

Summary
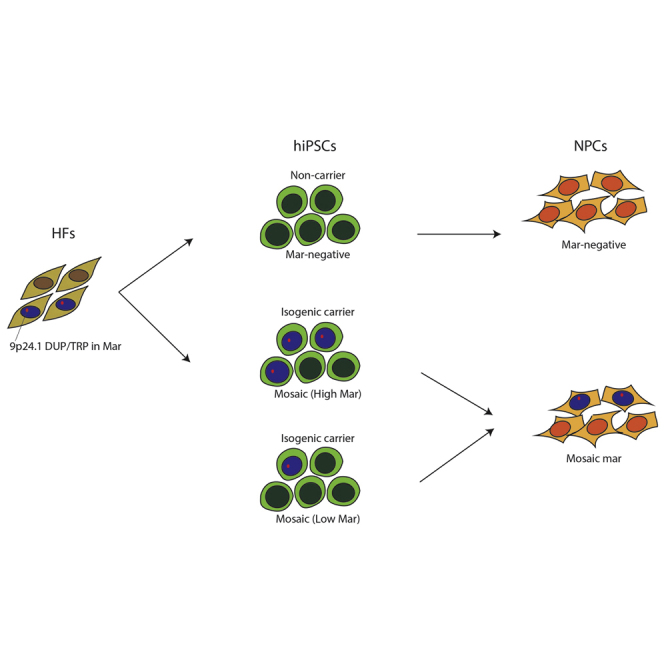
In the process of generating presumably clonal human induced pluripotent stem cells (hiPSCs) from two carriers of a complex structural rearrangement, each having a psychotic disorder, we also serendipitously generated isogenic non-carrier control hiPSCs, finding that the rearrangement occurs as an extrachromosomal marker (mar) element. All confirmed carrier hiPSCs and differentiated neural progenitor cell lines were found to be mosaic. We caution that mar elements may be difficult to functionally evaluate in hiPSC cultures using currently available methods, as it is difficult to distinguish cells with and without mar elements in live mosaic cultures.
Keywords: 9p24.1, marker chromosome, human induced pluripotent stem cell, neural progenitor cell, schizophrenia, bipolar disorder, isogenic, mosaic
Graphical Abstract
Highlights
-
•
Patients with mar chromosome yield carrier and non-carrier hiPSCs
-
•
Presumably clonal mar carrier hiPSCs show mosaicism in all lines
-
•
Neural differentiation of mosaic hiPSCs yields mosaic NPCs
Brennand, Levy, Carvalho, and colleagues present a systematic evaluation of marker chromosome mosaicism in somatic fibroblasts, hiPSCs, and NPCs. Because this mutation was initially identified as a complex genomic rearrangement rather than as a karyotypic abnormality, it represents a cautionary indication that the precise structure of any genetic mutation should be clarified before moving forward with hiPSC-based studies.
Introduction
Chromosomal trisomy disorders have long been associated with abnormal developmental outcomes (Oster-Granite, 1986). While early studies detected large chromosomal abnormalities via karyotype analysis or fluorescence in situ hybridization (FISH), more recently, genomic approaches have facilitated unbiased identification of microdeletions and microduplications (reviewed in Watson et al., 2014), identifying many that are significantly associated with schizophrenia and bipolar disorder (CNV and Schizophrenia Working Groups of the Psychiatric Genomics Consortium and Psychosis Endophenotypes International Consortium, 2016, Malhotra et al., 2011). Human induced pluripotent stem cell (hiPSC)-based models are an emerging strategy by which to evaluate the functional effects of such chromosomal aberrations in human neurons.
