

ABSTRACT
Hepatitis C virus (HCV) is an enveloped RNA virus belonging to the Flaviviridae family. It infects mainly human hepatocytes and causes chronic liver diseases, including cirrhosis and cancer. HCV encodes two envelope proteins, E1 and E2, that form a heterodimer and mediate virus entry. While E2 has been extensively studied, less has been done so for E1, and its role in the HCV life cycle still needs to be elucidated. Here we developed a new cell culture model for HCV infection based on the trans-complementation of E1. Virus production of the HCV genome lacking the E1-encoding sequence can be efficiently rescued by the ectopic expression of E1 in trans. The resulting virus, designated HCVΔE1, can propagate in packaging cells expressing E1 but results in only single-cycle infection in naive cells. By using the HCVΔE1 system, we explored the role of a putative fusion peptide (FP) of E1 in HCV infection. Interestingly, we found that the FP not only contributes to HCV entry, as previously reported, but also may be involved in virus morphogenesis. Finally, we identified amino acid residues in FP that are critical for biological functions of E1. In summary, our work not only provides a new cell culture model for studying HCV but also provides some insights into understanding the role of E1 in the HCV life cycle.
IMPORTANCE Hepatitis C virus (HCV), an enveloped RNA virus, encodes two envelope proteins, E1 and E2, that form a heterodimeric complex to mediate virus entry. Compared to E2, the biological functions of E1 in the virus life cycle are not adequately investigated. Here we developed a new cell culture model for single-cycle HCV infection based on the trans-complementation of E1. The HCV genome lacking the E1-encoding sequence can be efficiently rescued for virus production by the ectopic expression of E1 in trans. This new model renders a unique system to dissect functional domains and motifs in E1. Using this system, we found that a putative fusion peptide in E1 is a multifunctional structural element contributing to both HCV entry and morphogenesis. Our work has provided a new cell culture model to study HCV and provides insights into understanding the biological roles of E1 in the HCV life cycle.
KEYWORDS: E1, envelope proteins, fusion peptide, hepatitis C virus, trans-complementation, virus entry, virus morphogenesis
INTRODUCTION
Hepatitis C virus (HCV) is a major human pathogen that infects about 170 million people worldwide and can lead to chronic hepatitis and more severe liver diseases such as cirrhosis and hepatocellular carcinoma (1–3). For a long time, standard therapy for chronic HCV infection consisted of the administration of pegylated interferon alpha and ribavirin, which can achieve cure in only some patients (4, 5). The recent introduction of direct-acting antivirals (DAAs) that target the viral NS3, NS5B, and NS5A proteins dramatically improved cure rates for hepatitis C patients (6, 7). However, besides drug resistance, the high costs and limited access of DAAs in developing countries with high HCV prevalences impose large challenges to the eradication of HCV (6, 8). An effective preventive vaccine is necessary for the global control of HCV infection (9).
HCV is an enveloped virus belonging to the family Flaviviridae. Its 9.6-kb, single-stranded, positive-sense RNA genome encodes a single polyprotein that is co- or posttranslationally cleaved into structural proteins (core, E1, and E2) and nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (10, 11). The nonstructural proteins NS3 to NS5B are required for HCV genomic RNA replication, while the structural proteins core to E2 encapsidate and package the nascent viral RNA genome to form an infectious viral particle, a process that is also highly dependent upon the concerted action of nearly all nonstructural proteins (10).
The two envelope proteins E1 and E2 are cleaved from the HCV polyprotein precursor by cellular signal peptidases in the endoplasmic reticulum (12, 13). They are highly N-glycosylated and belong to the class of type I transmembrane proteins (12). Through their C-terminal transmembrane domains (TMDs), E1 and E2 form a stable noncovalent heterodimeric complex that mediates virus entry and determines viral pathogenicity and the host immune response (13–15). The HCV envelope proteins may also mediate the association of viral particles with host low-density lipoprotein (LDL) or very-low-density lipoprotein (VLDL), which plays important roles in virus entry, egress, and evasion of the host immune response (12, 16–18). E2 contains the major viral structural elements that are responsible for binding to host receptors, including CD81 and scavenger receptor class B type I (SR-BI) (13–16). After the virus enters the cells through clathrin-dependent endocytosis, the release of the viral genome into the cytosol requires a process of pH-dependent viral and endosomal membrane fusion (10, 12, 19). This membrane fusion process is usually mediated by a virus-encoded fusion protein or peptide that normally resides in viral envelope proteins and often undergoes a conformational change in order to trigger membrane fusion (20). Despite extensive research, the viral structural elements that mediate membrane fusion during HCV entry have not been fully determined. The recently solved crystal structure of the core domain of E2 (21, 22) and the N terminus of E1 (nE1) (residues 192 to 271) (23) failed to identify any class II viral fusion protein-like structures shared by other flaviviruses (24–26). It has been proposed that a conserved hydrophobic sequence, CSALYVGDLC (residues 272 to 281), in E1, which was not included in the structural analysis of nE1, may represent a truncated class II fusion peptide (27). The role of this sequence in virus entry and membrane fusion was supported by data from several studies (27–32). The potential role of this putative fusion peptide in E1 in other aspects of the HCV life cycle has not been explored.
The trans-complementation-based reverse-genetics approach has been used to study the role of individual HCV proteins in the viral life cycle independent of their cis-acting effects. Previously, we developed a trans-complementation system that allows the production of single-cycle infectious HCV particles lacking the E1 and E2 genes in the viral RNA genome (designated HCVΔE) (33). The production of HCVΔE was efficiently rescued by the expression of the E1 and E2 envelope proteins in trans. In this study, we developed a new cell culture model for HCV infection based on the trans-complementation of E1. The resulting virus, designated HCVΔE1, can propagate in packaging cells expressing E1 but results in only single-cycle infection in naive cells. By using the HCVΔE1 system, we explored the role of a putative fusion peptide (FP) of E1 in HCV infection. Interestingly, we found that the FP not only contributes to HCV entry, as previously reported, but also may be involved in virus morphogenesis. Finally, we identified amino acid residues in the FP that are critical for biological functions of E1. In summary, our work has provided not only a new cell culture model for studying HCV but also some insights into understanding the role of E1 in the virus life cycle.
RESULTS
Virus production of the HCV genome lacking the E1 gene can be rescued by the ectopic expression of E1 proteins in trans.
Previously, we developed a trans-complementation system allowing the production of single-cycle infectious HCV particles that lack the E1 and E2 genes (designated HCVΔE) (33). The production of HCVΔE could be efficiently rescued by the expression of the E1 and E2 envelope proteins in trans (33). To test whether the function of E1 alone could be complemented in trans, we constructed JFH1-delE1 (Fig. 1A), in which the E1-encoding sequence (amino acid positions 217 to 334) of a genotype 2a cell culture-infectious HCV (HCVcc) strain (JFH1 strain) was deleted. The in vitro-transcribed JFH1-delE1 RNA was transfected into Huh7.5.1 cells that stably express E1 (designated Huh7.5.1E1 cells) by electroporation. Infectious viruses were detected in the culture supernatants at day 3 posttransfection (∼100 focus-forming units [FFU]/ml) (Fig. 1C). This trans-complemented virus was designated HCVΔE1. Reverse transcription-PCR (RT-PCR) analysis using a primer set flanking the E1-encoding region confirmed the deletion of E1 in the HCVΔE1 genome isolated from the transfected culture supernatants (Fig. 1B). Next, we inoculated Huh7.5.1E1 cells with HCVΔE1 recovered from the above-described transfection experiment at a multiplicity of infection (MOI) of 0.1. To monitor the kinetics of virus expansion, culture supernatants at the indicated time points after infection were collected for the quantification of infectivity titers, and the infected cells were analyzed for HCV NS3 immunofluorescence. As shown in Fig. 1D, HCVΔE1 production reached a peak titer of 1,000 FFU/ml at day 4 postinfection in Huh7.5.1E1 cells, whereas no infectious progeny virions were produced in naive Huh7.5.1 cells. By day 6 postinfection, HCVΔE1 infection expanded to almost the entire Huh7.5.1E1 cell populations (Fig. 1E). Taken together, our data showed that the HCV genome with a deletion in the E1 gene could be packaged for virus production by the ectopic expression of E1 in trans.
