

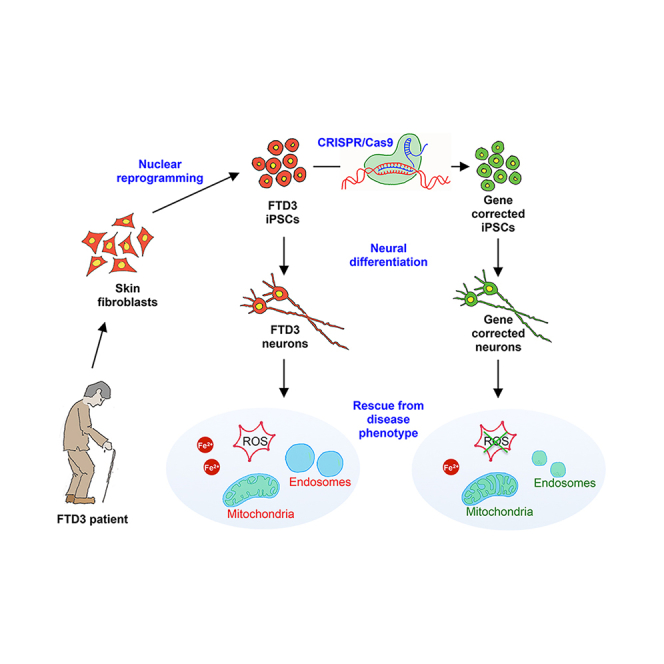
Summary
The truncated mutant form of the charged multivesicular body protein 2B (CHMP2B) is causative for frontotemporal dementia linked to chromosome 3 (FTD3). CHMP2B is a constituent of the endosomal sorting complex required for transport (ESCRT) and, when mutated, disrupts endosome-to-lysosome trafficking and substrate degradation. To understand the underlying molecular pathology, FTD3 patient induced pluripotent stem cells (iPSCs) were differentiated into forebrain-type cortical neurons. FTD3 neurons exhibited abnormal endosomes, as previously shown in patients. Moreover, mitochondria of FTD3 neurons displayed defective cristae formation, accompanied by deficiencies in mitochondrial respiration and increased levels of reactive oxygen. In addition, we provide evidence for perturbed iron homeostasis, presenting an in vitro patient-specific model to study the effects of iron accumulation in neurodegenerative diseases. All phenotypes observed in FTD3 neurons were rescued in CRISPR/Cas9-edited isogenic controls. These findings illustrate the relevance of our patient-specific in vitro models and open up possibilities for drug target development.
Keywords: frontotemporal dementia linked to chromosome 3 (FTD3), CHMP2B, iPSC-derived neuron, disease modeling, endosome, mitochondria, iron homeostasis, oxidative stress, neurodegeneration
Graphical Abstract
Highlights
-
•
FTD3 neurons show abnormalities in endosomes and mitochondria
-
•
Parkinson's and Alzheimer's disease core genes are altered in FTD3 neurons
-
•
Iron homeostasis is perturbed in FTD3 neurons
-
•
Impairments in FTD3 neurons are rescued in CRISPR/Cas9-edited isogenic controls
In this article, Freude, Zhang, and colleagues describe a patient iPSC-derived neuronal model for FTD3. This cellular model shows endosome abnormalities previously reported in patients. Furthermore, it provides insights into the role of impaired mitochondria function and imbalanced iron homeostasis in FTD3 pathology. All observed phenotypes were rescued in CRISPR/Cas9-edited isogenic controls.
Introduction
Frontotemporal dementia (FTD) is the second most common form of early-onset (<65 years) dementia, accounting for 5%–8% of total dementia cases. FTD is a neurodegenerative disorder with cognitive impairment affecting the frontal and/or temporal lobes of the brain associated with progressive brain atrophy (Rossor et al., 2010). FTD is clinically, neuropathologically, and genetically heterogeneous. One gene affected in familial cases is the charged multivesicular body protein 2B (CHMP2B) located on chromosome 3 (FTD3). Patients display global cortical and central brain atrophies, with no apparent amyloid plaque formation or conclusive hyperphosphorylated tau aggregates (Isaacs et al., 2011). Early behavioral changes include apathy, restlessness, disinhibition, and hyperorality. Late-stage behavioral changes include stereotype behavior, mutism, and dystonia (Isaacs et al., 2011). CHMP2B is a component of the endosomal sorting complex required for transport III (ESCRT-III) complex, which facilitates recycling or degradation of cell surface receptors (Chassefeyre et al., 2015). As such, the FTD3-causing mutation of CHMP2B affects functionality of the endosome. Mouse and Drosophila FTD3 animal models have yielded valuable in vivo insights into the dysfunction of the endosomal lysosomal system and pathologic progression (Ahmad et al., 2009, Ghazi-Noori et al., 2012). However, transgene integration and species-specific differences may contribute to observed phenotypes in such models. Hence, there is an emerging need for human FTD3 models. In addition, studying how CHMP2B, as a rare mutation, contributes to neurodegenerative disorders has not yet attracted broader attention. Consequently, a CHMP2B mutant cellular model could provide further insights into common underlying dysfunction of biological pathways, disrupted or disturbed in neurodegenerative diseases and thereby linking different forms of neurodegeneration.