Growing evidence suggests that hiPSCs are fundamentally similar regardless of reprogramming methods (Choi et al., 2015, Schlaeger et al., 2015) or donor cell types (Kyttala et al., 2016) and that reprogramming increases the number of genes with a detectable donor effect in disease models (Thomas et al., 2015). Mitochondrial heteroplasmy (Perales-Clemente et al., 2016), genetic (Gore et al., 2011, Hussein et al., 2011, Liu et al., 2014), and epigenetic (Mekhoubad et al., 2012, Nazor et al., 2012) differences all contribute to intra-individual variability between hiPSCs. While genetic errors likely reflect both pre-existing mutations in the source somatic cells (Abyzov et al., 2012, Young et al., 2012) and the stresses associated with cellular replication (Laurent et al., 2011, Lu et al., 2014), epigenetic aberrations occur in hiPSCs regardless of the somatic cell type of origin and likely arise during the reprogramming process (Ma et al., 2014). Genetic and epigenetic errors can distinguish hiPSC lines from the same individual. Since such mutations are assumed to arise at equal frequency in hiPSCs reprogrammed from cases and controls, they are not typically considered serious impediments to disease-modeling studies if multiple hiPSC lines are used in comparisons. Although hiPSC-based models are generally assumed to capture the genetic variants contributing to a disease state, notable exceptions have been reported. First, not only can trisomy correction be facilitated by selecting against a transgene (TKNEO) targeted to one copy of chromosome 21 (chr 21) (Li et al., 2012), but spontaneous derivation of isogenic controls can also occur in Down syndrome trisomy 21 hiPSCs (Weick et al., 2013). Second, in hiPSCs derived from a patient with Miller-Dieker syndrome (MDS; MIM #247200), patient fibroblasts lost an abnormal ring chr 17 during hiPSC derivation and duplicated the wild-type homolog, apparently via a compensatory uniparental disomy mechanism (Bershteyn et al., 2014).
We present evidence that although extrachromosomal marker (mar) elements are relatively stable during extended culture of mosaic patient-derived fibroblasts, instability during the reprogramming process led to the derivation of isogenic non-carrier hiPSC lines as well as mosaic carrier hiPSC lines. Because this mutation was initially identified as a complex genomic rearrangement (CGR), rather than as a karyotypic abnormality, it represents a cautionary indication that the precise structure of any genetic mutation should be clarified before moving forward with hiPSC-based studies.
Results
Identification of a Duplication/Triplication of 9p24.1 in Two Patients with Psychotic Disorders
A small family cohort was identified in which a CGR involving chr 9p24.1 occurred in two carriers, one with a diagnosis of schizoaffective disorder (DL3363, proband) and one with a diagnosis of bipolar disorder with psychotic features (DL5459, mother) (Figure 1); there were also two clinically unaffected non-carrier relatives (full brother and maternal half-sister of the proband) (Figure 1C). This 9p24.1 CGR is constituted by a duplication (DUP) of 1.18 Mb (comprising 12 genes) followed by a triplicated (TRP) segment of 0.672 Mb (comprising three genes: UHRF2, GLDC, and part of KDM4C) (Figure 1A). Originally identified by array comparative genomic hybridization (aCGH) (Malhotra et al., 2011), it was more fully characterized by custom high-resolution aCGH as a CGR involving 9p24.1 (Figure 1B) (unpublished data).
Figure 1.

Identification of 9p24.1 DUP/TRP and a Mar Chromosome in Two Patients with Psychotic Disorders
(A) Schematic of human chr 9, highlighting band 9p24.1, which contains the DUP/TRP region. Encoded genes are listed on the right; genes in the DUP region (red) and TRP region (blue) are indicated (GLDC is in the TRP region).
(B) From leukocytes, custom aCGH identified a CGR in the proband DL3363 (shown) and mother DL5459. This structural rearrangement, constituted by a DUP/TRP, spans 1.85 Mb.
(C) List of HF lines used in this study.
(D and E) Karyotype analysis of HF lines derived from the 9p24.1-carrier proband (DL3363) and mother (DL5459). Representative karyotype images of mar (M) (D) and percentage mosaicism (E) of HF lines derived from the 9p24.1-carrier proband (DL3363) and mother (DL5459). Red arrows indicate an additional t(1;18)(p22;q11.2) abnormality identified in 3/20 fibroblast cells (but not present in validated hiPSC lines).
(F and G) GLDC FISH analysis of HF lines derived from the 9p24.1-carrier proband (DL3363) and mother (DL5459). FISH images (600× magnification, images cropped and resized post capture) (F) from HFs from the proband (DL3363) (shown) and his mother (DL5459) with two (top), three (middle), and four (bottom) GLDC probes (red) within the DAPI-positive nucleus. Proportion of HF cells (G) with GLDC FISH probes in early passage (p5) and late passage (p20) from DL3363 and DL5459 (number of cells = 200 each, one assay per line).
