

Summary
Huntington disease (HD) is a dominant neurodegenerative disorder caused by a CAG repeat expansion in HTT. Here we report correction of HD human induced pluripotent stem cells (hiPSCs) using a CRISPR-Cas9 and piggyBac transposon-based approach. We show that both HD and corrected isogenic hiPSCs can be differentiated into excitable, synaptically active forebrain neurons. We further demonstrate that phenotypic abnormalities in HD hiPSC-derived neural cells, including impaired neural rosette formation, increased susceptibility to growth factor withdrawal, and deficits in mitochondrial respiration, are rescued in isogenic controls. Importantly, using genome-wide expression analysis, we show that a number of apparent gene expression differences detected between HD and non-related healthy control lines are absent between HD and corrected lines, suggesting that these differences are likely related to genetic background rather than HD-specific effects. Our study demonstrates correction of HD hiPSCs and associated phenotypic abnormalities, and the importance of isogenic controls for disease modeling using hiPSCs.
Keywords: human induced pluripotent stem cell (hiPSC), neurodegenerative disorders, Huntington disease, genetic editing, disease modeling, disease phenotypes, transcriptional dysrgulation, CHCHD2, mitochondrial dysfunction
Graphical Abstract
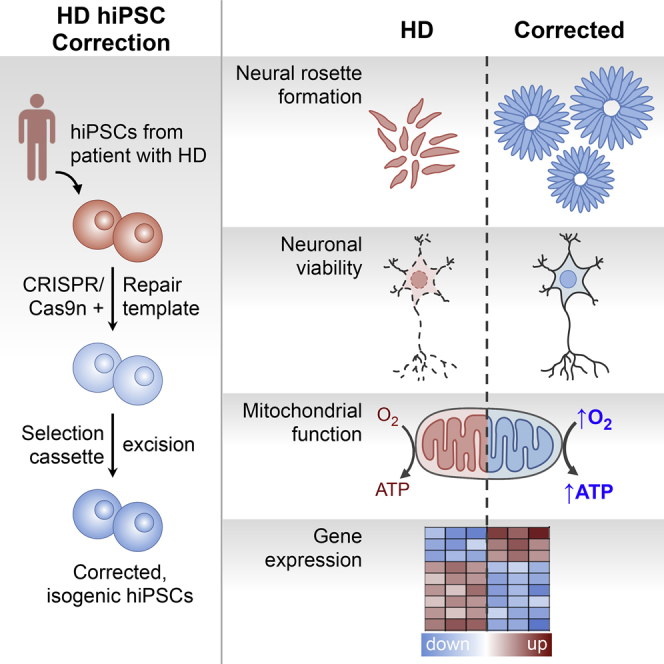
Highlights
-
•
A CRISPR-Cas9 and PiggyBac-based approach allows efficient correction of HD hiPSCs
-
•
The corrected hiPSCs can be differentiated into synaptically active neurons
-
•
Correction of HD gene mutation reverses a number of phenotypic abnormalities
-
•
Isogenic hiPSCs help distinguish mutation from genetic background-related effects
Pouladi and colleagues show that genetic correction of HD human induced pluripotent stem cells using a CRISPR-Cas9 and piggyBac transposon-based approach rescues a number of phenotypic abnormalities in derived neural cells, including increased susceptibility to growth factor withdrawal and deficits in mitochondrial respiration, and helps distinguish HD-specific from genetic background-related molecular and cellular phenotypes.
Introduction
Human induced pluripotent stem cells (hiPSCs) carry the whole genetic context of a patient and can be a virtually unlimited source of differentiated cell types of interest. Increasingly, hiPSCs are being applied for disease modeling and drug screening for a number of neurodegenerative disorders, including Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, and Huntington disease (HD) (Sterneckert et al., 2014). Indeed, a number of valuable disease phenotypes have been uncovered using differentiated neuronal subtypes of disease relevance (Ichida and Kiskinis, 2015).
However, one of the challenges of hiPSC-based disease modeling is the variability in differentiation potential due to variations in their genetic background (Kajiwara et al., 2012). This may result in inappropriate interpretation of disease phenotypes in vitro. Furthermore, the influence of genetic background on disease phenotype can be significant even for monogenic, dominant, and highly penetrant diseases such as HD. Indeed, a recent genome-wide association study identified a number of loci with putative disease-modifying variants that appear to influence the age of neurological onset in HD (GeM-HD Consortium, 2015). Therefore, the use of control hiPSC lines that are genetically identical is crucial to increase confidence in candidate disease phenotypes and mechanisms, and to minimize the chance of missing modifiable effects that are of relevance to the pathogenesis of disease.