The availability of viable neurons from patient brains, at least in part, limits the investigation of the mechanism of neurodegenerative pathogenesis. In this context, human-induced pluripotent stem cells (iPSCs) provide invaluable access to study the disease progression in neurons derived from patient iPSCs and facilitate the development of new therapies (Ehrlich et al., 2015, Rasmussen et al., 2014). Meanwhile, recent advances of state-of-the-art genome engineering technique CRISPR/Cas9 (Ran et al., 2013) have had a tremendous impact allowing for gene correction in patients who are carriers of disease-causing single-point mutations. Such genetically edited iPSCs are ideal isogenic controls for the patient-derived iPSCs, allowing to precisely dissect the significance of the disease-causing mutation while maintaining the patient's own genetic background.
To study the cellular and molecular events of FTD3, we derived a disease model using human iPSCs from patients carrying the 31449G > C mutation in CHMP2B and isogenic controls generated via the CRISPR/Cas9 system with subsequent in vitro neuronal differentiation. Our model not only confirmed partly prior-observed pathologies in animal models such as endosome dysregulation (Ghazi-Noori et al., 2012), but also revealed additional disease-relevant phenotypes in FTD3 by way of abnormal mitochondrial ultrastructure and function. Moreover, RNA sequencing (RNA-seq) uncovered that the CHMP2B mutation manifests in dysregulated expression of key genes associated with Parkinson’s disease (PD) and Alzheimer’s disease (AD) and indicated that perurbed iron homeostasis is one of the underlying disease mechanisms.
Results
Generation of FTD3 iPSCs and Genetically Corrected Isogenic Controls
Skin fibroblasts from two symptomatic FTD3 patients (H150 and H151) and one presymptomatic FTD3 patient (H242) were reprogrammed using non-integrative episomal plasmids. All of these individuals are from the same family, and one independent healthy control from an unrelated family was also included. Individual FTD3 iPSC lines were assessed for pluripotency features via qPCR (Figure S1A) and immunocytochemistry (ICC) (Figure S1B). Assessment of pluripotency potential was confirmed via embryoid body formation and subsequent spontaneous differentiation (Figure S1C). The FTD3-causing mutation 31449G > C (located at the splice acceptor site in CHMP2B, at the border of intron 5 and exon 6) was repaired in the FTD3 iPSC lines using the CRISPR/Cas9 system (Figure 1A). The presence of altered CHMP2B mRNA, as formerly shown in Urwin et al. (2010) was confirmed via qPCR and showed expression of the intron 5 and delta-10 isoforms only in the patient iPSC. These abnormal transcripts, generated via the CHMP2B mutation affecting the slice acceptor site, are completely absent in the isogenic controls (Figure 1A). All iPSC and genetically corrected isogenic lines were determined to be karyotypically normal (Figure S1D).
Figure 1.

Gene Correction of FTD3 iPSCs, In Vitro Neuronal Differentiation, and Morphometric Analyses
(A) CRISPR/Cas9-mediated gene correction of CHMP2B 31449G > C, ssODN with silent mutation (SacII), sequencing results, and quantification of CHMP2B mutant and wild-type mRNA transcripts. The expression values were calculated as relative amount of mRNA versus expression values of wild-type which was set to 100%.
(B) Schematic of neuronal differentiation protocol (see Experimental Procedures for details). Scale bar, 200 μm for all. BFGF, basic fibroblast growth factor; GDNF, glial cell line-derived neurotrophic factor; cAMP, cyclic AMP.
(C) Quantification of TUJ1, VGlut1, and FOXG1 mRNA expression levels in FTD3 neurons (H150, H151, H242) and isogenic controls (H150 ISO, H151 ISO, H242 ISO). n = 3 with three replicates in each experiment, no significant differences were found among the different lines.
(D) Morphometric analyses in FTD3 neurons and isogenic controls: Average neurite length and soma size. n = 3 with three replicates in each experiment, significant differences are indicated by ∗p < 0.05 and ∗∗∗p < 0.001.