The 9p24.1 DUP/TRP Exists on a Marker Chromosome
Human fibroblasts (HFs) were obtained from the two 9p24.1 DUP/TRP carriers; unexpectedly, G-band chromosomal karyotyping identified mosaicism for an abnormal mar element (DL3363, 56.2%; DL55459, 25.0%) (Figures 1D and 1E; Table S1). Mar elements are structurally abnormal (and typically small) chromosomes that cannot be resolved by conventional banding cytogenetics alone; their clinical significance varies depending on the specific genetic material (ISCN, 2013). Because karyotypic mosaicism can underestimate the prevalence of the mar element (false negatives occur due to the small size of the mar element), these results were confirmed by FISH in interphase cells for a probe to glycine decarboxylase (GLDC), a gene included in the 9p24.1 TRP region. FISH analysis indicated that in both carriers the mar element contained sequences from the 9p24.1 region, and also confirmed that HF lines from both carriers contain a similar level of mar mosaicism (55% ± 0.5%) (Figures 1F and 1G; Table S1).
Mar elements are often believed to have low stability, as they can be lost during cell division due to their small size. However, when we compared the frequency of the mar by FISH for GLDC in 9p24.1-carrier HFs at low (p5) (DL3363, 55.0%; DL5459, 54.5%) and high (p20) passage (DL3363, 54.0%; DL5459, 45.5%), we found only minimal decline in extra FISH signals for the GLDC locus in interphase cells (Figure 1G and Table S1).
Reprogramming of 9p24.1-Carrier HFs Yields Isogenic Carrier and Non-carrier hiPSCs; 9p24.1-Carrier hiPSCs Show Variable Mosaicism in All Lines
We generated genetically unmanipulated and presumably clonal hiPSCs from the two 9p24.1 carriers (Figures 2 and 3) and the two non-carrier relatives (data not shown) using Sendai viral vectors to reprogram subject HFs, as described previously (Topol et al., 2016). All hiPSCs showed robust self-renewal as well as mRNA and protein expression of NANOG, OCT4, TRA-1-81, and TRA-1-60 (Figures 2A, 2B, S2A, and S2B). Validated hiPSCs were identical to their original fibroblasts by DNA fingerprinting (Figures S2F and S2G). At least three validated hiPSCs were generated per person; moreover, three 9p24.1-carrier and three isogenic 9p24.1-non-carrier hiPSCs were derived from both 9p24.1 carriers’ HFs and confirmed by PCR and/or aCGH (Figures 2C and S2C–S2E). Karyotyping of 9p24.1 hiPSCs detected a high frequency of mar elements in otherwise karyotypically normal lines (Figure 2D and Table S2). PCR of one of the CGR breakpoint junctions confirmed that mar-positive hiPSCs were carriers of the 9p24.1 rearrangement; in contrast, the CGR was not present in mar-negative hiPSCs; this mosaic pattern was maintained throughout hiPSC passaging from low passage (p3) to higher passage (p10) in both mar-positive and mar-negative lines (Figures S2C and S2D).
Figure 2.

Serendipitous Generation of 9p24.1-Positive and 9p24.1-Negative hiPSCs from Two 9p24.1-Carrier Patients
(A and B) Validation of hiPSCs derived from the 9p24.1-carrier proband (DL3363) and mother (DL5459). Immunofluorescence for NANOG, OCT4, TRA1-81, and TRA-1-60 (A) and fluorescence-activated cell sorting histograms for TRA-1-60 and SSEA4 (B) from hiPSCs derived from the 9p24.1-carrier proband (DL3363) and mother (DL5459). Scale bar, 200 μm. Representative images per condition are selected from Figure S2A.
(C) Representative Agilent aCGH plots of the 9p24.1 region reveals the presence of the DUP/TRP rearrangement in some hiPSCs (9p24.1-positive) generated from the proband but not in others (9p24.1-negative isogenic hiPSCs).
(D and E) Karyotype analysis of hiPSCs derived from the 9p24.1-carrier proband (DL3363) and mother (DL5459). Representative karyotype images of mar (M) (D) and percentage mosaicism (E) of hiPSCs derived from the 9p24.1-carrier proband (DL3363) and mother (DL5459). Karyotype counts indicated variable levels of mosaicism of the mar element in all genotype-positive hiPSCs.
(F) Bright-field images of high mar (60%) and low mar (10%) 9p24.1-carrier hiPSCs. Scale bar, 200 μm.
Figure 3.