FIG 1.
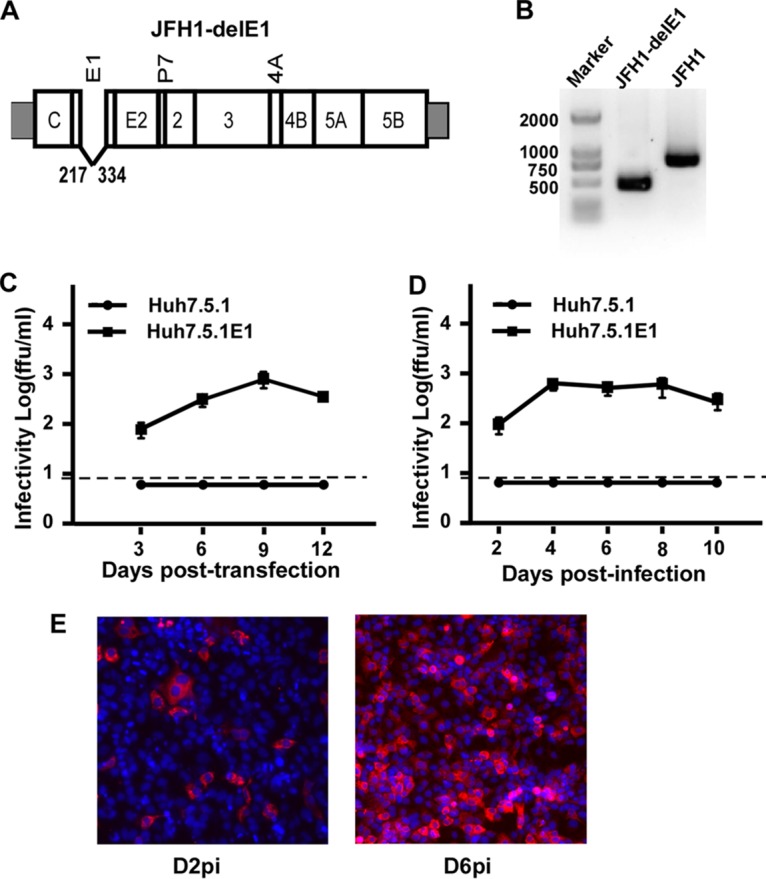
Virus production of the HCV genome with a deletion in E1 can be rescued by the ectopic expression of E1 proteins in trans. (A) Schematic representation of the JFH1-delE1 RNA genome carrying an in-frame deletion of 117 amino acids within the E1-encoding region. The amino acid positions of deletion sites within the envelope region are indicated as positions 217 to 334. (B) RT-PCR analysis of HCV genomic RNA of HCVΔE1 (lane 1) and HCVcc using a primer set flanking the E1 gene. (C) Kinetics of infectivity titers in the supernatants of naive Huh7.5.1 and Huh7.5.1E1 cells that were transfected with in vitro-transcribed JFH1-delE1 RNA. The dashed line indicates the detection limit of the titration assay. (D) Kinetics of infectivity titers in the supernatants of naive Huh7.5.1 and Huh7.5.1E1 cells that were infected with HCVΔE1 at an MOI of 0.1. The dashed line indicates the detection limit of the titration assay. Means and standard deviations from three independent tests are shown. (E) Immunofluorescence analysis of NS3 proteins (red) in Huh7.5.1E1 cells infected with HCVΔE1 at day 2 postinfection (D2pi) and day 6 postinfection. Nuclei (blue) were stained with Hoechst dye.
Adaptive mutations enhance HCVΔE1 production.
It was shown previously that cell culture-adaptive mutations, especially those in nonstructural proteins, enhance HCVcc production (34, 35). To improve the efficiency of HCVΔE1 production, two different sets of adaptive mutations were engineered into JFH1-delE1: one set contains an M1051T mutation in NS3 and a C2219R mutation in NS5A (designated JFH1-ΔE1-2M), which we previously demonstrated enhances the virus production of HCVcc (35) and HCVΔE (33), and the other set contains a deletion of 5 amino acids (EEDDT) at the C terminus of NS5A (positions 2435 to 2439) (designated JFH1-ΔE1-NS5AM), which we recently found also enhances HCVcc virus production (our unpublished data). The parental and the two mutant JFH1-ΔE1 RNAs were electroporated into Huh7.5.1E 1 cells, and the kinetics of virus expansion was determined. The NS3 immunofluorescence at day 2 posttransfection indicated similar efficiencies of transfection and HCV genome replication (Fig. 2A). However, as shown in Fig. 2B, both sets of adaptive mutations significantly enhanced the infectivity titers of HCVΔE1. Of note, the mutant with a deletion of 5 amino acids in NS5A (NS5AM) reached a peak titer of nearly 105 FFU/ml at day 9 posttransfection. Therefore, this adaptive HCVΔE1 mutant was used in subsequent studies.
FIG 2.
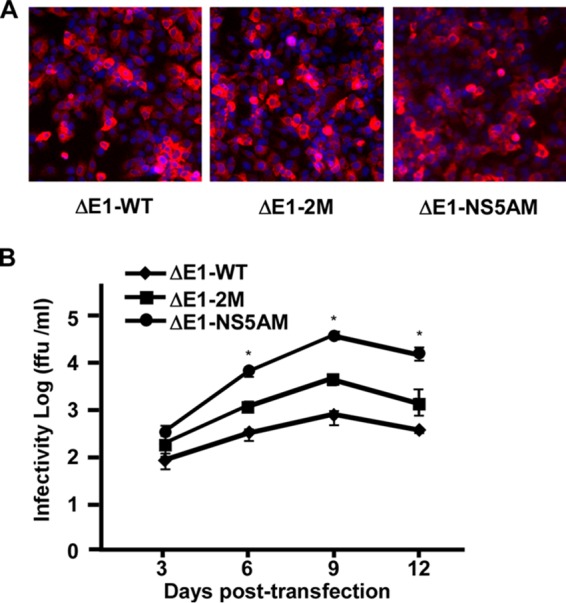
Adaptive mutations enhance HCVΔE1 production. Huh7.5.1E1 cells were transfected with wild-type JFH1-delE1 (WT), a mutant containing mutations M1051T in NS3 and C2219R in NS5A (2M), and a mutant containing a deletion from amino acid residues 2435 to 2439 (EEDDT) in the C terminus of NS5A (NS5AM). (A) Immunofluorescence analysis of NS3 proteins (red) in transfected Huh7.5.1E1 cells at day 2 posttransfection. Nuclei (blue) were stained with Hoechst dye. (B) Kinetics of infectivity titers in the supernatants of transfected Huh7.5.1E1 cells. Means and standard deviations from three independent tests are shown. *, P < 0.05.
FLAG-tagged E1 efficiently rescues HCVΔE1 production.
No good antibody against E1 of the JFH1 strain was available, which made it difficult to study the biological functions of E1. To overcome this problem, we attempted to add a tag to E1 and evaluated the effect of this modification on the biological function of E1. We constructed a Huh7.5.1 cell line that stably expresses E1 with a three-tandem-FLAG tag at its C terminus (designated Huh7.5.1E1-FLAG). The expression of the FLAG-tagged E1 protein was confirmed by Western blotting and immunofluorescence analyses (Fig. 3A and B). Next, we tested whether HCVΔE1 could be propagated in Huh7.5.1E1-FLAG cells. Naive Huh7.5.1, Huh7.5.1E1, and Huh7.5.1E1-FLAG cells were infected with HCVΔE1 at an MOI of 0.01, and virus titers in the culture supernatants were monitored at the indicated time points after infection. As shown in Fig. 3C, HCVΔE1 resulted in productive infection in Huh7.5.1E1-FLAG cells, with kinetics very similar to those in Huh7.5.1E1 cells. By day 6 postinfection, HCVΔE1 infection had expanded to all the cells in both the Huh7.5.1E1 and Huh7.5.1E1-FLAG cultures (Fig. 3D). Importantly, E1 was detected by using an anti-FLAG antibody in the culture supernatants (Fig. 3E), suggesting that the FLAG-tagged E1 proteins were successfully incorporated into infectious HCV virions. Furthermore, we showed that HCVΔE1 could be passaged in Huh7.5.1E1-FLAG cells multiple times without losing infectivity (data not shown). Taken together, our data showed that the ectopically expressed E1 protein can be tagged without impairing its ability to complement the production of HCVΔE1.