Differentiation of FTD3 iPSCs to Forebrain Cortical Neurons
We employed a modified dual SMAD protocol (Figure 1B) to generate forebrain-specific cortical neurons. Flow cytometry analyses demonstrated the neural progenitor cell (NPC) cultures to be comparable with respect to NESTIN and SOX1 expression (Figure S2A). All NPCs expressed the forebrain markers PAX6 and OTX2 (Figure S2B). ICC revealed the majority of the matured neurons to be glutamatergic (immunopositive for VGlut1 [Figure S2C] and co-positive for MAP2, TAU, and TUJ1 [Figure S2C]). Approximately 10% of neurons were GABAergic (Figure S2C), while approximately 5% were GFAP-positive astrocytes, with a minor population (2%–3%) comprising tyrosine hydroxylase-positive dopaminergic neurons (Figure S2C). Since quantification via ICC is challenging we performed qPCR for TUJ1, VGlut1, and FOXG1 in order to estimate the comparability in regard to the neural cell composition of the investigated FTD3 iPSC- and isogenic iPSC-derived neurons. All lines showed comparable amounts of expression with no significant differences, which is also indicative that the mutation in CHMP2B does not affect the differentiation potential toward neurons (Figure 1C). Neurons exhibited spiking activity and action potentials, which occurred both spontaneously and under current-clamp conditions (Figures S3B–S3D). Measurement of intracellular calcium levels also demonstrated that cultures exhibited neuronal functionality. The neurons were responsive to the neurotransmitters glutamate/glycine, GABA, and, to a lesser extent, acetylcholine (Figure S3A), and depolarization of the plasma membrane by addition of potassium resulted in a small increase in intracellular calcium (Figure S3A). Serotonin and dopamine did not induce a change in intracellular calcium levels. All lines showed similar responses.
FTD3 Neurons Display Aberrant Endosomes
Via transmission electron microscopy, we observed similarly enlarged endosome (assessed as endosome-occupied relative neuronal area, see the Supplemental Experimental Procedures) in our FTD3 neurons, as previously demonstrated in patients (Urwin et al., 2010) (Figure 2A). Such enlarged endosomes were absent from the isogenic controls, indicating a rescue of the phenotype by CRISPR/Cas9-mediated gene correction and is consistent with previous knockdown experiments (Nielsen et al., 2012). Specifically, the electron-lucent and, thus, vacant endosomes were significantly more abundant in the FTD3 neurons (Figures 2B and 2C), indicative of an effect on early-stage endosomes.
Figure 2.

FTD3 Neurons Exhibit Aberrant Endosome and Mitochondria
(A and B) Stereology and ultrastructure of endosome in FTD3 neurons (red bar), isogenic controls (gray bar), and an independent healthy control (black bar): (A) shows total size of endosome; (B) shows the ratio of endosome to cytoplasm and further subdivision by appearance into electron-lucent (Endo L), medium electron-dense (Endo M), and electron-dense (Endo D); n = 3 with three replicates in each experiment, significant differences are indicated by ∗p < 0.05. n.s., not significant.
(C) Transmission electron microscopy (TEM) images of Endo L, Endo M, and Endo (D) Scale bar, 1 μm for all.
(D) Total number of endosome-associated genes and the top ten differentially expressed genes (on the basis of fold-change) within a false discovery rate (FDR) of 5%.
(E) Stereology and ultrastructure of mitochondria in FTD3 neurons (red bar), isogenic controls (gray bar), and an independent healthy control (black bar): relative size of mitochondria compared with cytoplasmic area of all mitochondria (Mito total) and cristaeless mitochondria; n = 3 with three replicates in each experiment, significant differences are indicated by ∗∗p < 0.01, and ∗∗∗p < 0.001.
(F) TEM images of normal and cristaeless mitochondria. Scale bar, 1 μm for both.
(G) Images of neurons dyed with the mitochondrial probe MitoTracker (red) counterstained with DAPI (blue). Arrowheads indicate dyed mitochondria and the dashed line shows a representative distribution of mitochondria in an isogenic control neuron. Scale bar, 10 μm for all.
(H) Total number of mitochondria-associated genes and top ten differentially expressed genes (on the basis of fold-change) within an FDR of 5%.
FTD3 Neurons Display Abnormal Mitochondria
Given that mitochondrial defects are commonly observed in neurodegenerative diseases, we assessed whether such a phenotype is evident in FTD3 neurons. While we noted no apparent difference in size of mitochondria between FTD3 neurons and their isogenic controls, we did observe a significant increase in abnormal mitochondria with severe impairment of cristae formation in FTD3 neurons (Figures 2E and 2F). Strikingly, the distribution of mitochondria within neurons was also perturbed in FTD3 neurons: MitoTracker analyses revealed mitochondria in FTD3 neurons to preferentially localize to the perinuclear region, while isogenic controls exhibited a homogeneous distribution throughout axons and dendrites (Figure 2G). Consistent with previous studies correlating a lack of axonal mitochondria with axonal degradation (Rawson et al., 2014), we also observed disorganized microtubule organization in FTD3 neurons (Figure S4A). These findings were substantiated by the fact that the average neurite length and soma size were significantly reduced in FTD3 neurons versus isogenic controls (Figure 1D).
