

ABSTRACT
All retroviruses need to integrate a DNA copy of their genome into the host chromatin. Cellular proteins regulating and targeting lentiviral and gammaretroviral integration in infected cells have been discovered, but the factors that mediate alpharetroviral avian leukosis virus (ALV) integration are unknown. In this study, we have identified the FACT protein complex, which consists of SSRP1 and Spt16, as a principal cellular binding partner of ALV integrase (IN). Biochemical experiments with purified recombinant proteins show that SSRP1 and Spt16 are able to individually bind ALV IN, but only the FACT complex effectively stimulates ALV integration activity in vitro. Likewise, in infected cells, the FACT complex promotes ALV integration activity, with proviral integration frequency varying directly with cellular expression levels of the FACT complex. An increase in 2-long-terminal-repeat (2-LTR) circles in the depleted FACT complex cell line indicates that this complex regulates the ALV life cycle at the level of integration. This regulation is shown to be specific to ALV, as disruption of the FACT complex did not inhibit either lentiviral or gammaretroviral integration in infected cells.
IMPORTANCE The majority of human gene therapy approaches utilize HIV-1- or murine leukemia virus (MLV)-based vectors, which preferentially integrate near genes and regulatory regions; thus, insertional mutagenesis is a substantial risk. In contrast, ALV integrates more randomly throughout the genome, which decreases the risks of deleterious integration. Understanding how ALV integration is regulated could facilitate the development of ALV-based vectors for use in human gene therapy. Here we show that the FACT complex directly binds and regulates ALV integration efficiency in vitro and in infected cells.
KEYWORDS: ALV, FACT, integration
INTRODUCTION
All retroviruses must integrate their genomes into the host chromatin, a process that is catalyzed by the virally encoded integrase (IN) protein. This obligate part of the retroviral life cycle has made retroviral vectors an appealing approach for the delivery of therapeutic genes during human gene therapy (1). However, because the virus does integrate into the host genome, there is a substantial risk of insertional mutagenesis, a problem that was manifested in early therapeutic trials that made use of murine leukemia virus (MLV)-based vectors (2–4).
Retroviral integration into the host genome is not random and varies dramatically across genera. The lentivirus HIV-1 has been shown to exhibit strong integration site preferences within active gene units, whereas the gammaretrovirus MLV exhibits a strong preference for enhancers and transcription start sites (5–7). These biases have been attributed to interaction of IN in the context of the preintegration complex with their cognate host cell factors (8–10). For example, cellular chromatin-associated protein lens epithelial-derived growth factor (LEDGF/p75) interacts with HIV-1 IN and directs lentiviral integration into actively transcribed genes (11–13). Similarly, BET (bromodomain and extraterminal domain) proteins have been shown to interact with MLV IN and target MLV integration to transcription start sites, enhancers, and gene regulatory regions (14–18). These host cell factors bind their cognate viral IN and selected histone marks to act as a bimodal tether to recruit the preintegration complex to specific genomic regions surrounding the host factor binding sites (8–10, 19, 20). In addition to targeting integration events to specific genomic features, these factors also serve to significantly enhance integration efficiencies (13, 14, 21).
A recent study has identified cellular serine/threonine protein phosphatase 2A (PP2A) as a selective binding partner of deltaretroviral (human T cell lymphotropic virus type 1 and 2 and bovine leukemia virus) INs (22). Furthermore, the B′ subunit of PP2A has been shown to bind and stimulate concerted integration of deltaretroviral INs in vitro (22). However, unlike LEDGF/p75 and BET proteins, PP2A does not directly engage chromatin, and it remains to be seen whether this cellular protein can modulate deltaretroviral integration in infected cells.
Alpharetroviruses such as avian leukosis virus (ALV) exhibit a distinct integration pattern with a seemingly random distribution of integration sites throughout chromatin and with only a slight preference for integrating into gene regions (23–26). To understand how ALV integration is regulated by host cellular factors, we have performed affinity capture followed by mass spectrometry (MS)-based proteomics experiments. This approach has identified structure specific recognition protein 1 (SSRP1) and suppressor of Ty 16 (Spt16), the components of the heterodimeric FACT (facilitates chromatin transcription) complex (27), as the top protein hits that specifically bound to ALV but not HIV-1 IN.
The FACT complex is a highly conserved general histone chaperone protein that is essential for transcription and DNA replication (28–30). The complex has also been shown to play important roles in DNA damage responses, centromere deposition, recombination, and DNA methylation (31–34). The FACT complex is thought to destabilize the histone octamer, providing access to the DNA for various enzymes (35–37). The complex is also important for reassembling nucleosomes after polymerases have moved through the DNA to establish new chromatin (36).
In this report, we show that both components of the FACT complex, SSRP1 and Spt16, can individually bind ALV IN. Furthermore, we show that the C-terminal domain (CTD) of ALV IN is essential for the interaction with the FACT complex. In vitro integration activity assays revealed that the FACT complex, rather than its individual components, specifically stimulates ALV but not HIV-1 IN activity. Our findings also indicate that the FACT complex regulates ALV integration in infected cells, as the frequency of ALV proviral integration is directly correlated with the abundance of the FACT complex. The decrease in proviral integration when the FACT complex was depleted was accompanied by an increase in 2-long-terminal-repeat (2-LTR) circles, indicating that the FACT complex stimulates the integration step of the viral life cycle. Moreover, we show that the FACT complex specifically promotes ALV integration, as cells with depletion of the FACT complex had no inhibitory effect on either HIV-1 or MLV integration efficiencies.
RESULTS
The FACT complex specifically interacts with and stimulates catalytic activity of ALV IN.
To identify host cell factors that bind ALV IN, we performed affinity capture coupled with MS analysis using recombinant ALV and HIV-1 INs as baits and nuclear extracts of chicken DT40 and human Sup-T1 cells. Unique hits that were reproducible in both cell lines were identified through semiquantitative analysis of peptide spectral counts. This revealed SSRP1 and Spt16, the components of the FACT complex, to be the main binding partners of ALV IN (Fig. 1A). Using the taxonomic term Homo sapiens allowed us to identify these proteins from Sup-T1 cells. However, since the MASCOT search engine does not contain a chicken taxonomy, we used the higher-order classification “bony vertebrates” for analyzing DT40 proteins. In these samples, Spt16 (which is not annotated in chicken cells) was identified due to the high homology with its human counterpart. The confidence in the correct identification of Spt16 in DT40 cells is high due to identification of 16 unique peptides and 18% coverage of the protein (Fig. 1B). SSRP1 is functionally annotated in both species and was identified by MASCOT as of chicken or human origin depending on the cell type. No interacting peptides from either protein of the FACT complex were detected in parallel HIV-1 IN pulldown fractions. In contrast, as expected, LEDGF/p75 peptides were detected in HIV-1 but not with ALV IN pulldowns (Fig. 1A).
FIG 1.