With recent advances in genome-editing technologies, such as the zinc finger nucleases, transcription activator-like effector nuclease, and the CRISPR-Cas9 system, the establishment of isogenic control hiPSCs has become more feasible. Here, we describe the seamless correction of an HD hiPSC line. HD, the most common genetic cause of dementia, results from an expansion of a polymorphic CAG repeat tract in exon 1 of HTT (Group, 1993). Using a CRISPR-Cas9 and a piggyBac transposon-based selection system (Yusa, 2013), we corrected HD hiPSCs and established isogenic control hiPSCs with seamless excision of the selection cassette. Evaluation of the corrected lines demonstrates that a number of phenotypic abnormalities and gene expression changes in HD hiPSC-derived neural cells are rescued in isogenic controls. Our study highlights the utility of isogenic controls in distinguishing HD-specific molecular phenotypes from those related to the genetic background.
Results
Gene Correction of HD Patient-Derived hiPSCs
To correct the disease mutation in HD hiPSCs and generate isogenic control lines, we employed a CRISPR/Cas9 and piggyBac-based gene-editing approach. We chose one pair of sgRNAs (sgRNA-a and sgRNA-b) for a Cas9 nickase (Cas9n)-mediated cleavage (Ran et al., 2013) at the HTT locus to reduce off-target (OT) activity and enhance homology-dependent repair efficiency (Figure 1A). sgRNAs were cloned into Cas9n-expressing vectors and their cleavage activity was tested using a fluorescence-based surrogate reporter assay (Ramakrishna et al., 2014). Forty-eight hours after transfection, 3.28% RFP and GFP double-positive cells were detected by flow cytometry in cells co-transfected with CRISPR-Cas9n and surrogate reporter plasmids. This was 2.5-fold higher (1.3% double positive) than in cells transfected with the surrogate reporter only (Figure 1B), indicating efficient cleavage using this pair of Cas9n/sgRNAs.
Figure 1.

Correction of HD Patient-Derived hiPSCs Using piggyBac and CRISPR-Cas9
(A) Cas9 nickase gRNA binding sequences in HTT 5ʹ UTR and exon 1.
(B) Quantification of activity of HTT-targeted CRISPR-Cas9 using fluorescence-based surrogate reporter assay.
(C) Schematic depicting the donor DNA and piggyBac transposon-based selection strategy used for targeting the HTT locus.
(D) Overview of the targeting workflow, including selection, screening, excision, and rescreening.
(E) Junction PCR-based screening for successfully targeted iPSC clones.
(F) Verification of successful correction at the HTT locus in selected iPSC clones by western blotting.
(G) Summary of targeting efficiency by PCR screening.
(H) Junction PCR-based screening for selection cassette removal in targeted clones.
(I) Expression of normal HTT is maintained following removal of the selection cassette in corrected hiPSCs.
To establish isogenic controls for HD hiPSCs, we employed a piggyBac transposon (PB) selection cassette-based homologous recombination (HR) donor, which enables seamless transposase-mediated removal of the selection cassette from the targeted locus (Figure 1C). The PB selection cassette contains a puromycin-resistance gene (PuroR) for positive clone selection, and an hsvTK gene for negative selection. HD hiPSCs were transfected with the HR donor plasmid and the sgRNA-a and sgRNA-b Cas9n-expressing plasmids, followed by puromycin selection. Drug-resistant colonies were selected for further culture and screening by junction PCR (Figure 1D). Two pairs of primers were designed for HR screening (Figure 1C). Targeted clones were identified by positive PCR amplification using both primer pairs (Figure 1E). Successful correction of the mutant HTT allele was confirmed by western blot using antibodies for total HTT (MAB2166) and mutant HTT (1C2 and MW1) (Figure 1F). Of the 129 colonies screened, 14 were positive by junction PCR, and 6 of these were confirmed for correction by western blot (Figure 1G).
Because the integrated selection cassettes in targeted clones may affect HTT expression, corrected hiPSCs were transiently transfected with a PB-expressing plasmid, followed by negative selection with 0.2 μM fialuridine. Resistant colonies were screened by junction PCR, and clones with no positive PCR amplification with the F1/R1 and F2/R2 primers were determined as free of the PB selection cassette at the HTT locus (Figure 1H). Using fragment analysis, we verified that the expanded CAG tract present in the CAG180 parental line was absent in the corrected clones (Figure S1A). Furthermore, we performed Sanger sequencing analysis of the TTAA sites that flank the inverted terminal repeat sequences of the PB selection cassette and confirmed effective excision of the selection cassette (Figure S1B). Finally, using immunoblotting we verified expression of normal HTT in the corrected hiPSC clones post-excision (Figure 1I).
