Burke CA. Individuals with sessile serrated polyps express an aggressive colorectal phenotype. Dis Colon Rectum. 2011;54:1216–23. doi: 10.1097/DCR.0b013e318228f8a9. [DOI] [PubMed] [Google Scholar]
- 62.Schreiner MA, Weiss DG, Lieberman DA. Proximal and large hyperplastic and non dysplastic serrated polyps detected by colonoscopy are associated with neoplasia. Gastroenterology. 2010;139:1497–502. doi: 10.1053/j.gastro.2010.06.074. [DOI] [PubMed] [Google Scholar]
- 63.Chetty R, Hafezi-Bakhtiari S, Serra S, Colling R, Wang LM. Traditional serrated adenomas (TSAs) admixed with other serrated (so-called precursor) polyps and conventional adenomas: a frequent occurrence. J Clin Pathol. 2015;68:270–3. doi: 10.1136/jclinpath-2014-202827. [DOI] [PubMed] [Google Scholar]
- 64.Hiraoka S, Kato J, Fujiki S, Kaji E, Morikawa T, Murakami T, Nawa T, Kuriyama M, Uraoka T, Ohara N, Yamamoto K. The presence of large serrated polyps increases risk for colorectal cancer. Gastroenterology. 2010;139:1503–10. doi: 10.1053/j.gastro.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 65.Pancione M, Remo A, Colantuoni V. Genetic and epigenetic events generate multiple pathways in colorectal cancer progression. Patholog Res Int. 2012;2012:509348. doi: 10.1155/2012/509348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Michalopoulos G, Tzathas C. Serrated polyps of right colon: guilty or innocent? Ann Gastroenterol. 2013;26:212–19. [PMC free article] [PubMed] [Google Scholar]
- 67.Patai AV, Molnár B, Tulassay Z, Sipos F. Serrated pathway: alternative route to colorectal cancer. World J Gastroenterol. 2013;19:607–15. doi: 10.3748/wjg.v19.i5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fu X, Zhang X. Histological evidence of traditional serrated adenoma originating from a goblet cell hyperplastic polyp: a case report. Anal Quant Cytopathol Histopathol. 2014;36:351–4. [PubMed] [Google Scholar]
- 69.Bultman SJ. The microbiome and its potential as a cancer preventive intervention. Semin Oncol. 2016;43:97–106. doi: 10.1053/j.seminoncol.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Flemer B, Lynch DB, Brown JM, Jeffery IB, Ryan FJ, Claesson MJ, O'Riordain M, Shanahan F, O'Toole PW. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut. 2016 doi: 10.1136/gutjnl-2015-309595. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WK, Ng SC, Tsoi H, Dong Y, Zhang N, He Y, Kang Q, Cao L, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun. 2015;6:8727. doi: 10.1038/ncomms9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu J, Feng Q, Wong SH, Zhang D, Liang QY, Qin Y, Tang L, Zhao H, Stenvang J, Li Y, Wang X, Xu X, Chen N, Wu WK, et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. 2015 doi: 10.1136/gutjnl-2015-309800. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 73.Bultman SJ. Interplay between diet, gut microbiota, epigenetic events, and colorectal cancer. Mol Nutr Food Res. 2016 doi: 10.1002/mnfr.201500902. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe. 2014;15:317–28. doi: 10.1016/j.chom.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wei Z, Cao S, Liu S, Yao Z, Sun T, Li Y, Li J, Zhang D, Zhou Y. Could gut microbiota serve as prognostic biomarker associated with colorectal cancer patients' survival? A pilot study on relevant mechanism. Oncotarget. 2016;7:46158–46172. doi: 10.18632/oncotarget.10064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shen XJ, Rawls JF, Randall T, Burcal L, Mpande CN, Jenkins N, Jovov B, Abdo Z, Sandler RS, Keku TO. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes. 2010;1:138–47. doi: 10.4161/gmic.1.3.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanapareddy N, Legge RM, Jovov B, McCoy A, Burcal L, Araujo-Perez F, Randall TA, Galanko J, Benson A, Sandler RS, Rawls JF, Abdo Z, Fodor AA, et al. Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. ISME J. 2012;6:1858–68. doi: 10.1038/ismej.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marchesi JR, Dutilh BE, Hall N, Peters WH, Roelofs R, Boleij A, Tjalsma H. Towards the human colorectal cancer microbiome. PloS One. 2011;6:e20447. doi: 10.1371/journal.pone.0020447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, Holt RA. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, Baselga J, Liu C, Shivdasani RA, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292–98. doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McCoy AN, Araujo-Perez F, Azcarate-Peril A, Yeh JJ, Sandler RS, Keku TO. Fusobacterium is associated with colorectal adenomas. PloS One. 2013;8:e53653. doi: 10.1371/journal.pone.0053653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, Corthier G, J Tran Van Nhieu, Furet JP. Microbial dysbiosis in colorectal cancer (CRC) patients. PloS One. 2011;6:e16393. doi: 10.1371/journal.pone.0016393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY, Schloss PD. The gut microbiome modulates colon tumorigenesis. MBio. 2013;4:e00692–13. doi: 10.1128/mBio.00692-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nugent JL, McCoy AN, Addamo CJ, Jia W, Sandler RS, Keku TO. Altered tissue metabolites correlate with microbial dysbiosis in colorectal adenomas. J Proteome Res. 2014;13:1921–9. doi: 10.1021/pr4009783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ito M, Kanno S, Nosho K, Sukawa Y, Mitsuhashi K, Kurihara H, Igarashi H, Takahashi T, Tachibana M, Takahashi H, Yoshii S, Takenouchi T, Hasegawa T, et al. Association of Fusobacterium nucleatum with clinical and molecular features in colorectal serrated pathway. Int J Cancer. 2015;137:1258–68. doi: 10.1002/ijc.29488. [DOI] [PubMed] [Google Scholar]
- 86.Park CH, Han DS, Oh YH, Lee AR, Lee YR, Eun CS. Role of Fusobacteria in the serrated pathway of colorectal carcinogenesis. Sci Rep. 2016;6:25271. doi: 10.1038/srep25271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ohigashi S, Sudo K, Kobayashi D, Takahashi O, Takahashi T, Asahara T, Nomoto K, Onodera H. Changes of the intestinal microbiota, short chain fatty acids, and fecal pH in patients with colorectal cancer. Dig Dis Sci. 2013;58:1717–26. doi: 10.1007/s10620-012-2526-4. [DOI] [PubMed] [Google Scholar]
- 88.Weir TL, Manter DK, Sheflin AM, Barnett BA, Heuberger AL, Ryan EP. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS One. 2013;8:e70803. doi: 10.1371/journal.pone.0070803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X, Jia W, Cai S, Zhao L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012;6:320–9. doi: 10.1038/ismej.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.MJ1 O'Brien, Zhao Q, Yang S. Colorectal serrated pathway cancers and precursors. Histopathology. 2015;66:49–65. doi: 10.1111/his.12564. [DOI] [PubMed] [Google Scholar]
- 91.Shih IM, Wang TL, Traverso G, Romans K, Hamilton SR, Ben-Sasson S, Kinzler KW, Vogelstein B. Top-down morphogenesis of colorectal tumors. Proc Natl Acad Sci USA. 2001;98:2640–45. doi: 10.1073/pnas.051629398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schmitz KJ, Hey S, Schinwald A, Wohlschlaeger J, Baba HA, Worm K, Schmid KW. Differential expression of microRNA 181b and microRNA 21 in hyperplastic polyps and sessile serrated adenomas of the colon. Virchows Arch. 2009;455:49–54. doi: 10.1007/s00428-009-0804-0. [DOI] [PubMed] [Google Scholar]
- 93.Fassan M, Pizzi M, Giacomelli L, Mescoli C, Ludwig K, Pucciarelli S, Rugge M. PDCD4 nuclear loss inversely correlates with miR-21 levels in colon carcinogenesis. Virchows Arch. 2011;458:413–9. doi: 10.1007/s00428-011-1046-5. [DOI] [PubMed] [Google Scholar]
- 94.Ito M, Mitsuhashi K, Igarashi H, Nosho K, Naito T, Yoshii S, Takahashi H, Fujita M, Sukawa Y, Yamamoto E, Takahashi T, Adachi Y, Nojima M, et al. MicroRNA-31 expression in relation to BRAF mutation, CpG island methylation and colorectal continuum in serrated lesions. Int J Cancer. 2014;135:2507–15. doi: 10.1002/ijc.28920. [DOI] [PubMed] [Google Scholar]
- 95.Tsikitis VL, Potter A, Mori M, Buckmeier JA, Preece CR, Harrington CA, Bartley AN, Bhattacharyya A, Hamilton SR, Lance P, Thompson PA. MicroRNA signatures of colonic adenomas according to histology. Cancer Prev Res (Phila) 2016;9:942–9. doi: 10.1158/1940-6207.CAPR-16-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Slattery ML, Herrick JS, Pellatt DF, Stevens JR, Mullany LE, Wolff E, Hoffman MD, Samowitz WS, Wolff RK. MicroRNA profiles in colorectal carcinomas, adenomas and normal colonic mucosa: variations in miRNA expression and disease progression. Carcinogenesis. 2016;37:245–61. doi: 10.1093/carcin/bgv249. [DOI] [PMC free article] [PubMed] [Google Scholar]