MS-based proteomics analysis of cellular binding partners of ALV and HIV-1 INs. Two independent experiments with Sup-T1 (human) and DT40 (chicken) cells were performed. (A) Shown is the list of top unique protein hits (compiled from both cell lines) from nuclear extracts of DT40 or Sup-T1 cells. Semiquantitative values of peptide spectral counts for each identified protein are indicated. ND, no peptides from the indicated protein were detected. (B) Summary of identified peptides sequences from SSRP1 and Spt1, which are indicated in bold and highlighted in yellow. Total spectral counts for SSRP1 peptides were 85 in SupT1 cells, yielding 36% (254 out of 709) amino acid coverage. In DT40, total spectral counts for SSRP1 peptides were 32, yielding 19% (192 out of 1,006) amino acid coverage. In sharp contrast, no SSRP1 peptides were detected in parallel experiments with HIV-1 IN. Total spectral counts for Spt16 peptides in SupT1 and DT40 cells were 103 and 39, yielding 33% and 18% (341 out of 1,047) amino acid coverage, respectively. Spt16 in DT40 cells was identified based on its homology with the corresponding human gene. In sharp contrast, no Spt16 peptides were detected in parallel experiments with HIV-1 IN. Oxidation or other modifications are highlighted in green.
To validate our MS-based results, we next analyzed the affinity pulldown fractions by immunoblotting using antibodies directed against SSRP1 or Spt16 protein. The results in Fig. 2A show that ALV IN interacted with both components of the endogenous FACT complex from nuclear extracts of HEK293T cells. In contrast, in parallel reactions, HIV-1 and MLV INs failed to interact with either SSRP1 or Spt16 (Fig. 2A). Figure 2B shows the recombinant purified IN proteins used for Fig. 2A.
FIG 2.

The components of the FACT complex, SSRP1 and Spt16, bind ALV IN but not HIV-1 or MLV INs. (A) Affinity pulldown results from nuclear lysates of HEK293T cells (100 μg of total protein) with indicated 2 μM 6×His-tagged recombinant retroviral INs, followed by immunoblotting with SSRP1 or Spt16 antibodies. (B) Coomassie-stained SDS/PAGE gel of recombinant purified INs used in panel A. (C) Coomassie-stained SDS/PAGE gel of affinity pulldown results of recombinant purified GST-tagged INs (1 μM) with the FACT complex (0.6 μM). All images show representative results of triplicate experiments, with molecular masses indicated.
As our MS-based results and immunoblotting cannot distinguish between direct and indirect interactions, we next performed affinity pulldowns with recombinant purified glutathione S-transferase (GST)-tagged ALV and HIV-1 IN proteins. For these experiments, we used either purified recombinant FACT complex or LEDGF/p75. The FACT complex specifically interacted with ALV IN but not with HIV-1 IN (Fig. 2C). In parallel experiments, the expected interaction of HIV-1 IN with its known cellular cofactor, LEDGF/p75, but not the FACT complex was seen (Fig. 2C).
We next wanted to further dissect the contributions of individual proteins and/or domains responsible for interaction between the FACT complex and ALV IN. We first examined binding of C-terminally truncated fragments of ALV IN with the FACT complex. The results in Fig. 3A show that the CTD (consisting of amino acids 208 to 286) is essential for binding to the FACT complex, as the isolated N-terminal domain (NTD) and the two domain fragments containing the NTD and catalytic core domain (CCD) fail to bind the FACT complex.
FIG 3.

The FACT complex stimulates in vitro integration activity of ALV integrase. (A) Schematic of C-terminally truncated constructs of ALV IN, with domains and flexible linkers indicated, and Coomassie-stained SDS-PAGE analysis of affinity pulldown fractions using recombinant purified GST-tagged ALV IN, ALV NTD-CCD, and ALV NTD (1 μM) with the FACT complex (0.6 μM). The lower band in the ALV NTD preparation is GST alone. (B) Coomassie-stained SDS-PAGE analysis of affinity pulldown fractions using recombinant purified GST-tagged ALV IN (1 μM) with the FACT complex, SSRP1, or Spt16 (0.6 μM). All images depict representative results of triplicate experiments, with molecular masses indicated. (C) HTRF strand transfer integration activity assay of HIV-1 or ALV INs (400 nM) with the FACT complex, SSRP1, or Spt16 (1.0 μM). The results from triplicate experiments, with standard deviations, are indicated. Shown are the HTRF raw counts and the percent stimulation.
To elucidate the contributions of individual components of the FACT complex to binding and catalytic activity of ALV IN, we next utilized affinity pulldowns and homogenous time-resolved fluorescence (HTRF)-based integration assays. Figure 3B shows that both purified proteins are able to bind ALV IN individually. However, both components of the FACT complex were needed to effectively stimulate ALV integration activity (∼350%). In contrast, SSRP1 or Spt16 alone failed to enhance ALV IN activity (Fig. 3C). The level of stimulation of ALV IN activity by the FACT complex is similar to that seen for the addition of LEDGF/p75 to HIV-1 IN or Brd4 to MLV IN (14). In parallel experiments, HIV-1 IN activity was not significantly stimulated by the addition of SSRP1, Spt16, or the FACT complex. In fact, the addition of either protein suppressed HIV-1 IN activity (Fig. 3C).
ALV proviral integration frequency correlates directly with FACT complex expression levels in infected cells.
Since the FACT complex binds ALV IN and stimulates its activity in vitro, we hypothesized that the FACT complex could also play a role in regulating ALV integration in infected cells. To determine the effect of the FACT complex on ALV integration in infected cells, we employed a chicken cell line (DT40) with various levels of expression of the FACT complex. Previous research has shown that the expression and abundance of the FACT complex are regulated by a complex feedback loop in which SSRP1 mRNA plays a critical role (38). The presence of SSRP1 mRNA is essential for the stability of the Spt16 protein and the FACT complex as a whole. In the absence of SSRP1 mRNA, both protein components are depleted. Similarly, when SSRP1 mRNA is overexpressed, Spt16 protein levels also increase (38).
We used an SSRP1 conditional knockout engineered in the chicken B-cell line DT40 to investigate the functional consequences of the FACT complex on ALV integration. This cell line lacks both endogenous copies of the SSRP1 gene but has a wild-type SSRP1 gene expressed from a tetracycline (Tet)-repressible promoter (SSRP1−/− + SSRP1) (30) (Fig. 4A). Because of the demonstrated complex regulation of the FACT complex, this cell line, which allowed us to manipulate SSRP1 levels, is ideal for controlling the levels of SSRP1, Spt16, and the FACT complex as a whole.
FIG 4.