In 9p24.1-Carrier hiPSCs, the Mar Contains Sequence of Chr 9 and the Triplicated Gene GLDC
(A–C) Chr 9 FISH analysis of hiPSCs from the 9p24.1-carrier proband (DL3363) and mother (DL5459). FISH images (600× magnification, images cropped and resized post capture) (A) with two (left) or three (right) chr 9 probes (red) within the DAPI-positive nucleus as well as approximate correlation (B) between karyotypic mar mosaicism and chr 9 FISH mosaicism. FISH image (600× magnification, image cropped and resized post capture) (C) from a 9p24.1-carrier hiPSC showing overlap of the chr 9 probe with the mar element (arrow).
(D–F) GLDC FISH analysis of hiPSCs derived from the 9p24.1-carrier proband (DL3363) and mother (DL5459). FISH images (600× magnification, images cropped and resized post capture) (D) and quantitative analysis (number of cells = 200 each, one assay per line) (E) from high mar and low mar of 9p24.1-carrier (C1–C3) and non-carrier (NC1–NC3) hiPSCs at low (p12) and high (p24) passage with three (left) and four (right) GLDC probes (red) within the DAPI-positive nucleus. FISH image (600× magnification, image cropped and resized post capture) (F) from 9p24.1-carrier hiPSCs showing overlap of the GLDC probe with the mar element (arrow). C, carrier; NC, non-carrier.
(G) qRT-PCR for GLDC mRNA levels in HFs as well as carrier and non-carrier hiPSCs from the 9p24.1-carrier proband (DL3363) and mother (DL5459). Percentages of mar in each cell line are indicated. Data are shown as mean ± SD from three technical replicates of each individual line. One-way ANOVA with Tukey's multiple comparison test: n.s., not significant; ∗∗p < 0.01, ∗∗∗p < 0.001.
We analyzed hiPSCs from both the proband (DL3363) and mother (DL5459) for the percentage presence of the mar element. Although presumably clonal in origin, 9p24.1-positive hiPSCs possessed varying levels (10%–60%) of the mar element, typically below the rate detected in the source HFs (Figure 2E and Table S2). There was no noticeable difference in hiPSC morphology between isogenic high mar (60%), low mar (10%), and non-carrier (no mar) hiPSCs (0%) (Figures 2A and 2F).
Mar Element Contains Sequence of Chr 9 and the Triplicated Gene GLDC
FISH in interphase cells confirmed that 9p24.1-carrier hiPSCs from both the proband (DL3363) and mother (DL5459) showed evidence of cells with two (left) or three (right) chr 9 signals (within the DAPI-positive nuclei) (Figure 3A). Moreover, additional FISH images demonstrated overlap of the chr 9 signal with the mar element, indicating that the mar is likely the source of the chr 9 DUP/TRP sequence (Figure 3C). The percentage of mosaicism in two genotype-positive hiPSCs, one with high mar and one with low mar mosaicism, by G-banding karyotype analysis at p12 (DL3363 C1, 10%; DL5459 C5, 55%), was comparable with the level of mosaicism determined by FISH at p12 (DL3363 C1, 6.5%; DL5459 C5, 49.0%), despite being tested 6–12 months apart (Figure 3B and Table S3).
When we compared the frequency of mosaicism in high and low mar hiPSCs by GLDC FISH at p12 (DL5459 C5 [high mar], 44.0%; DL5459 C6 [low mar], 32.5%), following extended culture (p22) (DL5459 C5 [high mar], 42.5%; DL5459 C6 [low mar], 20.5%), we found variable decline in evidence of extra FISH signals in interphase cells for the GLDC locus (Figures 3D and 3E; Tables S3 and S4). Consistent with chr 9 FISH signal in the mar element (Figure 3C), FISH analysis also demonstrated overlap of the GLDC signal with the mar element (Figure 3F).
Perhaps reflecting the low expression of GLDC in HFs relative to NPCs, qPCR for GLDC expression detected significantly increased GLDC levels in only one carrier HF (DL3363) compared with a non-carrier relative (DL6463) (Figure S1B). GLDC levels in hiPSCs (from both 9p24.1-carrier and isogenic non-carrier lines derived from DL3363 and DL5459) were significantly higher than in HFs (p = 0.0004 for DL3363 and p = 0.0002 for DL5459) (Figure 3G). While we observed a significant increase in GLDC expression in three 9p24.1-carrier hiPSCs from DL5459 relative to three isogenic non-carrier hiPSCs (p = 0.004), there was considerable variability in GLDC expression among DL3363 hiPSCs independent of carrier status (Figure 3G).