FIG 3.

FLAG-tagged E1 efficiently rescues HCVΔE1 production. (A) Naive Huh7.5.1, Huh7.5.1E1, and Huh7.5.1E1-FLAG cell lysates were analyzed by Western blotting using anti-FLAG and anti-actin antibodies. (B) Immunofluorescence analysis of FLAG-tagged E1 proteins (green) in Huh7.5.1, Huh7.5.1E1, and Huh7.5.1E1-FLAG cells using the anti-FLAG antibody. Nuclei (blue) were stained with Hoechst dye. (C) Kinetics of infectivity titers in the supernatants of Huh.7.5.1, Huh7.5.1E1, and Huh7.5.1E1-FLAG cells that were infected with HCVΔE1-NS5AM at an MOI of 0.01. The dashed line indicates the detection limit of the titration assay. Means and standard deviations from three independent tests are shown. NS, nonsignificant (P > 0.05). (D) Immunofluorescence analysis of HCV NS3 proteins (red) in infected Huh7.5.1, Huh7.5.1E1, and Huh7.5.1E1-FLAG cells at day 6 postinfection. Nuclei (blue) were stained with Hoechst dye. (E) The cell lysates and 100-fold-concentrated culture supernatants of Huh7.5.1E1 and Huh7.5.1E1-FLAG cells infected with HCVΔE1 were analyzed by Western blotting using anti-FLAG and anti-E2 antibodies. The 100-fold-concentrated culture supernatants of mock-infected Huh7.5.1E1 and Huh7.5.1E1-FLAG cells were included as controls.
Buoyant densities of HCVΔE1 and HCVΔE1-FLAG.
Next, we analyzed the buoyant densities of HCVΔE1 and HCVΔE1-FLAG by sucrose density gradient analysis. After ultracentrifugation in a 20 to 60% sucrose gradient, the infectivity titers and HCV RNA contents of each density fraction were determined. As shown in Fig. 4, >40% of the HCV RNA contents in HCVcc, HCVΔE1, and HCVΔE 1-FLAG were found in fraction 6, with a mean density of 1.17 g/ml, while the infectivities of all three viruses were distributed over a broad range of density fractions (fractions 2 to 5), with a mean density of 1.08 g/ml. Importantly, HCVΔE1 and HCVΔE1-FLAG did not show any significant difference in their buoyant density profiles, suggesting that the FLAG tag inserted into the C termini of E1 proteins had no influence on the physical properties of HCVΔE1 particles.
FIG 4.
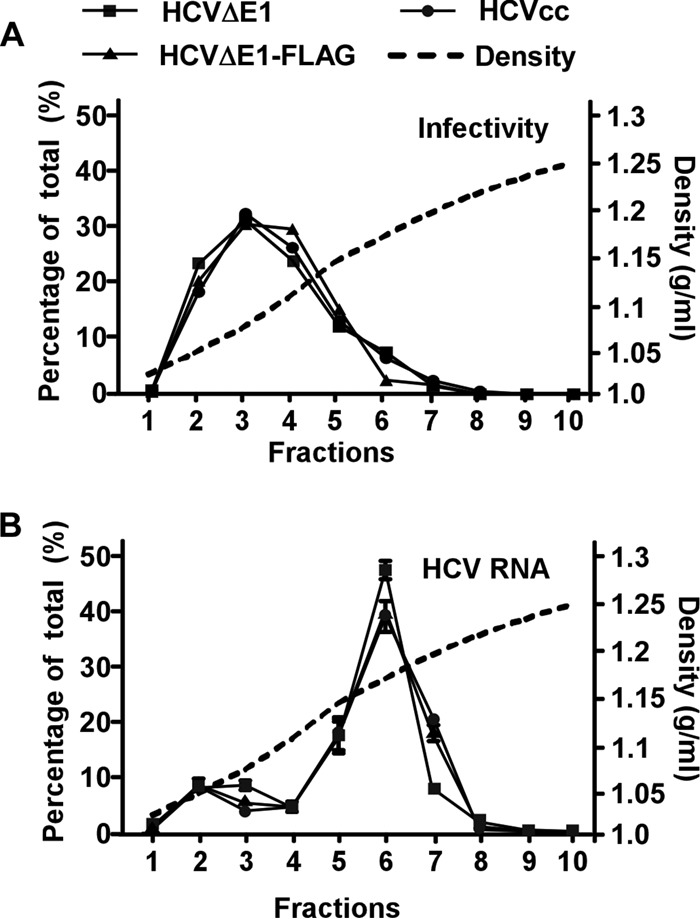
Characterization of the buoyant densities of HCVΔE1 and HCVΔE1-FLAG particles. The different HCV particles (HCVcc, HCVΔE1, and HCVΔE1-FLAG) were subjected to a 20% to 60% sucrose gradient. Ten fractions were collected from the top. The HCV RNA levels (A) and infectivity titers (B) of each fraction were determined by a titration assay and RT-qPCR, respectively. The results are expressed as the percentages of each fraction of the total. The density of each fraction was determined by measuring the mass of a 100-μl aliquot of the fraction. The data are representative of results from four independent experiments, and the error bars were calculated from duplicative RT-qPCR analyses.
Deletion of the E1 putative fusion peptide impairs HCV entry and morphogenesis.
It was reported previously that a region (amino acid residues 272 to 285) (Fig. 5A) in HCV E1 is a putative FP, which plays an important role in triggering the fusion of the viral envelope and endosomal membrane during the HCV entry process (27–32). We first examined the role of FP using lentivirus-based HCV pseudotype particles (HCVpp) that harbor the HCV envelope proteins E1 and E2 and mimic the HCV entry process (36, 37). As shown in Fig. 5B, the deletion of the FP in E1 had no apparent effect on the incorporation of HCV envelope proteins into HCVpp (Fig. 5B) but abolished HCVpp infection (Fig. 5C), confirming the role of the FP in HCV entry.
FIG 5.

Deletion of the E1 putative fusion peptide impairs HCV entry and morphogenesis. (A) Schematic of the putative FP (residues 272 to 285) and TMD (residues 353 to 381) of HCV E1 (residues 191 to 383). (B) Western blot analysis of HCV E2 and HIV P24 in HCVpp-Luc bearing JFH1 wild-type E1 (no tag) and E2 glycoproteins (WT), the mutant with a deletion in the E1 FP (del-FP), or no envelope protein (Env−). (C) HCVpp infection in Huh7.5.1 cells. Luciferase activities were measured at day 3 postinfection and are presented as a percentage of the values for the wild type. The error bars were derived from three independent experiments. (D to H) Huh7.5.1 cells expressing wild-type E1 (E1-WT), E1 with an FP deletion (E1-delFP), E1 with a TMD deletion (E1-delTMD), or the GFP control were infected with HCVΔE1 at an MOI of 2. The cells or culture supernatants were collected at day 3 postinfection for the following analyses. RLU, relative light units. (D) The quantities of intracellular HCV RNA were measured by RT-qPCR and are expressed as a percentage of E1-WT values. (E) Extracellular infectivity titers were measured by a titration assay. The dashed line indicates the detection limit. (F) The extracellular HCV RNA levels were measured by RT-qPCR and are expressed as a percentage of the E1-WT values. (G) The levels of extracellular HCV core proteins were measured by an ELISA. For panels C to F, the error bars were derived from three independent experiments. (H) The cell lysates were first immunoprecipitated using an anti-FLAG antibody and then subjected to Western blotting using an anti-E2 antibody. The amount of immunoprecipitated E2 relative to the amount of input E2 was quantified by using ImageJ software, normalized to the value for immunoprecipitated E1-FLAG, and expressed as a value relative to that of E1-WT and is shown at the top.