Validating the SSRP1 conditional knockout cell line. (A) Schematic of cell line. In a wild-type DT40 background, both endogenous loci of SSRP1 were targeted by homology constructs (indicated by red box) in order to knock down both copies of the gene. A wild-type copy of the SSRP1 gene was introduced into cells on a plasmid under the control of a Tet-repressible promoter. In the absence of doxycycline, SSRP1 is expressed (SSRP1−/− + SSRP1). In the presence of doxycycline, SSRP1 expression is ablated (SSRP1−/−). (B) Western blot showing that SSRP1 protein levels decrease to undetectable levels by 12 h after doxycycline addition. Relevant molecular mass markers are shown. (C) Growth curve of SSRP1 knockout and wild-type cells over 96 h after adding doxycycline. SSRP1 knockout (SSRP1−/−) does not significantly affect cell growth and proliferation until after 48 h.
In the presence of doxycycline, SSRP1−/− cells exhibited SSRP1 protein levels that declined to undetectable levels by 12 h posttreatment, resulting in a cell line with no functional FACT complex (Fig. 4B). Of note, FACT complex knockdown did not significantly affect cell growth during the initial 48 h after doxycycline addition (Fig. 4C). To analyze how various levels of SSRP1 could affect ALV integration, we compared infections in parental DT40 cells, cells expressing elevated levels of SSRP1 (SSRP1−/− + SSRP1), or cells expressing knockout (SSRP1−/−) levels of SSRP1. In the manipulated cell line (SSRP1−/− + SSRP1), SSRP1 is expressed from an exogenous promoter and is thus not expressed at wild-type levels. We observed a 5-fold increase in SSRP1 mRNA expression in SSRP1−/− + SSRP1 cells relative to that of the parental DT40 cell line.
SSRP1−/− cells express 65- and 10-fold-lower SSRP1 mRNA than do SSRP1−/− + SSRP1 and parental DT40 cells, respectively (Fig. 5A). By using these three conditions, we could assay for ALV proviral integration frequency at wild-type levels, overexpressed levels, and knockout levels of SSRP1 and hence the levels of the FACT complex.
FIG 5.

ALV proviral integration frequency correlates directly with SSRP1 mRNA expression levels. (A) SSRP1 expression in parental DT40 cells, SSRP1−/− cells, and SSRP1−/− + SSRP1 cells. The expression levels of SSRP1 mRNA were measured relative to a housekeeping gene, RPL30, by qRT-PCR. Under the knockout condition (SSRP1−/−), SSRP1 expression decreased 65- and 10-fold relative to that in SSRP1−/− + SSRP1 and parental DT40 cells, respectively (n = 6; P < 0.05). DT40 expression was approximately 6-fold lower than expression levels in SSRP1−/− + SSRP1 cells (n = 6; P < 0.05). (B) Analysis of integration frequency. Proviral integrations were measured by qPCR from gel-purified genomic DNA. In the SSRP1 knockout cell line, proviral integration frequency decreased 6.5-fold relative to that in cells expressing SSRP1 (SSRP1−/− + SSRP1) (n = 6; P < 0.05). DT40 cells exhibited approximately 2.5-fold-higher expression than knockout cells (n = 6; P < 0.05). (C) Proviral integrations were measured independently using a CR1-gag nested PCR approach. Integration frequency decreased 12-fold in the absence of SSRP1 expression (SSRP1−/−) relative to that in SSRP1−/− + SSRP1 cells (n = 5; P < 0.005). Consistent with previous data, DT40 cells had an intermediate phenotype, exhibiting approximately 3-fold more proviral integrations than SSRP1−/− cells.
Cells expressing the highest levels of SSRP1 also had the highest levels of ALV integration frequency, determined by quantitative PCR (qPCR) analysis of gel-purified DNA. We observed an approximately 2-fold increase in proviral integrations in the wild-type DT40 cells versus knockout cells (SSRP1−/−) and a 6-fold increase in integration frequency in cells overexpressing SSRP1 (SSRP1−/− + SSRP1) (Fig. 5B). Thus, the trend in integration frequency directly correlates with expression levels of SSRP1 as well as the levels of the FACT complex.
These trends were verified using a second, independent method. In this approach, proviral integration frequency was measured from genomic DNA collected from infected cells using nested PCR. A first round of PCR was performed to enrich for provirus-host genome junctions using a virus-specific primer and a consensus primer within the most abundant repeat element in the chicken genome (CR1 element). A second, quantitative PCR (qPCR) was then performed using primers within the virus. This method confirmed a significant (12-fold) decrease in proviral integrations in infected SSRP1 knockout cells relative to SSRP1−/− + SSRP1 cells (Fig. 5C). The parental DT40 cell line had an intermediate level of integration consistent with SSRP1 expression levels. These data show that ALV proviral integration frequency is directly correlated to FACT complex abundance and indicate that the FACT complex promotes ALV integration in infected cells.
The FACT complex regulates ALV at the level of integration.
Because the proteins of the FACT complex interacted with ALV IN, we hypothesized that this complex specifically regulates the ALV life cycle at the level of integration. However, the observed change in the number of detectable proviral integration events could be due to effects of the FACT complex at various levels of the retroviral life cycle preceding or during integration. For instance, if the FACT complex affects reverse transcription, nuclear import or integration, then one would expect to detect fewer integrants in the SSRP1 knockout (SSRP1−/−) cells.
To distinguish between these possibilities, we quantified various retroviral intermediates. Plus strand extension (PSE) products are an intermediate of the retroviral life cycle produced by the late steps of reverse transcription, and the abundance of PSE products can be used to assay variations in reverse transcription (39). There was no significant difference in the levels of PSE products between the knockout (SSRP1−/−) cells and cells expressing SSRP1 (SSRP1−/− + SSRP1), indicating that reverse transcription is not affected by the levels of the FACT complex (Fig. 6A).
FIG 6.

The FACT complex promotes ALV integration. To determine the step of the life cycle that the FACT complex affects, retroviral intermediates were assayed by qPCR. (A) The FACT complex does not disrupt reverse transcription. The abundance of plus strand extension (PSE) products, a product of late reverse transcription, was measured by qPCR using primers within gag. There was no significant difference in PSE product abundance observed between SSRP1 knockout cells (SSRP1−/−) and cells expressing SSRP1 (SSRP1−/− + SSRP1) (n = 3). (B) The FACT complex specifically promotes ALV integration. The abundance of 2-LTR circles was measured by qPCR in cells expressing SSRP1 (SSRP1−/− + SSRP1) and SSRP1 knockout cells. A 10-fold increase in 2-LTR circles was detected in the SSRP1 knockout cells (n = 4; P < 0.005), indicating that depletion of the FACT complex inhibits integration.
Once viral cDNA is reverse transcribed, it enters the nucleus of the host cell as part of the preintegration complex. Within the nucleus, the nonhomologous end-joining pathway circularizes unintegrated viral genomic DNA to generate 2-LTR circles. These circularized viral intermediates can be used as a proxy to measure the abundance of unintegrated nuclear viral genomes (40, 41). The unique LTR-LTR junction present in this intermediate makes it readily detectable and distinguishable by PCR (40). A 10-fold increase in 2-LTR circles was detected in the SSRP1 knockout (SSRP1−/−) cells over the level in cells expressing SSRP1 (SSRP1−/− + SSRP1) (Fig. 6B). A decrease in proviral integration accompanied by an increase in unintegrated nuclear viral products, specifically 2-LTR circles, indicates that nuclear import is not blocked and that the integration step is significantly impaired in the absence of the FACT complex.