Neural Differentiation of 9p24.1 High and Low Mar hiPSCs Can Yield NPCs with Similar Levels of Mar Elements
We tested the effect of neural differentiation on the frequency of mar mosaicism. When we compared NPCs differentiated from p15 high and low mar hiPSCs as determined by karyotyping at p12 (DL5459 C5 [high mar], 55.0%; DL5459 C6 [low mar], 10.0%), there were no obvious differences with respect to NPC cellular morphology or detection of NPC markers such as FOXP2, NESTIN, and PAX6 (Figures 4A and 4B). Interestingly, the percentage of mosaicism in carrier high and low mar hiPSC NPCs, evaluated by GLDC FISH, was similar (35% ± 2.5%) but differed from source hiPSCs (high mar, 55.0%; low mar, 10.0%), suggesting that the level of mosaicism in NPCs did not reflect the source hiPSCs (Figures 4C and 4D; Table S2).
Figure 4.

Neural Differentiation of 9p24.1 High and Low Mar hiPSCs Yields Similar Levels of Mar Elements
(A and B) Bright-field (A) and immunofluorescence for FOXP2, NESTIN, and PAX6 (B) images of NPCs from high (55.0%) and low mar (10.0%) 9p24.1-carrier hiPSCs (DL5459 C5 and C6, respectively). Scale bars, 500 μm.
(C and D) FISH images (600× magnification, images cropped and resized post capture) (C) and quantitative analysis (number of cells = 200 each, one assay per line) (D) from NPCs differentiated from high (55.0%) and low mar (10.0%) 9p24.1-carrier hiPSCs (DL5459 C5 and DL5459 C6) with three (left) and four (right) GLDC probes (red) within the DAPI-positive nucleus.
Discussion
Mar elements are rare in the general population (estimated to occur in 0.044% of newborn infants) (Liehr et al., 2004), but >30% mar element carriers are clinically abnormal (Liehr et al., 2006) and mar elements are enriched approximately 7-fold in patients with developmental delay (0.288% of cases) (Liehr and Weise, 2007). The size, genetic content, and extent of somatic mosaicism contribute to the clinical impact of a mar carrier case (Liehr et al., 2013). Somatic mosaicism is present in slightly more than 50% of mar carriers, and the extent of mosaicism can range from very low (<0.5%) to very high (>95%); nonetheless, it is difficult to directly correlate the extent of mosaicism to the magnitude of the clinical phenotype, likely because only one tissue is typically evaluated (Liehr et al., 2013). The mitotic stability of mar elements in vitro is dependent on their shape and telomeres (Hussein et al., 2014). Whether the 9p24.1 mar element described here affects the clinical phenotypes in these two carriers is unknown; the mosaicism in presumably clonal hiPSCs has made it difficult to functionally evaluate the causal impact of this mar element.
As future hiPSC-based studies include a growing number of patients with rare CGRs, they are likely to include individuals with mar elements; we recommend that the precise structure of novel CGRs be clarified by karyotyping before moving forward with hiPSC-based studies. Our work suggests that although mar elements can be maintained with relative stability in mosaic proliferating HF populations, they are frequently lost during the reprogramming process. More critically, the extent of mosaicism in patient HFs and hiPSCs is not particularly predictive of the extent of mosaicism present in patient-derived NPCs. While the generation of spontaneous isogenic carrier and non-carrier hiPSCs from the same patients is fortuitous, the difficulty resolving carrier status in individual living cells makes these hiPSCs a difficult platform for molecular and cellular phenotyping in disease-modeling studies.
Instability of chromosomal abnormalities has been well documented in vivo and in vitro. Among practicing clinical geneticists, it is widely understood that mar elements are unstable and likely to present as mosaic in vivo (Liehr et al., 2010). Similarly, trisomies found early in pregnancy during chorionic villus studies may undergo “trisomy-to-disomy” rescue via uniparental disomy (Spence et al., 1988). Moreover, high rates of aneuploidy in source cells do not always translate into similarly high rates in the derived hiPSCs, likely because aneuploidies may be subjected to selective pressures during reprogramming, resulting in different levels of tolerance in the reprogramming cells (Hamada et al., 2012).