Next, we took advantage of the above-described E1 trans-complementation system to study the role of this putative FP in the HCV life cycle. Huh7.5.1 cells that stably express FLAG-tagged wild-type E1 (E1-WT) or the FLAG-tagged E1 FP deletion mutant (E1-delFP) were generated by lentiviral vector-based transduction. Huh7.5.1 cells transduced with lentiviruses expressing green fluorescent protein (GFP) or the HCV E1 mutant lacking the C-terminal transmembrane domain (E1-delTMD) were also included as controls. The TMD dictates the membrane anchorage of E1 and is vital for E1 and E2 heterodimerization (38). Western blot analysis showed that the E1 expression levels were comparable in these cells (Fig. 5H and data not shown). These cells were then infected with HCVΔE1 at an MOI of 2, and intracellular HCV RNA levels and extracellular infectivity were determined at day 3 postinfection. As shown in Fig. 5D, the intracellular HCV RNA levels were comparable in all infected cells, indicating similar infection efficiencies among these groups. However, deletion of the FP or TMD completely abolished the infectivity titers (Fig. 5E). The lack of supernatant infectivity of the E1-delFP mutant can result from the effect of the FP deletion on virus entry or on virus production. In order to distinguish between these two possibilities, we then determined the levels of HCV RNA and core proteins in the culture supernatants by RT-quantitative PCR (RT-qPCR) and an enzyme-linked immunosorbent assay (ELISA), respectively. As shown in Fig. 5F and G, the levels of extracellular HCV RNA and core proteins in the E1-delFP or E1-delTMD mutant were decreased to the same levels as the GFP negative control, suggesting that the FP deletion impairs the morphogenesis of HCV virions. Interestingly, a coimmunoprecipitation experiment using an anti-FLAG antibody showed that the deletion of FP did not affect the heterodimerization of E1 and E2, whereas the deletion of the TMD severely reduced the interaction of E1 and E2 (Fig. 5H). This result implied that the FP deletion did not completely disrupt the global conformation of E1, and the effect of the FP deletion on virus entry and morphogenesis may be due to the critical roles of the FP in these two processes.
Identification of key amino acid residues within the putative fusion peptide of E1.
To identify key amino acid residues in the putative FP motif of E1, we performed an alanine-scanning mutagenesis study. Each amino acid residue in the FP between positions 272 and 285, except for position 274, which is alanine itself, was changed to alanine individually (Fig. 6A). First, a coimmunoprecipitation experiment using an anti-FLAG antibody showed that neither of these single-point-substitution mutations affected the heterodimerization of E1 and E2 (Fig. 6B). Next, we analyzed how these mutations affected HCV entry using the HCVpp system. As shown in Fig. 6C, neither mutation affected the incorporation of HCV envelope proteins into HCVpp. As shown in Fig. 6D and as also summarized in Table 1, mutations Y276A, G283A, and V284A increased HCVpp infection compared to infection by the wild type (Table 1). HCVpp containing the mutation C272A, L275A, V277A, or M285A retained about 30% to 60% infectivity (Table 1), while HCVpp containing the mutation S273A, D279A, or G282A showed strongly reduced infectivity, with about 10% to 15% of the infectivity of the wild type (Table 1). Importantly, HCVpp containing the G278A, L280A, or C281A mutation displayed no detectable infectivity (Table 1).
FIG 6.

Analysis of a single point mutation within the putative fusion peptide of E1. (A) Schematic of amino acid residues of the E1 putative fusion motif (residues 272 to 285) of JFH1. Each residue was mutated to alanine, except for that at position 274, which is alanine itself. (B) Coimmunoprecipitation experiment to determine the interaction of E1 and E2. Huh7.5.1 cells were transfected with plasmids expressing E2 and FLAG-tagged E1 containing a single point mutation in the FP for 2 days. The cell lysates were immunoprecipitated (Ip) with an anti-FLAG antibody and analyzed by Western blotting using anti-FLAG and anti-E2 antibodies. The amount of immunoprecipitated E2 relative to the amount of input E2 was quantified by using ImageJ software, normalized to the value for immunoprecipitated E1-FLAG, and expressed as a value relative to the value for E1-WT and is shown at the top. (C) Western blot analysis of HCV E2 and HIV P24 in HCVpp-Luc bearing JFH1 wild-type E1 (no tag) with a single point mutation in the FP. The amount of E2 was quantified by using ImageJ software, normalized to the amount of P24, and expressed as a value relative to that of E1E2-WT and is shown at the top. (D) Effect of each mutation in the E1 FP on HCVpp infection. Infection was quantified by a luciferase assay and is presented as a percentage of the value for the wild type. The error bars represent results from three independent experiments.
TABLE 1.
Summary of the phenotypes of E1 harboring a point mutation in the putative fusion peptide
| E1 construct | Phenotype |
||
|---|---|---|---|
| HCVpp infectiona | Infectivity titerb | Supernatant HCV RNAc | |
| WT | +++ | +++ | +++ |
| C272 | ++ | − | − |
| S273 | + | + | + |
| L275 | ++ | ++ | ++ |
| Y276 | +++ | + | ++ |
| V277 | ++ | +++ | +++ |
| G278 | − | − | − |
| D279 | + | − | − |
| L280 | − | ++ | + |
| C281 | − | − | − |
| G282 | + | + | ++ |
| G283 | +++ | +++ | +++ |
| V284 | +++ | +++ | +++ |
| M285 | ++ | ++ | +++ |
| NC | − | − | − |
Percent HCV entry, where +++ indicates >75% HCV entry, ++ indicates between 25% and 75%, + indicates between 5% and 25%, and − indicates <5%.
Percent supernatant infectivity, where +++ indicates >50% infectivity, ++ indicates between 15% and 50%, + indicates between 1% and 15%, and − indicates <1%.
Percent supernatant HCV RNA release, where +++ indicates >75% HCV RNA release, ++ indicates between 25% and 75%, + indicates between 5% and 25%, and − indicates <5%.
Next, we tested the effect of these mutations on the HCV life cycle using the HCVΔE1 system. Huh7.5.1 cells expressing FLAG-tagged E1 with an individual point mutation in the FP were generated by lentiviral transduction, and E1 expression was validated by Western blotting (Fig. 7A). These cells were then infected with HCVΔE1 at an MOI of 2, and the intracellular/extracellular HCV RNA levels and extracellular infectivity titers were determined at day 3 postinfection. As shown in Fig. 7B, the intracellular HCV RNA levels in all groups were similar, suggesting that none of these point mutations in E1 affected HCV genome replication. However, these mutations had differential effects on the infectivity titers of trans-complemented HCVΔE1 (Fig. 7C and Table 1). While the V277A, G283A, or V284A mutant displayed infectivity titers similar to those of the wild type (Table 1), no infectivity titer was detected in the culture supernatants of cells expressing E1 with the mutation C272A, G278A, G279A, or C281A (Table 1). The changes in HCV RNA levels in the culture supernatants were consistent with the changes in infectivity titers (Fig. 7D and Table 1). Of note, the extracellular HCV RNA levels in the C272A, G278A, G279A, or C281A mutant were similar to those in the GFP negative control. To test whether the loss of infectivity titers in the culture supernatants was not due to a blockade of virus release, we also examined the intracellular infectivity titers of FP mutants that either displayed no significant changes in the extracellular infectivity titers (V277A, G283A, and V284A) or had no detectable extracellular infectivity titer (C272A, G278A, G279A, and C281A). As shown in Fig. 7E, consistent with their extracellular infectivity titers, the V277A, G283A, and V284A mutants displayed intracellular infectivity titers that were similar to those of the wild type, whereas the intracellular infectivity titers of the C272A, G278A, G279A, and C281A mutants were below the detection limit.
FIG 7.

Identification of key amino acid residues within the putative fusion peptide of E1. (A) Western blot analysis of E1 expression in Huh7.5.1 cells that were transduced by a lentiviral vector expressing wild-type or mutant E1 proteins. The amount of E1-FLAG was quantified by using ImageJ software, normalized to the value for actin, and expressed as a value relative to that of the wild type and is shown on the top. (B to D) Huh7.5.1 cells expressing wild-type E1 (WT), E1 with an individual alanine point mutation within the FP, or the GFP control were infected with HCVΔE1 at an MOI of 2. The cells or culture supernatants were collected at day 3 postinfection for the following analyses. (B) The quantities of intracellular HCV RNA were measured by RT-qPCR and are expressed as a percentage of the values for the wild type. (C) Extracellular infectivity titers. The dashed line indicates the detection limit of the titration assay. (D) Extracellular HCV RNA levels were measured by RT-qPCR and are expressed as a percentage of the value for the wild type. (E) Intracellular infectivity titers of selected E1 mutants. The dashed line indicates the detection limit of the titration assay. For panels D to G, the error bars were derived from three independent experiments.