Knockdown of the FACT complex does not inhibit lentiviral or gammaretroviral integration.
We next wanted to know if the regulation of integration by the FACT complex was specific to ALV or could also affect other retroviruses. Therefore, we infected knockout cells (SSRP1−/−) or cells overexpressing SSRP1 (SSRP1−/− + SSRP1) with vesicular stomatitis virus glycoprotein (VSV-G)-pseudotyped MLV or HIV-1. There was no significant difference in the frequency of MLV or HIV-1 proviral integration in the knockout (SSRP1−/−) cells relative to that in cells expressing SSRP1 (SSRP1−/− + SSRP1) (Fig. 7). The levels of plus strand extension products did not significantly differ, nor did the abundance of 2-LTR circles (data not shown).
FIG 7.
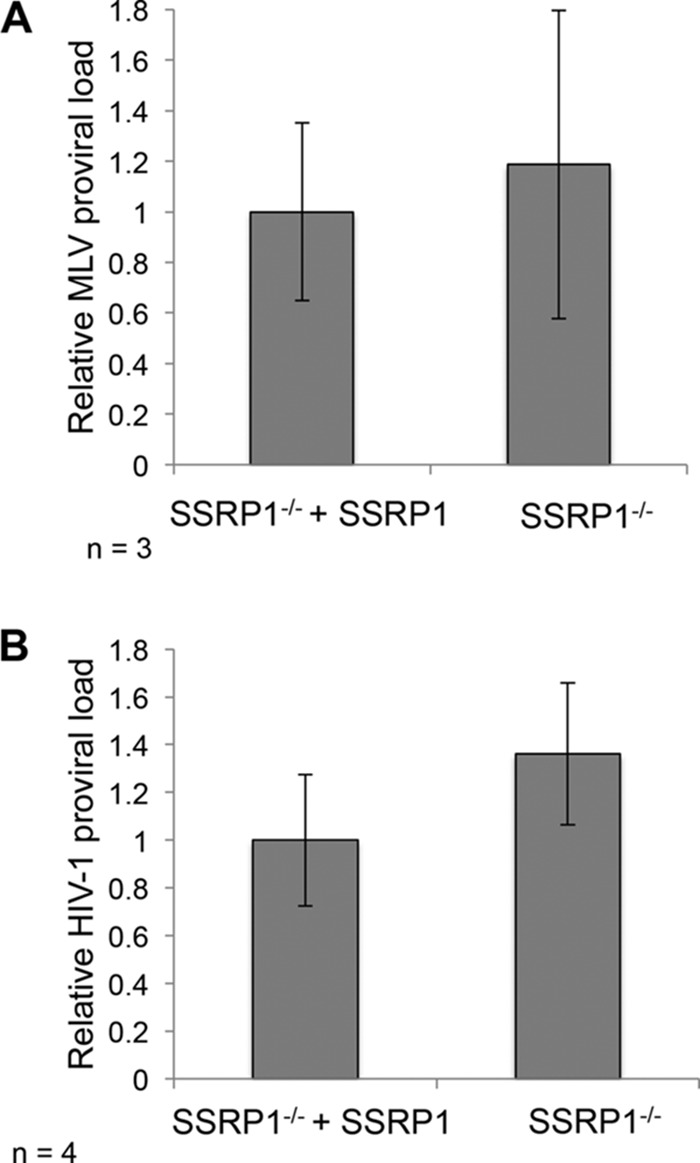
The FACT complex does not promote gammaretroviral or lentiviral integration. (A) The FACT complex does not affect MLV integration. MLV proviral load was measured in infected cells by qPCR using virus-specific primers relative to GAPDH. No significant difference in MLV integration frequency was observed in the SSRP1 knockout cells (SSRP1−/−) relative to that in cells expressing SSRP1 (SSRP1−/− + SSRP1). (B) HIV-1 proviral load was not affected by various abundances of SSRP1. HIV-1 integration frequency was measured by qPCR using virus-specific primers relative to GAPDH. No significant difference in HIV integration frequency was observed in the SSRP1 knockout cells relative to that in cells expressing SSRP1 (SSRP1−/− + SSRP1).
DISCUSSION
In this study, we have identified the FACT protein complex, which is comprised of SSRP1 and Spt16, as the principal cellular binding partner of ALV IN. While ALV IN interacts with both SSRP1 and Spt16 individually, the FACT complex as a whole is required to stimulate integration activity in vitro. Additionally, we have identified the importance of the ALV IN CTD for binding to the FACT complex. Furthermore, we show that the level of ALV integration positively correlates with the abundance of the FACT complex in infected cells. The levels of 2-LTR circles were elevated when the FACT complex was depleted, demonstrating that this complex is critical for the integration step of the ALV life cycle. The level of plus strand extension products was unaffected by the levels of the FACT complex, indicating that the FACT complex does not affect reverse transcription. Taken together, our results elucidate a key role of the FACT complex in promoting ALV integration in infected cells. This regulation is likely not species specific, as interactions of ALV IN and the FACT complex were detected in both human and chicken cells. This is similar to what is seen for other retroviruses, such as MLV IN, which interacts with BET proteins from human and murine cells due to the high degree of conservation in chromatin binding proteins (14).
Our findings indicate that the regulation of integration by the FACT complex is specific to alpharetroviral ALV. Unlike ALV IN, HIV-1 and MLV INs failed to bind the FACT complex. Moreover, MLV and HIV-1 integration was not significantly affected by altering the cellular levels of the FACT complex in infected cells.
HIV-1 and MLV exhibit strong integration site preferences for actively transcribed regions and gene regulatory regions, such as transcription start sites and enhancers, respectively (5–7). In sharp contrast, ALV does not seem to target such regions but does exhibit a slight preference for transcriptional units (23–26). Previous research has shown that the distinct integration site preferences of retroviruses can be linked to interaction of the virally encoded IN protein with various host cell factors (8–10). Mechanistically, these host cell factors act largely as a bimodal tether to recruit the preintegration complex to the chromatin, thereby targeting proviral integrations (10). For example, LEDGF/p75 engages HIV-1 IN through its C-terminal integrase binding domain and guides HIV-1 integration to active genes (11–13, 42, 43). The selection of the chromatin sites for integration is affected by the preferential binding of the N-terminal PWWP domain of LEDGF/p75 with the H3 histone tail containing trimethylated Lys36 (H3K36me3), a hallmark of actively transcribed genes (19). In a very similar manner, BET proteins, with their dual bromodomains, are able to guide MLV integration by bimodal interaction with both MLV IN and acetylated histone marks (14, 20, 44).