FTD3 Neurons Display Impaired Mitochondria Function and Increased Oxidative Stress
Prompted by our observations of aberrant mitochondrial morphology, we sought to test the functionality of such FTD3 mitochondria by assessing their basal respiration rate, ATP-linked respiration, proton leak, and reserve capacity. Our analyses uncovered a reduced basal respiration rate in FTD3 neurons as well as a reduced maximal capacity (Figure 3E). Consistent with mitochondrial dysfunction manifesting in oxidative stress, we observed up to 20% occurrence of oxidative stress in FTD3 neurons compared with only 7% in isogenic controls (Figure 3A). Thus, FTD3 neurons are exposed to higher levels of oxidative stress, and targeted gene editing can reverse the disease phenotype. Subsequently, we investigated whether the increased oxidative stress impacts cell viability, but we detected no evidence for increased apoptosis in FTD3 neurons (Figure 3B).
Figure 3.

Impaired Mitochondrial Function and Oxidative Stress in FTD3 Neurons
(A) CellROX green and SYTOX red flow cytometry assay of live oxidative stressed neurons. n = 3 with three replicates in each experiment, significant differences are indicated by ∗∗p < 0.01 and ∗∗∗p < 0.001.
(B) CellEvent Caspase-3/7 green and SYTOX red flow cytometry assay of apoptotic neurons.
(C) Total number of oxidative stress-associated genes and the top ten differentially expressed genes (on the basis of fold-change) within an FDR of 5%. n.s., not significant.
(D and E) Mitochondrial function profile of FTD3 neurons (H150, H151 and H242) and isogenic controls (H150 ISO, H151 ISO, H242 ISO). (D) oxygen consumption rate (OCR) was measured before (basal) and after the addition of oligomycin (oligom), carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), and a mixture of rotenone (rot) and antimycin A (antimA), respectively; (E) mitochondrial respiration parameters were calculated based on the OCR measurements shown in (D) as described in the Supplemental Experimental Procedures. n = 3 with three replicates in each experiment, significant differences are indicated by ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Global Gene Expression Analyses Confirm Defects in the Endosome, Mitochondrial Dysfunction, and Increased Oxidative Stress
To obtain an overview of gene expression changes in CHMP2B-dependent neurodegeneration, we performed RNA-seq analyses, which revealed 3,205 differentially expressed genes in FTD3 neurons compared with their isogenic controls. Heatmap analyses showed that neurons derived from iPSCs of all three FTD3 individuals clustered together, while isogenic controls clustered together with the independent healthy individual (Figure 4A), suggesting that the isogenic control lines reverted to a healthy control gene expression profile after repair of the CHMP2B mutation. To identify which cellular components are primarily affected in FTD3 neurons, we performed gene ontology (GO) term analysis. We found enrichment of the GO term for postsynaptic membrane, synaptic membrane, neuron projection, and synaptic vesicle membrane (Figure 4C), and depletion of early endosome membrane, lysosomal lumen, and endocytic vesicle membrane (Figure 4D).
Figure 4.

Transcriptional Analysis of FTD3 Neurons Versus Isogenic Controls and Quantification of Intracellular Fe2+ Content
(A) Heatmap and hierarchical clustering analysis of gene expression profile of FTD3 neurons (H150, H151, H242) and isogenic controls (H150 ISO, H151 ISO, H242 ISO) as well as one independent healthy control (H256).
(B) qPCR validation of genes of interest from RNA-seq results and western blot relative quantification of TRPC6 protein expression.
(C and D) GO term enrichment analysis of differentially expressed genes via ClueGO v1.4 software. Functionally grouped network with terms as nodes linked based on their kappa score level (≥0.5), where only the label of the most significant term per group is shown. The node size represents the term enrichment significance. Functionally related groups partially overlap.
(E) Diagram of key dysregulated genes involved in intracellular iron homeostasis.
(F) Fe2+ assay and concentration normalized to total cellular protein.n = 3 with three replicates in each experiment, significant differences are indicated by ∗p < 0.05.