Finally, we determined the effects of the selected FP point mutations on viral membrane fusion using a previously developed fusion assay (Fig. 8A) (39). HEK293T cells were transfected with plasmids expressing the Cre recombinase and HCV E1-E2 containing the wild-type protein; the C272A, Y276A, G278A, or G284A mutation in the FP; or the FP deletion mutation. After coculture of transfected HEK293T cells and Huh7.5.1 cells expressing an inactive luciferase disrupted by the loxP sequences, membrane fusion can be triggered by a low-pH shock to transfer the Cre recombinase from donor HEK293T cells to recipient Huh7.5.1 cells to activate luciferase activity. Western blot analysis indicated similar levels of E2 expression among all groups (Fig. 8B). As shown in Fig. 8C, the Y276A and G284A mutants displayed levels of membrane fusion comparable to those of the wild type, while the G278A and the FP deletion mutants had significantly reduced levels of membrane fusion, confirming that G278 plays a critical role in HCV entry. The C272A mutant had an intermediate level of membrane fusion, consistent with the observation that the C272A mutant supported a certain level of HCV entry.
FIG 8.
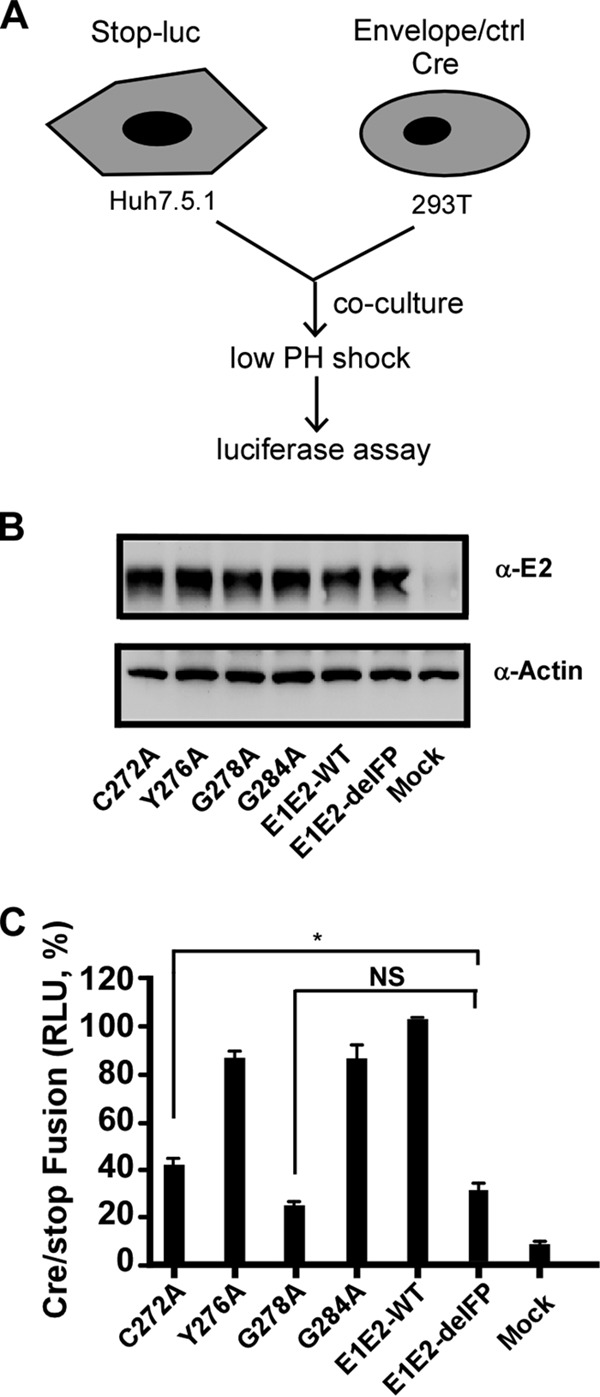
Membrane fusion assay. (A) Schematic of the membrane fusion assay. Donor HEK293T cells were transfected with plasmids expressing the Cre recombinase and HCV envelope proteins, and recipient Huh7.5.1 cells were transfected with plasmids expressing an inactive luciferase disrupted by the loxP sequences. During coculture, a low-pH shock triggers membrane fusion between two cells that allows the transfer of the Cre recombinase from HEK293T cells to Huh7.5.1 cells to activate luciferase activity. (B) Western blot analysis of E2 expression in HEK293T cells transfected with wild-type E1 or E1 mutants. (C) Luciferase activity upon triggering of membrane fusion. NS, nonsignificant (P > 0.05); *, P < 0.05.
DISCUSSION
In this study, we developed a cell culture model for single-cycle HCV infection based on the trans-complementation of the envelope protein E1 (HCVΔE1). Virus production of the HCV genome lacking the E1-encoding sequence can be efficiently rescued by the ectopic expression of E1. Adaptive mutations, particularly the 5-amino-acid deletion (EEDDT) in the C terminus of NS5A (NS5AM), significantly enhance HCVΔE1 production. HCVΔE1 can efficiently propagate in packaging cells (Huh7.5.1E1 or Huh7.5.1E1-FLAG) but results in only single-cycle infection in naive Huh7.5.1 cells. This HCVΔE1 model renders a unique system to study the biology of E1 in the HCV life cycle and to dissect functional domains and motifs in E1. Since E1 can be expressed alone in trans, it is convenient to perform mutagenesis on E1. Moreover, the introduction of mutations in trans-expressed E1 will avoid the cis effects of the mutations, for example, the disruption of critical RNA secondary or tertiary structures in the HCV genome, or interfere with proteolytic cleavage at a junction site between E1 and adjacent proteins (core/E1 and E1/E2), thereby providing a more specific system to evaluate the biological roles of domains, motifs, or amino acid residues within E1.
The viral structural elements responsible for the fusion of the HCV envelope and host cell endosomal membrane have not been fully determined. The recently solved crystal structure of the core domain of E2 failed to identify any fusion-triggering domains/motifs in E2 (21, 22, 26). It has been proposed that E1 may contain a putative FP (27–32). Recently the 3.5-Å-resolution crystal structure of nE1 (residues 192 to 271) was solved (23). Unfortunately, the FP (residues 272 to 285) was not included in the sequence of nE1, and thus, no 3-dimensional structure information on the FP is available. By using the HCVΔE1 system, we analyzed the roles of the FP in the HCV life cycle. We found that the deletion of the FP not only abolishes viral infectivity but also reduces the levels of HCV RNA and core proteins in the supernatants. This result suggests that the FP may also play an important role in virus morphogenesis in addition to its contributions to HCV entry, as previously reported (29, 40). To our knowledge, our finding is the first evidence to suggest that the FP may be a multifunctional motif in the HCV life cycle.