While our evidence only points to a role for the FACT complex in promoting ALV integration efficiency, we suggest that it may also be involved in guiding ALV integration site selection in much the same way as LEDGF/p75 and BET proteins do for HIV-1 and MLV. Conceptually, the FACT complex is an ideal candidate for targeting ALV integration. The FACT complex is a highly conserved general histone chaperone protein that is essential for DNA replication and transcription (28–30). The complex is widely distributed across the genome due to its role in replication, with a slight enrichment in transcriptionally active regions (29), which is highly consistent with the previously observed integration pattern of ALV. Both proteins of the FACT complex are involved in chromatin interactions with histones, DNA, and intact nucleosomes (45). Spt16 binds to histones H2A/H2B and H3/H4, making it a much more general chromatin binding protein than either LEDGF/p75 or BET protein (27, 46–48). SSRP1 has been shown to bind H2A/H2B histone dimers, H3/H4 histones, and DNA (49, 50). These interactions are relatively general, which could potentially explain the wide distribution of ALV integration sites across the genome. Interestingly, recruitment of the FACT complex to chromatin is enhanced by H3K36 trimethylation, a hallmark of actively transcribed genes (51). This could explain the slight preference of ALV to integrate into active genes.
The FACT complex is believed to destabilize the histone octamer providing access to the chromosomal DNA for various enzymes (35–37). In particular, the C-terminal tail of Spt16 displaces nucleosomal DNA, allowing for access to the histone octamer (45). This capability to make chromatin more accessible by loosening or releasing the chromosomal DNA could allow for more effective integration. The complex is also important for reassembling nucleosomes after polymerases have moved through the DNA to establish new chromatin (36), a function that would facilitate the establishment of chromatin at the newly integrated provirus. Further studies are necessary to elucidate the exact mechanism for regulation of ALV integration by the FACT complex, which could, in turn, facilitate the development of ALV-based vectors for their potential application in human gene therapy.
MATERIALS AND METHODS
Recombinant proteins, affinity pulldown, and MS-based proteomics.
6×His-tagged HIV-1 IN, GST-tagged HIV-1 IN, and 6×His-tagged MLV IN were purified as previously described (20, 52). Full-length GST-tagged ALV IN was made synthetically in pGEX-6P-1 by GenScript and truncated by site-directed mutagenesis to add a stop codon at codons 51 and 208 (generating GST-tagged ALV NTD IN and GST-tagged ALV NTD/CCD IN, respectively). ALV IN constructs were purified similarly to HIV-1 IN with either a HisTrap HP column followed by a heparin column or a glutathione Sepharose column (all from GE Healthcare). The FACT proteins were purified as described previously (45).
To identify cellular proteins selectively interacting with ALV IN, in parallel reactions we used recombinant ALV and HIV-1 INs as baits to capture their binding partners from cellular extracts. Affinity pulldown experiments were performed with GST- and 6×His-tagged proteins using glutathione Sepharose 4B and nickel affinity beads (GE Healthcare), respectively. Buffer conditions were 25 mM Tris (pH 8.0), 200 mM NaCl, 0.1% Nonidet P-40, 2 mM β-mercaptoethanol, and 1× complete protease mixture (Roche) or 50 mM Tris-HCl (pH 7.5), 250 mM NaCl, 2 mM β-mercaptoethanol, and 1× complete protease mixture (Roche), respectively, for nickel and GST beads. Sup-T1, DT40, or 293T cell nuclear extracts were prepared using an NE-PER nuclear and cytoplasmic kit (Thermo Scientific) and incubated with the prebound beads, and the bound proteins were separated by SDS-PAGE. Samples were either subjected to immunoblotting using SSRP1 (ab137034; Abcam) or Spt16 (sc-28734; Santa Cruz) antibody or analyzed by MS.
For MS experiments, entire lanes were excised and subjected to in-gel trypsin digestion, and the resulting peptides were analyzed with capillary-liquid chromatography and MS/MS using a Thermo Finnigan LTQ Orbitrap mass spectrometer equipped with a microspray source (Michrom Bioresources). We performed two sets of pulldowns from nuclear extracts of DT40 and Sup-T1 cells for the MS experiments. Human Sup-T1 cells were used in addition to chicken DT40 cells because the MASCOT search engine allows for peptide mass fingerprinting using Homo sapiens but not chicken taxonomy. This is because the chicken genome is currently not completely structurally and functionally annotated (based on Gene Ontology) and many of the genes have not yet been assigned standard nomenclature. Therefore, to identify the peptides from DT40 cells, we used the higher-order bony-vertebrate classification. For both sets of pulldowns, unique proteins (those with a spectral count greater than 5) that bound either HIV-1 or ALV IN were identified and were compared between cell types, and only those that were reproducible unique hits were selected for further analysis. LEDGF/p75 served as a control, as it is known to selectively bind HIV-1 IN.
Recombinant protein pulldowns were performed with purified GST-tagged HIV-1 or ALV INs as well as the ALV IN domains (1 μM) prebound to glutathione Sepharose 4B beads (GE Healthcare) in 50 mM Tris-HCl (pH 7.5), 250 mM NaCl, 2 mM β-mercaptoethanol, and 1× complete protease mixture (Roche). Purified SSRP1, Spt16, or FACT complex (0.6 μM) was added to the beads, and the bound proteins were separated by SDS-PAGE and visualized by Coomassie staining.
HTRF.
HIV-1 or ALV IN strand transfer activities were assayed using similar homogenous time-resolved fluorescence (HTRF)-based strand transfer assays developed for HIV-1 and MLV IN (14, 53). The assays contained 5′-Cy5-labeled viral donor DNA (200 nM) (ALV Don1, /5Cy5/ACGAGCACAGGAGTATGGATGACGACAACATT; ALV Don2, /5Cy5/AATGTTGTCGTCATCCATACTCCTGTGCTCGT), biotin-labeled target DNA (20 nM) (Ace1, ACAGGCCTAGCACGCGTCG/3′Bio/; Ace2, CGACGCGTGCTAGGCCTGT/3′Bio/), purified recombinant His-tagged ALV IN or HIV-1 IN (400 nM), and purified recombinant SSRP1, Spt16, or FACT complex (1 μM) added to the respective reaction mixtures containing IN, donor (HIV-1 or ALV specific), and target DNA substrates. The strand transfer products were detected after addition of europium chelate-streptavidin Lance reagent (2 nM; PerkinElmer). The HTRF signal was recorded using a Perkin-Elmer multimode Enspire plate reader using 314 nm for the excitation wavelength and 668 and 620 nm for the wavelengths of the acceptor and donor emissions, respectively.
Cell lines and viruses.