With the aim to identify additional genes associated with the main phenotypes observed in FTD3 neurons, we extracted the top ten differentially regulated genes functionally linked to neurodegeneration and associated with endosomes (Figure 2D), mitochondria (Figure 2H), and oxidative stress (Figure 3C). Strikingly, APOE ranked among the ten most downregulated endosome-associated genes (Figure 2D). APOE4, an isoform of APOE, is a well-known risk factor for sporadic AD (sAD) (Corder et al., 1993) and, consistent with a putative role in FTD3 pathophysiology, some sAD patients present with enlarged early endosomes (Cataldo et al., 2000).
Most interestingly, we observed an upregulation of leucine-rich repeat kinase 2 (LRRK2) among the mitochondria-related genes (Figure 2H). LRRK2 is one of the five genes mutated in familial forms of PD. Previous studies reported mitochondrial fragmentation due to increased expression of LRRK2 (Wang et al., 2012), pointing to an association between upregulated LRRK2 expression and cristae dysgenesis in FTD3 neurons. LRRK2 was also detected in the oxidative stress group (Figure 3C) and is upregulated in some cases of sporadic PD (Cho et al., 2013) accompanied by elevated levels of oxidative stress (Nguyen et al., 2011). MPO and APOD, both implicated in AD (Figure 3C) (Maki et al., 2009, Martinez et al., 2012), were strongly downregulated in FTD3 neurons.
Imbalance of Iron Homeostasis Represents a Unifying Factor Linking Endosome, Mitochondrial Dysfunction, and Oxidative Stress
Via our in silico analyses, we identified mis-expression of genes encoding proteins or receptors known to be involved in iron homeostasis. Among these were TRPC6 (12-fold upregulated), HFE (8-fold downregulated), and ABCG2, HP, and CP (all 2-fold downregulated) (Figure 4B). A potential imbalance of iron homeostasis might plausibly induce intracellular ferrous iron (Fe2+) accumulation, damaging mitochondria and inducing stress, to be further exacerbated by the defects in the endosome compartment contributing to increased amounts of trapped Fe3+ within endosomes (Figure 4E). Supporting this hypothesis, cytoplasmic Fe2+ levels are significantly increased in FTD3 neurons versus isogenic control neurons (Figure 4F). This increase in intracellular iron was consistent in several independent neural differentiation experiments, and the increased expression in TRCP6 was also confirmed via western blot analysis (Figure 4B), further substantiating our hypothesis for impaired iron homeostasis triggered by altered expression and protein amounts of key regulators.
Taken together we have shown that FTD3 neurons have enlarged early endosomes, display mitochondria dysfunction and perturbed iron homeostasis, with subsequent increase in oxidative stress.
Discussion
The biological function of CHMP2B has primarily been investigated in animal models, with the attendant caveat that observed phenotypes are potentially species specific or contributed to by transgene integration. Here, to preclude such issues, we developed an FTD3 patient-derived disease model by way of iPSCs generation, genome editing, and in vitro neuronal differentiation techniques.
We identified enlargements of particularly the early endosome population in FTD3 neurons, indicating that perturbed endo-lysosomal fusion and degradation of substrates is key to the underlying FTD3 pathophysiology. Cellular components, including mitochondria, are degraded in the endosome; in amyotrophic lateral sclerosis, a progressive lysosomal deficit is causative for impaired degradation of damaged mitochondria in motor neurons (Xie et al., 2015). Defective recycling of large organelles, such as mitochondria, is the earliest detectable impairment in a chain of events initiated by defective recycling (Cannizzo et al., 2012), which clearly links endosome and mitochondrial dysfunction.
FTD3 neurons displayed abnormal mitochondrial localization and cristae formation, manifesting in impaired functionality typical for neurodegenerative diseases. Defective mitochondria show impaired ATP generation and release of deleterious chemicals such as reactive oxygen species (ROS), potentially inducing cell death. We contend that increased ROS release contributes to increased organelle damage, which cannot be appropriately ameliorated through degradation owing to the observed endosome defects, resulting in a self-perpetuating cycle of cellular damage.
Strikingly, in FTD3 neurons we identified a robust, significant upregulation of LRRK2 encoding the dardarin protein. One function of dardarin is the modulation of intracellular trafficking via recruitment of RAB7 (Greggio et al., 2006). Mutant CHMP2B impairs the recruitment of RAB7, crucial for endosome-lysosome fusion (Urwin et al., 2010). This relationship between CHMP2B, RAB7, and dardarin is supported by studies showing that dardarin negatively regulates RAB7-dependent localization of the lysosome (Dodson et al., 2012). Therefore, upregulation of LRRK2 as a consequence of CHMP2B mutation might contribute to deficient endo-lysosomal trafficking and fusion. Dardarin also plays a role in mitochondrial functionality and positioning, since induced expression of wild-type LRRK2 induces mitochondrial fragmentation via increased fission and upregulation of DLP1, which is critical for correct mitochondrial distribution (Wang et al., 2012).