Using alanine-scanning mutagenesis, we analyzed the role of each amino acid residue in the FP (positions 272 to 285) in virus entry and virus morphogenesis. None of these substitution mutations impairs the interaction between E1 and E2 (Fig. 6B), suggesting that the mutations do not destroy the structure of E1. The effect of each point mutation on HCV entry was evaluated for HCVpp infection as well as for HCVΔE1 infection by comparing the infectivity titers and virus genome contents in the culture supernatants, while the effect of the mutation on HCV morphogenesis was evaluated by both the infectivity titers and virus genome contents in the culture supernatants. As summarized in Table 1, C272 and four consecutive amino acids in the middle of the FP (G278, D279, L280, and C281) play essential roles in either HCV entry, morphogenesis, or both. Mutations of other residues (residues 273 to 277 and 282 to 285) had no or only marginal effects on the HCV life cycle. G278 and C281 seem critical for both virus entry and morphogenesis, since an alanine point mutation at these two positions completely abolishes HCVpp infection, HCVΔE1 extracellular infectivity, and HCV RNA contents. Mutations at C272 and D279 completely abolish HCVΔE1 extracellular infectivity and HCV RNA content but maintain some activity in HCVpp infection. In addition, the C272A mutation did not completely abolish the activity of E1 in mediating membrane fusion (Fig. 8). These results suggest that the role of C272 and D279 in the virus life cycle may be more toward HCV morphogenesis rather than virus entry. The roles of C272 and C281 were previously investigated by Wahid et al. using an HCVcc system in which E1 is expressed in cis from the viral genome (41). Those researchers found that an alanine mutation at these two positions significantly reduced but did not completely abolish HCV infectivity (41). In contrast, we found that mutations at these two positions completely abolished HCV infectivity in our study. To determine whether this difference may result from E1 expression in cis or in trans, we introduced the C272A mutation into the JFH1 genome, which contains the same NS5AM adaptive mutation (EEDDT deletion in NS5A). Our results showed that no infectivity was detected after electroporation of the JFH1 genome containing the C272A mutation even in cis (data not shown), consistent with the results obtained from the HCVΔE1 study, where the C272A mutation was evaluated in trans. The exact reason for this discrepancy between our study and that of Wahid et al. (41) is not known at the moment. It is possible that this might be due to the different adaptive mutations in the virus genomes used in these two studies. While we used a JFH1 genome that contains an adaptive mutation in NS5A (EEDDT deletion), Wahid et al. used a JFH1 genome that contains two adaptive mutations in the core protein (F172C and P173S) as well as A4 epitope swapping in E1 to aid in the detection of E1 (41). More studies will be needed to address this question.
In order to detect E1 expression, we inserted a three-tandem-FLAG tag at the C terminus of E1. Unlike the hemagglutinin (HA) tag reported previously (42), our results demonstrated that the FLAG tag was not cleaved out from E1 by the cellular signal peptidase that is responsible for cleavage between E1 and E2 (Fig. 3B and E). To further confirm the integrity of E1-FLAG, we constructed an Huh7.5.1 cell line that stably expresses FLAG-tagged E1 containing an A4 epitope derived from E1 of the H77 strain (43, 44). The insertion of the A4 epitope would allow analysis of the size and intensity of E1-FLAG by using an antibody specific for the A4 epitope together with the anti-FLAG antibody. Western blot analysis demonstrated that E1-FLAG in cells and in the supernatants was indeed intact (data not shown). Therefore, we did not have to modify the C terminus of E1 to avoid the cleavage of the FLAG tag from E1 by the signal peptidase. Importantly, the insertion of the FLAG tag did not impair the ability of E1 to rescue HCVΔE1 production. With this FLAG tag, E1 can be detected by Western blot and immunofluorescence analyses (Fig. 3). Furthermore, free E1 proteins can be immunoprecipitated by using an anti-FLAG antibody (Fig. 5H), providing a new tool to study host factors and viral proteins that may interact with E1 during HCV infection. It is worth pointing out that this FLAG-tagged E1 protein will not be accessible to anti-FLAG antibodies after it is incorporated into mature virions, because the FLAG tag is added to the C terminus of E1 and should be located on the inner side of the HCV virion envelope. Consistently, HCVΔE1 infection was insensitive to neutralization by the anti-FLAG antibody (data not shown).
In conclusion, we developed a new cell culture model of HCV single-cycle infection based on the trans-complementation of E1. By using this system, we found that a putative fusion peptide in E1 may be a multifunctional structural element contributing to both HCV entry and morphogenesis. Our study provides new insights into understanding the biological roles of E1 in the HCV life cycle.
MATERIALS AND METHODS
Cell culture and preparation.
Huh7.5.1 hepatic cells and derivatives were maintained in complete Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum, 10 mM HEPES buffer, 100 U/ml penicillin, and 100 mg/ml streptomycin.
Huh7.5.1E1 packaging cells that stably express E1 were produced by transfecting 2 μg of the pcDNA3-JFH1-E1 plasmid into 8 × 105 Huh7.5.1 cells, followed by 3 weeks of selection with 500 μg/ml of G418. One cell clone (A3) that was highly permissive for HCVΔE1 production was expanded and used for studies.
To generate Huh7.5.1 cells stably expressing mutant E1 proteins, a lentiviral vector encoding wild-type E1 or each mutant E1 protein was cotransfected with plasmids encoding compatible packaging proteins into HEK293T cells, as described previously (45). Briefly, HEK293T cells were transfected with the vesicular stomatitis virus (VSV) glycoprotein expression plasmid and HIV gag-pol (pLP1), HIV rev (pLP2), and transfer pLenti6 vectors encoding wild-type or mutant E1 by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). At 72 h posttransfection, cell supernatants were filtered through 0.45-μm-pore-size membranes and used to transduce Huh7.5.1 cells. A control cell line expressing GFP was generated in the same way.
Plasmid construction.
Plasmid pUC-JFH1-delE1 containing an in-frame deletion (amino acid residues 217 to 334) in the region of JFH1 encoding E1 was constructed by fusion PCR to amplify an amplicon containing the deletion mutation using the PrimerStar PCR system (TaKaRa, Dalian, China), followed by restriction digestion with FspAI and NotI and ligation into the same sites of pUC-JFH1 (46, 47). Plasmid pUC-JFH1-delE1-2M containing two previously reported point mutations, M1051T and C2219R (33), and plasmid pUC-JFH1-delE1-NS5AM containing a 5-amino-acid deletion (EEDDT) (positions 2435 to 2439) at the C terminus of NS5A were constructed by using the same fusion PCR strategy. Plasmid pLenti-E1, expressing JFH1 E1, was constructed by PCR to amplify the E1 open reading frame (ORF) plus 21 amino acid residues of the C terminus of the core protein as the signal peptide (37). The amplicon was digested and inserted into the BamHI and SalI sites of the pLenti vector (45). Plasmid pLenti-E1-FLAG, expressing JFH1 E1 with 3 tandem FLAG tags at its C terminus, was constructed by using the same strategy. Plasmids pLenti-E1-FLAG-delTMD containing a deletion in the E1 transmembrane domain (amino acid residues 353 to 381), pLenti-E1-FLAG-delFP containing a deletion in the E1 putative fusion peptide (amino acid residues 272 to 285), and pLenti-E1-FLAG containing a single-alanine point mutation in the FP were constructed by fusion PCR followed by restriction and ligation into pLenti-E1-FLAG. Plasmid pcDNA3-JFH1-E1/E2 expressing wild-type or mutant JFH1 envelope glycoproteins was constructed by PCR amplification of the JFH1 E1 and E2 regions (amino acid residues 171 to 750) and insertion of the PCR product into pcDNA3.1. All constructed plasmids were verified by DNA sequencing.
HCVcc preparation.
HCVcc (JFH1 strain, genotype 2a) (GenBank accession number AB047639) (46, 47) was used in these studies. The protocols for the generation of HCVcc or HCVΔE1, including in vitro RNA transcription and electroporation, were described previously (18, 33, 47).
Indirect immunofluorescence.
Intracellular immunostaining was performed as described previously (33). Briefly, cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. HCV E2, NS3, and FLAG were stained by using a human monoclonal anti-E2 antibody (48), a mouse monoclonal anti-NS3 antibody (produced in our laboratory), and a monoclonal anti-FLAG antibody (M2; Sigma, St. Louis, MO), respectively. Bound primary antibodies were detected by using Alexa Fluor 488- or Alexa Fluor 555-conjugated secondary antibodies (Molecular Probes, Eugene, OR). Nuclei were stained with Hoechst dye.
Western blot analysis.
Western blot analysis was performed as described previously (33). Briefly, cell lysate proteins were separated by 10% SDS-polyacrylamide gel electrophoresis and then transferred to a polyvinylidene difluoride membrane (Millipore, Bedford, MA). Membranes were probed first with a primary monoclonal antibody against FLAG (Sigma), against HCV E2 (1C9; produced by our laboratory), or against actin (Sigma) and then with horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary antibodies (Promega, Madison, WI). Proteins were visualized by using an ECL kit (Millipore).