The presence of mar mosaicism in source HFs, presumably clonal hiPSCs and hiPSC NPCs, indicates that these lines will not be readily amenable to functional evaluation via in vitro modeling using current methods, as one cannot distinguish 9p24.1 and control cells in live cultures. Standard phenotypic comparisons in these variably mosaic lines would be expected to yield large inter-experiment and intra-individual variation, requiring comparisons of increased numbers of cells and/or biological replicates to reach statistically significant conclusions. While phenotypic assays could theoretically be combined with stringent molecular techniques to resolve the 9p24.1 status of individual neural cells, combining post hoc FISH with many standard hiPSC-based assays of neuronal function (dendritic branching, synaptic imaging, multi-electrode array [MEA], electrophysiology, neurotransmitter release) and/or global transcriptomic, proteomic, or epigenetic approaches would likely prove practically difficult. FISH sample processing methodologies may be technically difficult to pair with some sensitive synaptic staining protocols, many commercial MEA plates are not amenable to imaging, and post hoc FISH would dramatically reduce the throughput of dendritic tracing or electrophysiological comparisons, approaches already limited by the small number of cells that can be evaluated. For population-wide studies, from neurotransmitter release to “-omics” approaches, each experiment would have to be normalized to reflect the current extent of mosaicism in each culture, dramatically confounding already variable analyses.
The reversion of the mutation to wild-type may reflect a selection disadvantage to 9p24.1 cells in vitro, particularly during the process of reprogramming and neural differentiation; however, our inability to distinguish and/or purify 9p24.1 and control fibroblasts, or to repeatedly obtain skin biopsies from these two individuals, makes this a difficult hypothesis to test. We present this cautionary evidence of a heritable but unstable genetic mutation to alert hiPSC researchers to the possibility that some genetic variations may be particularly intransigent to hiPSC-based modeling at the present time. Thus, confirmation of the presence of the genetic variant in source HFs, clonal hiPSCs, and differentiated cells is essential prior to beginning phenotypic characterizations.
Experimental Procedures
Patients, hiPSC Derivation, and NPC Differentiation
Human subjects work was approved by the McLean Hospital Institutional Review Board. HFs were expanded from skin biopsies and hiPSCs were derived using Sendai viral vectors (Life Technologies). NPCs were generated using the 7-day neural induction protocol (Life Technologies). See Supplemental Experimental Procedures for detailed descriptions of cohort, HF culture, hiPSC derivation, and NPC differentiation protocols.
Molecular Analysis
aCGH was conducted using a custom 4 × 180 K microarray (Agilent Technologies); genotypes were confirmed by long-range PCR. Karyotyping and FISH were performed by Wicell Cytogenetics (Madison, WI). See Supplemental Experimental Procedures for detailed descriptions of aCGH, karyotype, FISH, PCR, DNA fingerprinting, and immunocytochemical techniques.
Author Contributions
J.TCW and K.J.B. designed, performed, and analyzed the experiments, with technical support provided by A.N.A. D.L.L. and U.R. identified and recruited the family and initiated the collaboration to perform hiPSC-based modeling of this mutation; A.J.S. performed the skin biopsies. C.M.B.C., B.Y., S.G., J.R.L., S.M., D.M., and J.S. genotyped the carriers. K.J.B., J.TCW, D.L.L., C.M.B.C., and J.R.L. wrote the manuscript. All authors have reviewed the manuscript and are in agreement with the content.
Acknowledgments
Alison Goate kindly offered use of sequencing machine and reagents for fingerprinting of HFs and hiPSCs. Jano Roze assisted in the expansion of some hiPSC lines. All karyotype and FISH analyses were performed by the Cytogenetic Laboratory of WiCell Research Institute. K.J.B. is a New York Stem Cell Foundation - Robertson Investigator. This work was supported in part by NIH grants R01 MH101454 (K.J.B.), R21 MH097470 (D.L.L.), R21 MH1057321 (D.L.L.), RO1NS058529 (J.R.L.), R21 MH104505 (U.R.), New York Stem Cell Foundation (K.J.B.), Brain and Behavior Young Investigator Grant (K.J.B.), the Ellison Foundation (D.L.L., K.J.B., J.R.L., C.M.B.C.), Anonymous Foundation (D.L.L.), the Carmela and Menachem Abraham Fund (D.L.L.), and Team Daniel (D.L.L.).
Published: February 16, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.01.010.
Contributor Information
Claudia M.B. Carvalho, Email: cfonseca@bcm.edu.
Deborah L. Levy, Email: dlevy@mclean.harvard.edu.
Kristen J. Brennand, Email: kristen.brennand@mssm.edu.