Furthermore, our analysis revealed APOE to be significantly downregulated in FTD3 neural cells. The cells analyzed via RNA-seq are a mixed population of neurons and glial cells, with an up to 90% enrichment of VGlut1+ neurons. Due to this mixed population of cells we were still able to detect APOE via RNA-seq, even though it is mainly expressed in astrocytes. APOE is a major cholesterol carrier in neurons that supports lipid transport and brain injury repair, and there is a key requirement for APOE in facilitating the clearance of soluble amyloid beta (Abeta) (Jiang et al., 2008). Furthermore, APOE suppresses microglia activation (Laskowitz et al., 2001), and reduced APOE levels might potentially trigger inflammatory events. Together, this suggests that downregulation of APOE in FTD3 neurons might be a contributing factor in the microglia activation seen in postmortem FTD3 brains. Interestingly, we observed another phenotype restricted to glial cells. It has previously been reported for FTD3 mouse models and patient brains that these display p62 inclusions, which are indicative for impaired autophagy, oxidative stress, and neurotoxicity (Ghazi-Noori et al., 2012, Holm et al., 2007). We identified these p62 inclusions only in the S100β+ glial cell population and not in our TUJ1+ neurons (Figure S4B). Both the APOE and the p62 phenotypes clearly show that, even though several disease features can be replicated in our FTD3 neurons, other neural cell types need to be investigated in the future to understand the full complexity and pathophysiology of the disease.
Based on the differentially expressed key components and increase of intracellular iron in FTD3 neurons, we present an additional pathological feature of FTD3, which is associated with an imbalance in iron homeostasis (Figure 4E). Iron uptake principally proceeds through iron bound to transferrin mediated via the transferrin receptor or via direct uptake of Fe2+ facilitated by a variety of receptors including TRPC6 (Mwanjewe and Grover, 2004). We propose that upregulation of TRPC6 manifests in increased uptake of Fe2+, while downregulation of HFE (which competes with Tf), results in uptake of more ferric iron which can further be reduced into Fe2+. In contrast, export of heme-bound iron via ABCG2 or unbound Fe2+ through FPN can be reduced. Concordant with this model, we confirmed in several individual differentiation experiments a small but significant increase of intracellular iron within 5 weeks of neuronal differentiation in FTD3 neurons. Excessive amounts of intracellular Fe2+ are toxic to cells and trigger the production of ROS. Moreover, Fe2+ and hydrogen peroxide engage in the so-called Fenton reaction, producing ferric iron and highly reactive hydroxide, which damages DNA, proteins, and lipids in the cell (Altamura and Muckenthaler, 2009).
FTD3 neurons present with enlarged early endosomes, increased cristaeless mitochondria, intracellular iron accumulation, and oxidative stress. All of these phenotypes were rescued via genetic correction of the CHMP2B mutation.
We propose that in FTD3 neurons two events occur in parallel: (1) mutant CHMP2B causes an imbalance in iron homeostasis triggering mitochondrial impairment, which consequently triggers ROS and neuronal damage, and (2) mutant CHMP2B-dependent enlargement of endosomes, which causes defects of the endo-lysosomal degradation and recycling pathway, including recycling of receptors for iron uptake, results in accumulation of defective mitochondria, imbalanced iron homeostasis, ROS, and neuronal damage. Future studies will determine the exact role of mutant CHMP2B on a molecular level within these cellular events.
In conclusion, our findings indicate a possibility of targeting iron imbalances therapeutically in efforts to intervene in this cycle of damage to attempt treatment of FTD3. Furthermore, the CHMP2B-dependent human iPSC-derived neurons can be added to the list of in vitro cellular model systems that serve as a tool to study not only common mechanisms of pathogenesis, such as mitochondrial defects and oxidative stress, but also the effects of iron accumulation in neurodegenerative diseases.
Experimental Procedures
Cell Culture, iPSC Generation, and Neuronal Differentiation
iPSCs were generated via episomal reprogramming (Addgene, 27077, 27078, 27080) from human dermal fibroblasts obtained from skin biopsies. Neuronal differentiation followed a modified dual SMAD protocol.
Genome Editing
Generation of isogenic controls from the three FTD3 iPSC lines was achieved via the CRISPR/Cas9 system (Addgene, 62988) using single-strand donor oligonucleotides as a template.
Transmission Electron Microscopy
Cells were seeded on 13 mm Thermanox plastic coverslips (Nunc, 174950) coated with poly-O-Lysine/laminin. Ultra-thin (50–70 nm) sections were prepared, contrasted with 2% uranyl acetate (PolyScience, 21447) and lead citrate, and examined using a Philips CM100 transmission electron microscope equipped with a Morada camera.