To investigate potential OT CRISPR/Cas9n activity, we screened ten of the top-ranked OT sites predicted by in silico analysis using the Surveyor assay (Figure S1C). Our analysis revealed no detectable mutations at all ten regions examined (Figure S1D).
To further investigate potential OT effects beyond the top-ranked sites, we performed whole-exome sequencing on three isogenic control hiPSCs and compared their sequences with that of the parental CAG180 line (Table S4). While a low number of single nucleotide variants (SNVs) were detected in each of the corrected hiPSC lines (Table S5), no single SNV was common to all three isogenic corrected lines (Table S6). This strongly suggests that the SNVs detected represent de novo mutations acquired during normal passaging of the hiPSCs and not from OT activity of the CRISPR-Cas9n. These results are consistent with previous reports indicating low OT activity following CRISPR-Cas9-mediated genome editing (Suzuki et al., 2014).
Characterization of Pluripotency in HD iPSC-Derived Corrected Isogenic hiPSCs
We then examined whether the pluripotent characteristics of the parental HD hiPSC line, previously shown to express pluripotency markers and a normal karyotype (HD iPSC Consortium, 2012), were maintained in the corrected hiPSC lines. Indeed, all corrected hiPSC clones stained positive for OCT4 (Figure 2A), and had similar mRNA levels of the pluripotency genes OCT4 and LIN28 compared with the parental HD hiPSC line, CAG180 (Figure 2B). We focused on three corrected clones, HD-C#1, HD-C#2, and HD-C#3, for further characterization. The HD-C#1 and HD-C#2 clones were assayed using the PluriTest, a genome-wide gene expression-based bioinformatic assay for pluripotency (Müller et al., 2011), and both demonstrated very high scores (Figures 2C and S2). All three corrected clones showed potential to differentiate into the three germ layers, as shown by positive immunostaining for AFP (endoderm), Tuj-1 (ectoderm), and ASM-1 (mesoderm) (Figure 2D). Furthermore, karyotyping and G-banding analysis showed that all three clones maintained a normal 46,XY karyotype (Figure 2E).
Figure 2.

Gene-Corrected hiPSCs Maintain Pluripotency and Normal Karyotype
(A–C) The gene-corrected hiPSCs maintain pluripotency as shown by (A) positive immunostaining for the pluripotency marker OCT4; (B) expression of pluripotency genes OCT4 and LIN28 (n = 6 for CAG33, 3 for CAG180 and HD-C#1–3; values for independent biological replicates shown as mean ± SEM); and (C) high pluripotency score on the PluriTest (Müller et al., 2011). Scale bar, 25 μm. See also Figure S1.
(D) The gene-corrected hiPSCs maintain the potential to differentiate into all three germ layers as shown by positive immunostaining for AFP (endoderm), Tuj-1 (ectoderm), and ASM-1 (mesoderm).
(E) Karyotyping and g-band analysis show all gene-corrected iPSC clones have a normal 46,XY karyotype.
Differentiation of HD and Isogenic Control hiPSCs into Forebrain Neural Cells
The cardinal neuropathological feature of HD is preferential loss of striatal and cortical neurons (Macdonald and Halliday, 2002, Reiner et al., 1988). Therefore, studying human forebrain neurons derived from hiPSCs may shed light on pathogenic mechanisms contributing to HD. To assess the potential of our hiPSC lines to generate neural progenitor cells (NPCs) and neurons with forebrain identity, we used previously published dual SMAD inhibition protocols with some modifications (Delli Carri et al., 2013, Maroof et al., 2013, Xu et al., 2013) (Figure 3A). After 15 days of neural induction, CAG180, isogenic control, and the non-isogenic healthy control CAG33 hiPSCs each were efficiently differentiated into forebrain NPCs, as shown by positive staining for Nestin (Figure 3B) and FOXG1 (Figure 3C). Moreover, the CNS NPC markers, PAX6 and SOX1, were also highly expressed on day 15 (Figure 3D). Forebrain NPCs were subsequently differentiated into GABAergic neurons expressing mature neuronal marker MAP2, neurotransmitter gamma-aminobutyric acid (GABA), and presynaptic marker synaptophysin (SYP) on day 48 (Figure 3E). The transcription factor NKX2.1, a marker of the human medial and lateral ganglionic eminence regions, which represent the predominant developmental origin of striatal tissue (Maroof et al., 2013, Onorati et al., 2014), was also highly elevated at 34 and 48 days of neuronal differentiation (Figure 3F). Consistent with previous reports (HD iPSC Consortium, 2012), no mutant HTT-containing aggregates were detected in neurons differentiated from HD hiPSCs (Figure S3).