Supplemental Information
References
- Abyzov A., Mariani J., Palejev D., Zhang Y., Haney M.S., Tomasini L., Ferrandino A.F., Rosenberg Belmaker L.A., Szekely A., Wilson M. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012;492:438–442. doi: 10.1038/nature11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershteyn M., Hayashi Y., Desachy G., Hsiao E.C., Sami S., Tsang K.M., Weiss L.A., Kriegstein A.R., Yamanaka S., Wynshaw-Boris A. Cell-autonomous correction of ring chromosomes in human induced pluripotent stem cells. Nature. 2014;507:99–103. doi: 10.1038/nature12923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Lee S., Mallard W., Clement K., Tagliazucchi G.M., Lim H., Choi I.Y., Ferrari F., Tsankov A.M., Pop R. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat. Biotechnol. 2015;33:1173–1181. doi: 10.1038/nbt.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CNV and Schizophrenia Working Groups of the Psychiatric Genomics Consortium, Psychosis Endophenotypes International Consortium Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 2016;49:27–35. doi: 10.1038/ng.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore A., Li Z., Fung H.L., Young J.E., Agarwal S., Antosiewicz-Bourget J., Canto I., Giorgetti A., Israel M.A., Kiskinis E. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada M., Malureanu L.A., Wijshake T., Zhou W., van Deursen J.M. Reprogramming to pluripotency can conceal somatic cell chromosomal instability. PLoS Genet. 2012;8:e1002913. doi: 10.1371/journal.pgen.1002913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein S.M., Batada N.N., Vuoristo S., Ching R.W., Autio R., Narva E., Ng S., Sourour M., Hamalainen R., Olsson C. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62. doi: 10.1038/nature09871. [DOI] [PubMed] [Google Scholar]
- Hussein S.S., Kreskowski K., Ziegler M., Klein E., Hamid A.B., Kosyakova N., Volleth M., Liehr T., Fan X., Piaszinski K. Mitotic stability of small supernumerary marker chromosomes depends on their shape and telomeres - a long term in vitro study. Gene. 2014;552:246–248. doi: 10.1016/j.gene.2014.09.041. [DOI] [PubMed] [Google Scholar]
- ISCN . Karger Medical and Scientific Publishers; 2013. An International System for Human Cytogenetic Nomenclature. [Google Scholar]
- Kyttala A., Moraghebi R., Valensisi C., Kettunen J., Andrus C., Pasumarthy K.K., Nakanishi M., Nishimura K., Ohtaka M., Weltner J. Genetic variability overrides the impact of parental cell type and determines iPSC differentiation potential. Stem Cell Rep. 2016;6:200–212. doi: 10.1016/j.stemcr.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent L.C., Ulitsky I., Slavin I., Tran H., Schork A., Morey R., Lynch C., Harness J.V., Lee S., Barrero M.J. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell. 2011;8:106–118. doi: 10.1016/j.stem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.B., Chang K.H., Wang P.R., Hirata R.K., Papayannopoulou T., Russell D.W. Trisomy correction in Down syndrome induced pluripotent stem cells. Cell Stem Cell. 2012;11:615–619. doi: 10.1016/j.stem.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liehr T., Weise A. Frequency of small supernumerary marker chromosomes in prenatal, newborn, developmentally retarded and infertility diagnostics. Int. J. Mol. Med. 2007;19:719–731. [PubMed] [Google Scholar]
- Liehr T., Claussen U., Starke H. Small supernumerary marker chromosomes (sSMC) in humans. Cytogenet. Genome Res. 2004;107:55–67. doi: 10.1159/000079572. [DOI] [PubMed] [Google Scholar]
- Liehr T., Mrasek K., Weise A., Dufke A., Rodriguez L., Martinez Guardia N., Sanchis A., Vermeesch J.R., Ramel C., Polityko A. Small supernumerary marker chromosomes—progress towards a genotype-phenotype correlation. Cytogenet. Genome Res. 2006;112:23–34. doi: 10.1159/000087510. [DOI] [PubMed] [Google Scholar]
- Liehr T., Karamysheva T., Merkas M., Brecevic L., Hamid A.B., Ewers E., Mrasek K., Kosyakova N., Weise A. Somatic mosaicism in cases with small supernumerary marker chromosomes. Curr. Genomics. 2010;11:432–439. doi: 10.2174/138920210793176029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liehr T., Klein E., Mrasek K., Kosyakova N., Guilherme R.S., Aust N., Venner C., Weise A., Hamid A.B. Clinical impact of somatic mosaicism in cases with small supernumerary marker chromosomes. Cytogenet. Genome Res. 2013;139:158–163. doi: 10.1159/000346026. [DOI] [PubMed] [Google Scholar]