HCV infectivity titer and genome RNA quantification.
HCV infectivity titers were determined in Huh7.5.1 cells by endpoint dilution and immunostaining as described previously (33, 47). HCV RNA levels were determined by RT-qPCR as described previously (33, 48).
HCV infection kinetics assay.
HCV infection kinetics were determined as described previously (34). Briefly, 8 × 104 Huh7.5.1 or packaging cells were seeded into 12-well plates overnight and then inoculated with the virus at an MOI of 0.01 or 0.1. The infected cells reached confluence on day 4 postinfection and were then split at a ratio of 1:3 into 12-well plates (harvested on day 6), 6-well plates (harvested on day 8), and T25 flasks (harvested on day 10). Culture supernatants were collected at the indicated time points, and infectivity titers were determined as described above.
Measurement of HCV intracellular infectivity titers.
HCV intracellular infectivity titers were determined as described previously (41). Briefly, infected cells were washed with phosphate-buffered saline (PBS), harvested by trypsin treatment, and pelleted at 100 × g for 5 min. The cell pellets were resuspended in complete medium and mechanically lysed in a Dounce homogenizer (30 strokes). The cell lysates were clarified by centrifugation at 10,000 × g for 5 min, and the infectivity titers were then determined by a titration assay as described above.
Density gradient ultracentrifugation.
Gradients were formed by overlaying 2 ml of 20%, 30%, 40%, 50%, and 60% sucrose solutions in TNE buffer (10 mM Tris-HCl [pH 8], 150 mM NaCl, 2 mM EDTA) as described previously (33, 48). Equilibrium was reached by ultracentrifugation for 16 h at 154,000 × g in an SW41 Ti rotor at 4°C in a Beckman Optima L-80XP preparative ultracentrifuge. Ten gradient fractions of 1,150 μl were collected from the top, and their infectivity titers and HCV RNA levels were determined as described above. The density of each fraction was determined by measuring the mass of 100-μl aliquots of each sample.
HCV core ELISA.
The core proteins in culture supernatants were quantified by an ELISA according to the manufacturer's instructions (HCV core antigen Elisa detect kit; LaiBo, China). Absorbance values were determined at 450 nm after the reaction was stopped with 0.5 M sulfuric acid. Quantities were calculated based on a standard curve.
Infection by pseudotyped virus.
HCVpp were generated as described previously (36, 45). Briefly, HEK293T cells were cotransfected with plasmids expressing HCV Ε1-Ε2 glycoproteins, a retroviral core packaging component, and luciferase. The medium was refreshed at 6 h posttransfection. Supernatants were collected 72 h later and filtered through 0.45-μm-pore-size membranes. For infection experiments, 8,000 Huh7.5.1 cells seeded into 96-well plates were infected with 50 μl of HCVpp. Three days after infection, the firefly luciferase activity was measured by using a luciferase assay system according to the manufacturer's instructions (Promega).
Membrane fusion assay.
A cell-cell fusion assay was performed as described previously (39). Briefly, HEK293T “donor” cells were seeded into 6-well plates (8 × 105 cells/per well) and transfected with plasmids expressing wild-type or mutant E1E2 together with a Cre recombinase-expressing plasmid for 12 h. The cells were then dissociated from the wells by using a nonenzymatic cell dissociation solution (Sigma), according to the manufacturer's instructions, and suspended in DMEM containing 10% fetal bovine serum (FBS) prior to coculture. Huh7.5.1 “target” cells were seeded into a 24-well plate at 1.5 × 105 cells/well for 24 h and transfected with the pCMV-Stop-Luc plasmid for 12 h. The donor cells were placed at a density of 5 × 105 cells/well on top of the target cells and cocultured for 4 to 6 h at 37°C. To initiate fusion, the medium was removed and replaced with fusion buffer (135 mM NaCl, 15 mM sodium citrate, 10 mM morpholineethanesulfonic acid, 5 mM HEPES, 1 mM EDTA) for <1 min. The low-pH buffer was replaced with DMEM containing 10% FBS. After incubation of the cells for 12 h at 37°C, the cells lysates were analyzed for luciferase activity (Promega).
ACKNOWLEDGMENTS
We thank Rui Li, Ying He, Wanyin Tao, Qiang Ding, and Yongfeng Gao (Institut Pasteur of Shanghai, Chinese Academy of Sciences) for technical assistance and Jean Dubuisson (Institut Pasteur de Lille, France) for sharing the anti-A4 antibody and insightful discussions and suggestions.
We declare no conflict of interest.
This study was supported by grants from the National Natural Science Foundation of China (31170149 and 31270203) and the Chinese National 973 Program (2015CB554300) to J.Z. as well as from the National Natural Science Foundation of China (31300147), the Postdoctoral Science Foundation of China (20110490755), the Postdoctoral Science Foundation of Shanghai Municipality (13R21417500), and the Post-Doctor Research Program of Shanghai Institutes for Biological Sciences (2012KIP517) to Y.T. We gratefully acknowledge the support of a Sanofi-Aventis Shanghai Institutes for Biological Sciences scholarship to Y.T.
REFERENCES
- 1.Wiley. 1999. Global surveillance and control of hepatitis C. Report of a WHO Consultation organized in collaboration with the Viral Hepatitis Prevention Board, Antwerp, Belgium. J Viral Hepat 6:35–47. [PubMed] [Google Scholar]
- 2.Lauer GM, Walker BD. 2001. Hepatitis C virus infection. N Engl J Med 345:41–52. doi: 10.1056/NEJM200107053450107. [DOI] [PubMed] [Google Scholar]
- 3.Wasley A, Alter MJ. 2000. Epidemiology of hepatitis C: geographic differences and temporal trends. Semin Liver Dis 20:1–16. doi: 10.1055/s-2000-9506. [DOI] [PubMed] [Google Scholar]
- 4.Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht JK. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958–965. doi: 10.1016/S0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- 5.McHutchison JG, Lawitz EJ, Shiffman ML, Muir AJ, Galler GW, McCone J, Nyberg LM, Lee WM, Ghalib RH, Schiff ER, Galati JS, Bacon BR, Davis MN, Mukhopadhyay P, Koury K, Noviello S, Pedicone LD, Brass CA, Albrecht JK, Sulkowski MS, IDEAL Study Team . 2009. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N Engl J Med 361:580–593. doi: 10.1056/NEJMoa0808010. [DOI] [PubMed] [Google Scholar]
- 6.Liang TJ, Ghany MG. 2013. Current and future therapies for hepatitis C virus infection. N Engl J Med 368:1907–1917. doi: 10.1056/NEJMra1213651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pawlotsky JM. 2014. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology 146:1176–1192. doi: 10.1053/j.gastro.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. 2013. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 57:1333–1342. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 9.Li DP, Huang Z, Zhong J. 2015. Hepatitis C virus vaccine development: old challenges and new opportunities. Natl Sci Rev 2:285–295. doi: 10.1093/nsr/nwv040. [DOI] [Google Scholar]
- 10.Lindenbach BD, Rice CM. 2013. The ins and outs of hepatitis C virus entry and assembly. Nat Rev Microbiol 11:688–700. doi: 10.1038/nrmicro3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scheel TK, Rice CM. 2013. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med 19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vieyres G, Dubuisson J, Pietschmann T. 2014. Incorporation of hepatitis C virus E1 and E2 glycoproteins: the keystones on a peculiar virion. Viruses 6:1149–1187. doi: 10.3390/v6031149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Voisset C, Dubuisson J. 2004. Functional hepatitis C virus envelope glycoproteins. Biol Cell 96:413–420. doi: 10.1016/j.biolcel.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 14.Fafi-Kremer S, Fauvelle C, Felmlee DJ, Zeisel MB, Lepiller Q, Fofana I, Heydmann L, Stoll-Keller F, Baumert TF. 2012. Neutralizing antibodies and pathogenesis of hepatitis C virus infection. Viruses 4:2016–2030. doi: 10.3390/v4102016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Hahn T, Rice CM. 2008. Hepatitis C virus entry. J Biol Chem 283:3689–3693. doi: 10.1074/jbc.R700024200. [DOI] [PubMed] [Google Scholar]