DT40 cells were cultured in Dulbecco's modified eagle medium (Thermo Fisher Scientific), 10% fetal calf serum, 5% chicken serum, 5% tryptose phosphate, and 1% antibiotic at 37°C and 5% CO2 (54). Chicken embryo fibroblasts were cultured in medium 199 (Thermo Fisher Scientific) supplemented with 2% tryptose phosphate, 1% fetal calf serum, 1% chicken serum, and 1% antibiotic at 39°C and 5% CO2. HEK293T cells were cultured in Dulbecco's modified eagle medium, 10% fetal bovine serum (FBS), and 1% antibiotic at 37°C and 5% CO2.
ALV was generated by transfecting chicken embryo fibroblasts with RCASBP(B) plasmid using electroporation (55). Viral supernatant was collected and filtered through a 0.22-μm filter. To generate MLV and HIV-1 pseudotyped with vesicular stomatitis virus glycoprotein (VSV-G), NIH 3T3 or HEK293T cells were cotransfected using Lipofectamine with pMD.G (VSV-G envelope plasmid) (56) and either MLV (pNCS) (57) or HIV plasmid (pNL4-3ΔE-GFP), respectively (58). Viral supernatant was collected after 48 h, filtered through a 0.22-μm filter, and concentrated by polyethylene glycol (PEG) precipitation (10% PEG 8000) (59).
SSRP1 knockout.
SSRP1 conditional knockout cells were obtained as a gift from Takemi Enomoto, Tohoku University (30). This cell line was generated by knocking out both endogenous SSRP1 loci in an otherwise wild-type DT40 background. A Flag-tagged wild-type copy of the chicken SSRP1 gene was introduced under the control of a Tet-repressible promoter. To induce knockout, cells were pretreated with 1 μg/ml of doxycycline for 24 h. Knockout was verified by reverse transcription-PCR (RT-PCR) or Western blotting prior to infection using a Flag antibody (8146; Cell Signaling Technology). Cells were cultured for an additional 24 h postinfection in the presence or absence of drug. Cell proliferation was monitored at 12-h intervals up to 96 h after doxycycline treatment. Cells were collected and counted using a Bio-Rad automated cell counter (TC20). Population doublings were calculated from total live cell count relative to that at time zero.
Quantification of SSRP1 expression.
RNA was purified from cells using RNA-Bee reagent (Tel-Test, Inc., Friendswood, TX). Reverse transcription was performed with Maxima H reverse transcriptase (Thermo Fisher Scientific) using an oligo(dT)18 primer. Quantitative PCRs were performed with the CFX96 real-time system (Bio-Rad) and prepared using iQ SYBR green Supermix (Bio-Rad). Expression levels of SSRP1 (primers GGCTCACCAAGAACATGTCA and AGTCCTGAGCTGGCCTTGTA) were normalized to RPL30 (60).
Genomic DNA purification.
Cells were harvested 24 h postinfection. DNA was isolated using a DNeasy blood and tissue kit (Qiagen). Genomic DNA was further purified for integration analysis by gel purification. Total DNA was loaded on a 0.5% low-melting-point agarose gel and run at 100 V for 3 h. The high-molecular-weight band was then purified using a QiaEx II gel purification kit (Qiagen).
Quantitative PCR analysis of proviral integrations and retroviral intermediates.
All quantitative PCRs were performed with the CFX96 real-time system (Bio-Rad) and prepared using iQ SYBR green Supermix (Bio-Rad). Reactions were normalized to GAPDH (61). ALV proviral integrations were detected either from gel-purified genomic DNA using virus-specific primers (ACATCCTTCTGACCGACCCA and CAATTCTGTCTCATTTGGGAGCAA) or from unpurified DNA using a CR1-gag nested PCR approach. In this approach, a PCR was performed using a forward primer in the CR1 repeat element (N[8]ATTCTRTGATTCTRT) and a reverse primer in the gag gene of ALV (TAGGTTTTACACGCGGACGA). The product of this reaction was used as a template for qPCRs with virus specific primers located in the LTR (ACCGTTGATTCCCTGACGAC and TGGCCGACCACTATTCCCTA). 2-LTR circles were detected using primers spanning the LTR-LTR junction (GACTACGAGCACCTGCATGA and TCTCCTTGTAAGGCATGTTGCT). Plus strand extension products were quantified with gag forward and reverse primers (CTTGGGGAGTCCAACTCCAG and AGCCGGGCAACTTCTCTAAA). MLV and HIV-1 integrations were quantified with virus-specific primers from gel-purified genomic DNA (MLV gag, TCAGGTCGGGCCACAAAAAC and ACTAGCTCTGTATCTGGCGGA; HIV-1 eGFP, ATCATGGCCGACAAGCAGAA and TCTCGTTGGGGTCTTTGCTC). Relative quantification was performed using the threshold cycle (2−ΔΔCT) method.
ACKNOWLEDGMENTS
We thank Takemi Enomoto, Tohoku University, Miyagi, Japan, for the SSRP1 conditional knockout cell line.
This work was supported by NIH grant RO1CA124596 to K.L.B., R01 AI062520 to M.K., and NIH training grant T32GM007321, which partially supported S.W.