- Liu P., Kaplan A., Yuan B., Hanna J.H., Lupski J.R., Reiner O. Passage number is a major contributor to genomic structural variations in mouse iPSCs. Stem Cells. 2014;32:2657–2667. doi: 10.1002/stem.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., Li H., Hu M., Sasaki T., Baccei A., Gilbert D.M., Liu J.S., Collins J.J., Lerou P.H. The distribution of genomic variations in human iPSCs is related to replication-timing reorganization during reprogramming. Cell Rep. 2014;7:70–78. doi: 10.1016/j.celrep.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H., Morey R., O'Neil R.C., He Y., Daughtry B., Schultz M.D., Hariharan M., Nery J.R., Castanon R., Sabatini K. Abnormalities in human pluripotent cells due to reprogramming mechanisms. Nature. 2014;511:177–183. doi: 10.1038/nature13551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D., McCarthy S., Michaelson J.J., Vacic V., Burdick K.E., Yoon S., Cichon S., Corvin A., Gary S., Gershon E.S. High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron. 2011;72:951–963. doi: 10.1016/j.neuron.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekhoubad S., Bock C., de Boer A.S., Kiskinis E., Meissner A., Eggan K. Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell. 2012;10:595–609. doi: 10.1016/j.stem.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazor K.L., Altun G., Lynch C., Tran H., Harness J.V., Slavin I., Garitaonandia I., Muller F.J., Wang Y.C., Boscolo F.S. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell. 2012;10:620–634. doi: 10.1016/j.stem.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oster-Granite M.L. The neurobiologic consequences of autosomal trisomy in mice and men. Brain Res. Bull. 1986;16:767–771. doi: 10.1016/0361-9230(86)90073-0. [DOI] [PubMed] [Google Scholar]
- Perales-Clemente E., Cook A.N., Evans J.M., Roellinger S., Secreto F., Emmanuele V., Oglesbee D., Mootha V.K., Hirano M., Schon E.A. Natural underlying mtDNA heteroplasmy as a potential source of intra-person hiPSC variability. EMBO J. 2016;35:1979–1990. doi: 10.15252/embj.201694892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaeger T.M., Daheron L., Brickler T.R., Entwisle S., Chan K., Cianci A., DeVine A., Ettenger A., Fitzgerald K., Godfrey M. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015;33:58–63. doi: 10.1038/nbt.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence J.E., Perciaccante R.G., Greig G.M., Willard H.F., Ledbetter D.H., Hejtmancik J.F., Pollack M.S., O'Brien W.E., Beaudet A.L. Uniparental disomy as a mechanism for human genetic disease. Am. J. Hum. Genet. 1988;42:217–226. [PMC free article] [PubMed] [Google Scholar]
- Thomas S.M., Kagan C., Pavlovic B.J., Burnett J., Patterson K., Pritchard J.K., Gilad Y. Reprogramming LCLs to iPSCs results in recovery of donor-specific gene expression signature. PLoS Genet. 2015;11:e1005216. doi: 10.1371/journal.pgen.1005216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topol A., Zhu S., Hartley B.J., English J., Hauberg M.E., Tran N., Rittenhouse C.A., Simone A., Ruderfer D.M., Johnson J. Dysregulation of miRNA-9 in a subset of schizophrenia patient-derived neural progenitor cells. Cell Rep. 2016;15:1024–1036. doi: 10.1016/j.celrep.2016.03.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson C.T., Marques-Bonet T., Sharp A.J., Mefford H.C. The genetics of microdeletion and microduplication syndromes: an update. Annu. Rev. Genomics Hum. Genet. 2014;15:215–244. doi: 10.1146/annurev-genom-091212-153408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick J.P., Held D.L., Bonadurer G.F., 3rd, Doers M.E., Liu Y., Maguire C., Clark A., Knackert J.A., Molinarolo K., Musser M. Deficits in human trisomy 21 iPSCs and neurons. Proc. Natl. Acad. Sci. USA. 2013;110:9962–9967. doi: 10.1073/pnas.1216575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young M.A., Larson D.E., Sun C.W., George D.R., Ding L., Miller C.A., Lin L., Pawlik K.M., Chen K., Fan X. Background mutations in parental cells account for most of the genetic heterogeneity of induced pluripotent stem cells. Cell Stem Cell. 2012;10:570–582. doi: 10.1016/j.stem.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.