- 16.Felmlee DJ, Hafirassou ML, Lefevre M, Baumert TF, Schuster C. 2013. Hepatitis C virus, cholesterol and lipoproteins—impact for the viral life cycle and pathogenesis of liver disease. Viruses 5:1292–1324. doi: 10.3390/v5051292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazumdar B, Banerjee A, Meyer K, Ray R. 2011. Hepatitis C virus E1 envelope glycoprotein interacts with apolipoproteins in facilitating entry into hepatocytes. Hepatology 54:1149–1156. doi: 10.1002/hep.24523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tao W, Xu C, Ding Q, Li R, Xiang Y, Chung J, Zhong J. 2009. A single point mutation in E2 enhances hepatitis C virus infectivity and alters lipoprotein association of viral particles. Virology 395:67–76. doi: 10.1016/j.virol.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 19.Haid S, Pietschmann T, Pecheur EI. 2009. Low pH-dependent hepatitis C virus membrane fusion depends on E2 integrity, target lipid composition, and density of virus particles. J Biol Chem 284:17657–17667. doi: 10.1074/jbc.M109.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan AG, Whidby J, Miller MT, Scarborough H, Zatorski AV, Cygan A, Price AA, Yost SA, Bohannon CD, Jacob J, Grakoui A, Marcotrigiano J. 2014. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 509:381–384. doi: 10.1038/nature13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kong L, Giang E, Nieusma T, Kadam RU, Cogburn KE, Hua Y, Dai X, Stanfield RL, Burton DR, Ward AB, Wilson IA, Law M. 2013. Hepatitis C virus E2 envelope glycoprotein core structure. Science 342:1090–1094. doi: 10.1126/science.1243876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El Omari K, Iourin O, Kadlec J, Sutton G, Harlos K, Grimes JM, Stuart DI. 2014. Unexpected structure for the N-terminal domain of hepatitis C virus envelope glycoprotein E1. Nat Commun 5:4874. doi: 10.1038/ncomms5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fauvelle C, Felmlee DJ, Baumert TF. 2014. Unraveling hepatitis C virus structure. Cell Res 24:385–386. doi: 10.1038/cr.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan AG, Miller MT, Marcotrigiano J. 2015. HCV glycoprotein structures: what to expect from the unexpected. Curr Opin Virol 12:53–58. doi: 10.1016/j.coviro.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sabahi A, Uprichard SL, Wimley WC, Dash S, Garry RF. 2014. Unexpected structural features of the hepatitis C virus envelope protein 2 ectodomain. J Virol 88:10280–10288. doi: 10.1128/JVI.00874-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garry RF, Dash S. 2003. Proteomics computational analyses suggest that hepatitis C virus E1 and pestivirus E2 envelope glycoproteins are truncated class II fusion proteins. Virology 307:255–265. doi: 10.1016/S0042-6822(02)00065-X. [DOI] [PubMed] [Google Scholar]
- 28.Douam F, Dao Thi VL, Maurin G, Fresquet J, Mompelat D, Zeisel MB, Baumert TF, Cosset FL, Lavillette D. 2014. Critical interaction between E1 and E2 glycoproteins determines binding and fusion properties of hepatitis C virus during cell entry. Hepatology 59:776–788. doi: 10.1002/hep.26733. [DOI] [PubMed] [Google Scholar]
- 29.Drummer HE, Boo I, Poumbourios P. 2007. Mutagenesis of a conserved fusion peptide-like motif and membrane-proximal heptad-repeat region of hepatitis C virus glycoprotein E1. J Gen Virol 88:1144–1148. doi: 10.1099/vir.0.82567-0. [DOI] [PubMed] [Google Scholar]
- 30.Lavillette D, Bartosch B, Nourrisson D, Verney G, Cosset FL, Penin F, Pecheur EI. 2006. Hepatitis C virus glycoproteins mediate low pH-dependent membrane fusion with liposomes. J Biol Chem 281:3909–3917. doi: 10.1074/jbc.M509747200. [DOI] [PubMed] [Google Scholar]
- 31.Li HF, Huang CH, Ai LS, Chuang CK, Chen SS. 2009. Mutagenesis of the fusion peptide-like domain of hepatitis C virus E1 glycoprotein: involvement in cell fusion and virus entry. J Biomed Sci 16:89. doi: 10.1186/1423-0127-16-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perin PM, Haid S, Brown RJ, Doerrbecker J, Schulze K, Zeilinger C, von Schaewen M, Heller B, Vercauteren K, Luxenburger E, Baktash YM, Vondran FW, Speerstra S, Awadh A, Mukhtarov F, Schang LM, Kirschning A, Muller R, Guzman CA, Kaderali L, Randall G, Meuleman P, Ploss A, Pietschmann T. 2016. Flunarizine prevents hepatitis C virus membrane fusion in a genotype-dependent manner by targeting the potential fusion peptide within E1. Hepatology 63:49–62. doi: 10.1002/hep.28111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li R, Qin Y, He Y, Tao W, Zhang N, Tsai C, Zhou P, Zhong J. 2011. Production of hepatitis C virus lacking the envelope-encoding genes for single-cycle infection by providing homologous envelope proteins or vesicular stomatitis virus glycoproteins in trans. J Virol 85:2138–2147. doi: 10.1128/JVI.02313-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaul A, Woerz I, Meuleman P, Leroux-Roels G, Bartenschlager R. 2007. Cell culture adaptation of hepatitis C virus and in vivo viability of an adapted variant. J Virol 81:13168–13179. doi: 10.1128/JVI.01362-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhong J, Gastaminza P, Chung J, Stamataki Z, Isogawa M, Cheng G, McKeating JA, Chisari FV. 2006. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J Virol 80:11082–11093. doi: 10.1128/JVI.01307-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med 197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci U S A 100:7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ciczora Y, Callens N, Penin F, Pecheur EI, Dubuisson J. 2007. Transmembrane domains of hepatitis C virus envelope glycoproteins: residues involved in E1E2 heterodimerization and involvement of these domains in virus entry. J Virol 81:2372–2381. doi: 10.1128/JVI.02198-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chi X, Niu Y, Cheng M, Liu X, Feng Y, Zheng F, Fan J, Li X, Jin Q, Zhong J, Li YP, Yang W. 2016. Identification of a potent and broad-spectrum hepatitis C virus fusion inhibitory peptide from the E2 stem domain. Sci Rep 6:25224. doi: 10.1038/srep25224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lavillette D, Pecheur EI, Donot P, Fresquet J, Molle J, Corbau R, Dreux M, Penin F, Cosset FL. 2007. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J Virol 81:8752–8765. doi: 10.1128/JVI.02642-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wahid A, Helle F, Descamps V, Duverlie G, Penin F, Dubuisson J. 2013. Disulfide bonds in hepatitis C virus glycoprotein E1 control the assembly and entry functions of E2 glycoprotein. J Virol 87:1605–1617. doi: 10.1128/JVI.02659-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cocquerel L, Op de Beeck A, Lambot M, Roussel J, Delgrange D, Pillez A, Wychowski C, Penin F, Dubuisson J. 2002. Topological changes in the transmembrane domains of hepatitis C virus envelope glycoproteins. EMBO J 21:2893–2902. doi: 10.1093/emboj/cdf295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dubuisson J, Hsu HH, Cheung RC, Greenberg HB, Russell DG, Rice CM. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J Virol 68:6147–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Helle F, Vieyres G, Elkrief L, Popescu CI, Wychowski C, Descamps V, Castelain S, Roingeard P, Duverlie G, Dubuisson J. 2010. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J Virol 84:11905–11915. doi: 10.1128/JVI.01548-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tong Y, Zhu Y, Xia X, Liu Y, Feng Y, Hua X, Chen Z, Ding H, Gao L, Wang Y, Feitelson MA, Zhao P, Qi ZT. 2011. Tupaia CD81, SR-BI, claudin-1, and occludin support hepatitis C virus infection. J Virol 85:2793–2802. doi: 10.1128/JVI.01818-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gastaminza P, Kapadia SB, Chisari FV. 2006. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J Virol 80:11074–11081. doi: 10.1128/JVI.01150-06. [DOI] [PMC free article] [PubMed] [Google Scholar]