REFERENCES
- 1.Barquinero J, Eixarch H, Pérez-Melgosa M. 2004. Retroviral vectors: new applications for an old tool. Gene Ther 11(Suppl 1):S3–S9. doi: 10.1038/sj.gt.3302363. [DOI] [PubMed] [Google Scholar]
- 2.Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, Brugman MH, Pike-Overzet K, Chatters SJ, de Ridder D, Gilmour KC, Adams S, Thornhill SI, Parsley KL, Staal FJT, Gale RE, Linch DC, Bayford J, Brown L, Quaye M, Kinnon C, Ancliff P, Webb DK, Schmidt M, von Kalle C, Gaspar HB, Thrasher AJ. 2008. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest 118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biasco L, Baricordi C, Aiuti A. 2012. Retroviral integrations in gene therapy trials. Mol Ther 20:709–716. doi: 10.1038/mt.2011.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCormack MP, Rabbitts TH. 2004. Activation of the T-cell oncogene LMO2 after gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 350:913–922. doi: 10.1056/NEJMra032207. [DOI] [PubMed] [Google Scholar]
- 5.Wu X, Li Y, Crise B, Burgess SM. 2003. Transcription start regions in the human genome are favored targets for MLV integration. Science 300:1749–1751. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- 6.Schröder ARW, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. 2002. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110:521–529. doi: 10.1016/S0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 7.Lewinski MK, Yamashita M, Emerman M, Ciuffi A, Marshall H, Crawford G, Collins F, Shinn P, Leipzig J, Hannenhalli S, Berry CC, Ecker JR, Bushman FD. 2006. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathog 2:e60. doi: 10.1371/journal.ppat.0020060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craigie R, Bushman FD. 2014. Host factors in retroviral integration and the selection of integration target sites. Microbiol Spectr 2(6):MDNA3-0026-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Debyser Z, Christ F, De Rijck J, Gijsbers R. 2015. Host factors for retroviral integration site selection. Trends Biochem Sci 40:108–116. doi: 10.1016/j.tibs.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Kvaratskhelia M, Sharma A, Larue RC, Serrao E, Engelman A. 2014. Molecular mechanisms of retroviral integration site selection. Nucleic Acids Res 42:10209–10225. doi: 10.1093/nar/gku769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maertens G, Cherepanov P, Pluymers W, Busschots K, De Clercq E, Debyser Z, Engelborghs Y. 2003. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J Biol Chem 278:33528–33539. doi: 10.1074/jbc.M303594200. [DOI] [PubMed] [Google Scholar]
- 12.Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, Bushman F. 2005. A role for LEDGF/p75 in targeting HIV DNA integration. Nat Med 11:1287–1289. doi: 10.1038/nm1329. [DOI] [PubMed] [Google Scholar]
- 13.Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. 2006. An essential role for LEDGF/p75 in HIV integration. Science 314:461–464. doi: 10.1126/science.1132319. [DOI] [PubMed] [Google Scholar]
- 14.Sharma A, Larue RC, Plumb MR, Malani N, Male F, Slaughter A, Kessl JJ, Shkriabai N, Coward E, Aiyer SS, Green PL, Wu L, Roth MJ, Bushman FD, Kvaratskhelia M. 2013. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc Natl Acad Sci U S A 110:12036–12041. doi: 10.1073/pnas.1307157110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Rijck J, de Kogel C, Demeulemeester J, Vets S, El Ashkar S, Malani N, Bushman FD, Landuyt B, Husson SJ, Busschots K, Gijsbers R, Debyser Z. 2013. The BET family of proteins targets moloney murine leukemia virus integration near transcription start sites. Cell Rep 5:886–894. doi: 10.1016/j.celrep.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El Ashkar S, De Rijck J, Demeulemeester J, Vets S, Madlala P, Cermakova K, Debyser Z, Gijsbers R. 2014. BET-independent MLV-based vectors target away from promoters and regulatory elements. Mol Ther Nucleic Acids 3:e179. doi: 10.1038/mtna.2014.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aiyer S, Swapna GVT, Malani N, Aramini JM, Schneider WM, Plumb MR, Ghanem M, Larue RC, Sharma A, Studamire B, Kvaratskhelia M, Bushman FD, Montelione GT, Roth MJ. 2014. Altering murine leukemia virus integration through disruption of the integrase and BET protein family interaction. Nucleic Acids Res 42:5917–5928. doi: 10.1093/nar/gku175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta SS, Maetzig T, Maertens GN, Sharif A, Rothe M, Weidner-Glunde M, Galla M, Schambach A, Cherepanov P, Schulz TF. 2013. Bromo- and extraterminal domain chromatin regulators serve as cofactors for murine leukemia virus integration. J Virol 87:12721–12736. doi: 10.1128/JVI.01942-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eidahl JO, Crowe BL, North JA, McKee CJ, Shkriabai N, Feng L, Plumb M, Graham RL, Gorelick RJ, Hess S, Poirier MG, Foster MP, Kvaratskhelia M. 2013. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res 41:3924–3936. doi: 10.1093/nar/gkt074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larue RC, Plumb MR, Crowe BL, Shkriabai N, Sharma A, DiFiore J, Malani N, Aiyer SS, Roth MJ, Bushman FD, Foster MP, Kvaratskhelia M. 2014. Bimodal high-affinity association of Brd4 with murine leukemia virus integrase and mononucleosomes. Nucleic Acids Res 42:4868–4881. doi: 10.1093/nar/gku135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shun M-C, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, Cherepanov P, Engelman A. 2007. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev 21:1767–1778. doi: 10.1101/gad.1565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maertens GN. 2016. B′-protein phosphatase 2A is a functional binding partner of delta-retroviral integrase. Nucleic Acids Res 44:364–376. doi: 10.1093/nar/gkv1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narezkina A, Taganov KD, Litwin S, Stoyanova R, Hayashi J, Seeger C, Skalka AM, Katz RA. 2004. Genome-wide analyses of avian sarcoma virus integration sites. J Virol 78:11656–11663. doi: 10.1128/JVI.78.21.11656-11663.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barr SD, Leipzig J, Shinn P, Ecker JR, Bushman FD. 2005. Integration targeting by avian sarcoma-leukosis virus and human immunodeficiency virus in the chicken genome. J Virol 79:12035–12044. doi: 10.1128/JVI.79.18.12035-12044.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Withers-Ward ES, Kitamura Y, Barnes JP, Coffin JM. 1994. Distribution of targets for avian retrovirus DNA integration in vivo. Genes Dev 8:1473–1487. doi: 10.1101/gad.8.12.1473. [DOI] [PubMed] [Google Scholar]
- 26.Mitchell RS, Beitzel BF, Schroder ARW, Shinn P, Chen H, Berry CC, Ecker JR, Bushman FD. 2004. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol 2:e234. doi: 10.1371/journal.pbio.0020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orphanides G, Wu WH, Lane WS, Hampsey M, Reinberg D. 1999. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400:284–288. doi: 10.1038/22350. [DOI] [PubMed] [Google Scholar]
- 28.Orphanides G, LeRoy G, Chang CH, Luse DS, Reinberg D. 1998. FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 92:105–116. doi: 10.1016/S0092-8674(00)80903-4. [DOI] [PubMed] [Google Scholar]