RNA-Seq Analysis
For deep RNA-seq, libraries of FTD3 neurons, isogenic controls, and an independent healthy control were prepared and sequenced on a HiSeq 2000 Sequencing System (Illumina) following the manufacturer's protocol.
Statistical Analysis
Data are presented as mean ± SE. n is the number of independent differentiations of NPCs (from the cryobank) to neurons. Significance of data was evaluated by Student's t test or two-way ANOVA with Bonferroni post hoc test. Unless noted otherwise, p < 0.05 was considered statistically significant.
For further details on Experimental Procedures, see the Supplemental Information.
Author Contributions
Y.Z. and K.K.F. designed the study. Y.Z., B.S., N.K.N., M.A.R., B.I.A., M.A., K.C., T.C.S., H.M.L., T.T.N., J.H., F.X., X.L., L.B., M.M., L.K.B., H.S.W., Y.L., J.E.N., The FReJA Consortium, B.H., C.C., P.H., and K.K.F. conducted the experiments and interpreted the results. Y.Z. and K.K.F. wrote the manuscript.
Acknowledgments
This work was supported by awards from: EU FP7 Marie Curie Industry-Academia Partnerships and Pathways (IAPP) grant (STEMMAD, PIAPP-GA-2012-324451), Innovation Fund Denmark (BrainStem, 4108-00008B), Lundbeck Foundation (R151-2013-14439) (L.B.), Danish Research Council for Independent Research (DFF-1337-00128 and DFF-1335-00763) (Y.L.), China Scholarship Council (Y.Z.), and Ministry of Science, Technology and Innovation of Mexico (B.I.A.). We are grateful to the FTD3 family for their support of this work. Finally, we would like to thank Ms. Hanne Holm and Tina Christoffersen from the University of Copenhagen, Ms. Ulla Poulsen from Bioneer A/S, and Mr. Lingfei Ye from BGI-Shenzhen for expert technical assistance.
Published: February 16, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.01.012.
Contributor Information
Yu Zhang, Email: yu.zhang@sund.ku.dk.
Kristine K. Freude, Email: kkf@sund.ku.dk.
Accession Numbers
The RNA-seq data described in this study have been deposited in the NCBI GEO database under accession number GEO: GSE92340.
Supplemental Information
References
- Ahmad S.T., Sweeney S.T., Lee J.A., Sweeney N.T., Gao F.B. Genetic screen identifies serpin5 as a regulator of the toll pathway and CHMP2B toxicity associated with frontotemporal dementia. Proc. Natl. Acad. Sci. USA. 2009;106:12168–12173. doi: 10.1073/pnas.0903134106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altamura S., Muckenthaler M.U. Iron toxicity in diseases of aging: Alzheimer's disease, Parkinson's disease and atherosclerosis. J. Alzheimers Dis. 2009;16:879–895. doi: 10.3233/JAD-2009-1010. [DOI] [PubMed] [Google Scholar]
- Cannizzo E.S., Clement C.C., Morozova K., Valdor R., Kaushik S., Almeida L.N., Follo C., Sahu R., Cuervo A.M., Macian F. Age-related oxidative stress compromises endosomal proteostasis. Cell Rep. 2012;2:136–149. doi: 10.1016/j.celrep.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo A.M., Peterhoff C.M., Troncoso J.C., Gomez-Isla T., Hyman B.T., Nixon R.A. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000;157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassefeyre R., Martinez-Hernandez J., Bertaso F., Bouquier N., Blot B., Laporte M., Fraboulet S., Coute Y., Devoy A., Isaacs A.M. Regulation of postsynaptic function by the dementia-related ESCRT-III subunit CHMP2B. J. Neurosci. 2015;35:3155–3173. doi: 10.1523/JNEUROSCI.0586-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H.J., Liu G., Jin S.M., Parisiadou L., Xie C., Yu J., Sun L., Ma B., Ding J., Vancraenenbroeck R. MicroRNA-205 regulates the expression of Parkinson's disease-related leucine-rich repeat kinase 2 protein. Hum. Mol. Genet. 2013;22:608–620. doi: 10.1093/hmg/dds470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder E.H., Saunders A.M., Strittmatter W.J., Schmechel D.E., Gaskell P.C., Small G.W., Roses A.D., Haines J.L., Pericak-Vance M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Dodson M.W., Zhang T., Jiang C., Chen S., Guo M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 2012;21:1350–1363. doi: 10.1093/hmg/ddr573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M., Hallmann A.L., Reinhardt P., Arauzo-Bravo M.J., Korr S., Ropke A., Psathaki O.E., Ehling P., Meuth S.G., Oblak A.L. Distinct neurodegenerative changes in an induced pluripotent stem cell model of frontotemporal dementia linked to mutant TAU protein. Stem Cell Rep. 2015;5:83–96. doi: 10.1016/j.stemcr.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazi-Noori S., Froud K.E., Mizielinska S., Powell C., Smidak M., de Marco M.F., O'Malley C., Farmer M., Parkinson N., Fisher E.M.C. Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain. 2012;135:819–832. doi: 10.1093/brain/aws006. [DOI] [PubMed] [Google Scholar]
- Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., van der Brug M.P., Beilina A., Blackinton J., Thomas K.J. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Holm I.E., Englund E., Mackenzie I.R., Johannsen P., Isaacs A.M. A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J. Neuropathol. Exp. Neurol. 2007;66:884–891. doi: 10.1097/nen.0b013e3181567f02. [DOI] [PubMed] [Google Scholar]
- Isaacs A.M., Johannsen P., Holm I., Nielsen J.E. Frontotemporal dementia caused by CHMP2B mutations. Curr. Alzheimer Res. 2011;8:246–251. doi: 10.2174/156720511795563764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q., Lee C.Y., Mandrekar S., Wilkinson B., Cramer P., Zelcer N., Mann K., Lamb B., Willson T.M., Collins J.L. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowitz D.T., Thekdi A.D., Thekdi S.D., Han S.K., Myers J.K., Pizzo S.V., Bennett E.R. Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp. Neurol. 2001;167:74–85. doi: 10.1006/exnr.2001.7541. [DOI] [PubMed] [Google Scholar]
- Maki R.A., Tyurin V.A., Lyon R.C., Hamilton R.L., DeKosky S.T., Kagan V.E., Reynolds W.F. Aberrant expression of myeloperoxidase in astrocytes promotes phospholipid oxidation and memory deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2009;284:3158–3169. doi: 10.1074/jbc.M807731200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez E., Navarro A., Ordonez C., Del Valle E., Tolivia J. Amyloid-beta25-35 induces apolipoprotein D Synthesis and growth arrest in HT22 hippocampal cells. J. Alzheimers Dis. 2012;30:233–244. doi: 10.3233/JAD-2012-112102. [DOI] [PubMed] [Google Scholar]
- Mwanjewe J., Grover A.K. Role of transient receptor potential canonical 6 (TRPC6) in non-transferrin-bound iron uptake in neuronal phenotype PC12 cells. Biochem. J. 2004;378:975–982. doi: 10.1042/BJ20031187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H.N., Byers B., Cord B., Shcheglovitov A., Byrne J., Gujar P., Kee K., Schule B., Dolmetsch R.E., Langston W. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 2011;8:267–280. doi: 10.1016/j.stem.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen T.T., Mizielinska S., Hasholt L., Isaacs A.M., Nielsen J.E. Reversal of pathology in CHMP2B-mediated frontotemporal dementia patient cells using RNA interference. J. Gene Med. 2012;14:521–529. doi: 10.1002/jgm.2649. [DOI] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M.A., Holst B., Tumer Z., Johnsen M.G., Zhou S.L., Stummann T.C., Hyttel P., Clausen C. Transient p53 suppression increases reprogramming of human fibroblasts without affecting apoptosis and DNA damage. Stem Cell Rep. 2014;3:404–413. doi: 10.1016/j.stemcr.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson R.L., Yam L., Weimer R.M., Bend E.G., Hartwieg E., Horvitz H.R., Clark S.G., Jorgensen E.M. Axons degenerate in the absence of mitochondria in C. elegans. Curr. Biol. 2014;24:760–765. doi: 10.1016/j.cub.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossor M.N., Fox N.C., Mummery C.J., Schott J.M., Warren J.D. The diagnosis of young-onset dementia. Lancet Neurol. 2010;9:793–806. doi: 10.1016/S1474-4422(10)70159-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urwin H., Authier A., Nielsen J.E., Metcalf D., Powell C., Froud K., Malcolm D.S., Holm I., Johannsen P., Brown J. Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum. Mol. Genet. 2010;19:2228–2238. doi: 10.1093/hmg/ddq100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Yan M.H., Fujioka H., Liu J., Wilson-Delfosse A., Chen S.G., Perry G., Casadesus G., Zhu X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012;21:1931–1944. doi: 10.1093/hmg/dds003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y., Zhou B., Lin M.Y., Wang S., Foust K.D., Sheng Z.H. Endolysosomal deficits augment mitochondria pathology in spinal motor neurons of asymptomatic fALS mice. Neuron. 2015;87:355–370. doi: 10.1016/j.neuron.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.