- 29.Belotserkovskaya R, Reinberg D. 2004. Facts about FACT and transcript elongation through chromatin. Curr Opin Genet Dev 14:139–146. doi: 10.1016/j.gde.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Abe T, Sugimura K, Hosono Y, Takami Y, Akita M, Yoshimura A, Tada S, Nakayama T, Murofushi H, Okumura K, Takeda S, Horikoshi M, Seki M, Enomoto T. 2011. The histone chaperone facilitates chromatin transcription (FACT) protein maintains normal replication fork rates. J Biol Chem 286:30504–30512. doi: 10.1074/jbc.M111.264721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumari A, Mazina OM, Shinde U, Mazin AV, Lu H. 2009. A role for SSRP1 in recombination-mediated DNA damage response. J Cell Biochem 108:508–518. doi: 10.1002/jcb.22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okada M, Okawa K, Isobe T, Fukagawa T. 2009. CENP-H-containing complex facilitates centromere deposition of CENP-A in cooperation with FACT and CHD1. Mol Biol Cell 20:3986–3995. doi: 10.1091/mbc.E09-01-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oliveira DV, Kato A, Nakamura K, Ikura T, Okada M, Kobayashi J, Yanagihara H, Saito Y, Tauchi H, Komatsu K. 2014. Histone chaperone FACT regulates homologous recombination by chromatin remodeling through interaction with RNF20. J Cell Sci 127:763–772. doi: 10.1242/jcs.135855. [DOI] [PubMed] [Google Scholar]
- 34.Ikeda Y, Kinoshita Y, Susaki D, Ikeda Y, Iwano M, Takayama S, Higashiyama T, Kakutani T, Kinoshita T. 2011. HMG domain containing SSRP1 is required for DNA demethylation and genomic imprinting in Arabidopsis. Dev Cell 21:589–596. doi: 10.1016/j.devcel.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 35.Reinberg D, Sims RJ. 2006. de FACTo nucleosome dynamics. J Biol Chem 281:23297–23301. doi: 10.1074/jbc.R600007200. [DOI] [PubMed] [Google Scholar]
- 36.Formosa T. 2012. The role of FACT in making and breaking nucleosomes. Biochim Biophys Acta 1819:247–255. doi: 10.1016/j.bbagrm.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winkler DD, Luger K. 2011. The histone chaperone FACT: structural insights and mechanisms for nucleosome reorganization. J Biol Chem 286:18369–18374. doi: 10.1074/jbc.R110.180778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Safina A, Garcia H, Commane M, Guryanova O, Degan S, Kolesnikova K, Gurova KV. 2013. Complex mutual regulation of facilitates chromatin transcription (FACT) subunits on both mRNA and protein levels in human cells. Cell Cycle 12:2423–2434. doi: 10.4161/cc.25452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karageorgos L, Li P, Burrell CJ. 1995. Stepwise analysis of reverse transcription in a cell-to-cell human immunodeficiency virus infection model: kinetics and implications. J Gen Virol 76:1675–1686. doi: 10.1099/0022-1317-76-7-1675. [DOI] [PubMed] [Google Scholar]
- 40.Butler SL, Hansen MS, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat Med 7:631–634. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- 41.Mandal D, Prasad VR. 2009. Analysis of 2-LTR circle junctions of viral DNA in infected cells. Methods Mol Biol 485:73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cherepanov P, Maertens G, Proost P, Devreese B, Van Beeumen J, Engelborghs Y, De Clercq E, Debyser Z. 2003. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J Biol Chem 278:372–381. [DOI] [PubMed] [Google Scholar]
- 43.Cherepanov P, Ambrosio ALB, Rahman S, Ellenberger T, Engelman A. 2005. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc Natl Acad Sci U S A 102:17308–17313. doi: 10.1073/pnas.0506924102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crowe BL, Larue RC, Yuan C, Hess S, Kvaratskhelia M, Foster MP. 2016. Structure of the Brd4 ET domain bound to a C-terminal motif from γ-retroviral integrases reveals a conserved mechanism of interaction. Proc Natl Acad Sci U S A 113:2086–2091. doi: 10.1073/pnas.1516813113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winkler DD, Muthurajan UM, Hieb AR, Luger K. 2011. Histone chaperone FACT coordinates nucleosome interaction through multiple synergistic binding events. J Biol Chem 286:41883–41892. doi: 10.1074/jbc.M111.301465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kemble DJ, McCullough LL, Whitby FG, Formosa T, Hill CP. 2015. FACT disrupts nucleosome structure by binding H2A-H2B with conserved peptide motifs. Mol Cell 60:294–306. doi: 10.1016/j.molcel.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kemble DJ, Whitby FG, Robinson H, McCullough LL, Formosa T, Hill CP. 2013. Structure of the Spt16 middle domain reveals functional features of the histone chaperone FACT. J Biol Chem 288:10188–10194. doi: 10.1074/jbc.C113.451369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stuwe T, Hothorn M, Lejeune E, Rybin V, Bortfeld M, Scheffzek K, Ladurner AG. 2008. The FACT Spt16 “peptidase” domain is a histone H3-H4 binding module. Proc Natl Acad Sci U S A 105:8884–8889. doi: 10.1073/pnas.0712293105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoffmann C, Neumann H. 2015. In vivo mapping of FACT-histone interactions identifies a role of Pob3 C-terminus in H2A-H2B binding. ACS Chem Biol 10:2753–2763. doi: 10.1021/acschembio.5b00493. [DOI] [PubMed] [Google Scholar]
- 50.Zhang W, Zeng F, Liu Y, Shao C, Li S, Lv H, Shi Y, Niu L, Teng M, Li X. 2015. Crystal structure of human SSRP1 middle domain reveals a role in DNA binding. Sci Rep 5:18688. doi: 10.1038/srep18688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carvalho S, Raposo AC, Martins FB, Grosso AR, Sridhara SC, Rino J, Carmo-Fonseca M, de Almeida SF. 2013. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res 41:2881–2893. doi: 10.1093/nar/gks1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McKee CJ, Kessl JJ, Shkriabai N, Dar MJ, Engelman A, Kvaratskhelia M. 2008. Dynamic modulation of HIV-1 integrase structure and function by cellular lens epithelium-derived growth factor (LEDGF) protein. J Biol Chem 283:31802–31812. doi: 10.1074/jbc.M805843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kessl JJ, Jena N, Koh Y, Taskent-Sezgin H, Slaughter A, Feng L, De Silva S, Wu L, Le Grice SFJ, Engelman A, Fuchs JR, Kvaratskhelia M. 2012. Multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J Biol Chem 287:16801–16811. doi: 10.1074/jbc.M112.354373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winding P, Berchtold MW. 2001. The chicken B cell line DT40: a novel tool for gene disruption experiments. J Immunol Methods 249:1–16. doi: 10.1016/S0022-1759(00)00333-1. [DOI] [PubMed] [Google Scholar]
- 55.Hughes SH. 2004. The RCAS vector system. Folia Biol (Praha) 50:107–119. [DOI] [PubMed] [Google Scholar]
- 56.Burns JC, Friedmann T, Driever W, Burrascano M, Yee JK. 1993. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci U S A 90:8033–8037. doi: 10.1073/pnas.90.17.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao G, Goff SP. 1998. Replication defect of Moloney murine leukemia virus with a mutant reverse transcriptase that can incorporate ribonucleotides and deoxyribonucleotides. J Virol 72:5905–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang H, Zhou Y, Alcock C, Kiefer T, Monie D, Siliciano J, Li Q, Pham P, Cofrancesco J, Persaud D, Siliciano RF. 2004. Novel single-cell-level phenotypic assay for residual drug susceptibility and reduced replication capacity of drug-resistant human immunodeficiency virus type 1. J Virol 78:1718–1729. doi: 10.1128/JVI.78.4.1718-1729.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cepko C. 2001. Large-scale preparation and concentration of retrovirus stocks. Curr Protoc Mol Biol Chapter 9:Unit9.12. [DOI] [PubMed] [Google Scholar]
- 60.Yue H, Lei X, Yang F, Li M, Tang C. 2010. Reference gene selection for normalization of PCR analysis in chicken embryo fibroblast infected with H5N1 AIV. Virol Sin 25:425–431. doi: 10.1007/s12250-010-3114-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heidari M, Zhang HM, Sharif S. 2008. Marek's disease virus induces Th-2 activity during cytolytic infection. Viral Immunol 21:203–214. doi: 10.1089/vim.2007.0078. [DOI] [PubMed] [Google Scholar]