Figure 3.

Differentiation of HD and Isogenic Control hiPSCs Cells into Neurons
(A) Scheme of neuronal differentiation protocol.
(B–D) Neural induction results in robust expression of neural stem cell markers (B) Nestin (day 10) and (C) FOXG1 (day 15) as assessed by immunocytochemistry, and (D) PAX6, SOX1, and FOXG1 (day 15) as measured by qRT-PCR (n = 3 per clone; values for independent biological replicates shown as mean ± SEM).
(E and F) Further differentiation results in neurons expressing (E) MAP2ab, GABA, and SYP (synaptophysin) protein at day 48 and (F) MAP2AB, SYP, NKX2.1, and GAD65 mRNA at days 34 and 48 (n = 3 per clone; values for independent biological replicates shown as mean ± SEM).
Furthermore, the differentiated neurons were found to exhibit electrical activity. First, using multi-electrode array (MEA) recordings to measure population-level electrical activity, we found that spontaneous activity, including both individual spikes and spike bursts, was evident in cultures of neurons by day 50 (Figure 4A). Application of tetrodotoxin (1 μM) completely abolished these responses (data not shown). In addition, we compared the activity of neurons cultured in two different media, N2B27 medium and BrainPhys medium (Bardy et al., 2015). Neurons cultured in BrainPhys medium were more active than those cultured in N2B27 medium (Figure S4) and, as such, were considered healthier and used in subsequent electrophysiological assessments.
Figure 4.

Electrophysiological Assessment of Neurons Differentiated from HD and Isogenic Control hiPSCs
(A) Raster plots of spike time stamps indicating spontaneous activity in differentiated neurons as measured by multi-electrode arrays. Raster plots representative of n = 3 biological replicates for CAG180 and HD-C#1 and #2, n = 2 biological replicates for CAG33. See also Figure S4.
(B) Representative image of dye-filled neuron from isogenic control hiPSCs.
(C) Action potential recordings from the isogenic control hiPSC-derived neurons shown in (B). While small current pulses evoked a single action potential (left), a larger current pulse evoked a train of action potentials (right) In both cases, the first action potential was delayed relative to the onset of the depolarizing current pulse. Resting membrane potential was −85 mV.
(D) Representative example of spontaneous action potentials (sAPs) recorded from an isogenic control hiPSC-derived neurons.
(E) Probability of observing sAPs measured in CAG33, CAG180, and HD-C#3 neurons.
(F) Frequencies of sAPs for CAG33, CAG180, and HD-C#3 neurons.
(G–I) Passive membrane properties of hiPSC-derived neurons. Similar values were observed for membrane capacitance (G), input resistance (H), and resting membrane potential (I) across all three experimental groups. Sample sizes: CAG33 = 24, CAG180 = 31, and HD-C#3 = 28.
Next, we used whole-cell patch-clamp recording techniques to measure the intrinsic electrical characteristics of these differentiated cells. Filling the neurons with fluorescent dye, via diffusion from the patch pipette, revealed that many cells had structures characteristic of neurons, namely a central cell body and many fine processes (Figure 4B). To determine whether these cells exhibited the excitability properties of neurons, depolarizing current pulses (1 s duration) were applied (Figure 4C). In hiPSC-derived CAG33 control (24 out of 26), CAG180 HD (31 out of 32), and isogenic control (HD-C#3; 28 out of 28) cells, action potentials (APs) could be evoked by depolarization. Thus, the majority of cells examined in all three hiPSC-derived cell lines were excitable, as expected for neurons. The small number of cells that did not generate APs in response to depolarizing current pulses were most likely astrocytes, which are unexcitable; these were not included in our analyses.
The majority of neurons fired APs repetitively during 1 s long depolarizing current pulses (Figure 4C, right). However, their maximum firing frequency was variable, ranging from 6 to 33 Hz. This indicates considerable heterogeneity in the types of neurons derived from the hiPSCs. In 37 of the 83 cells examined, including the example shown in Figure 4C, the minimum delay between stimulus onset and AP firing was longer than 200 ms (336 ± 17 ms; mean ± SEM). Such delayed firing is characteristic of medium spiny neurons (MSNs) (Arber et al., 2015, Klapstein et al., 2001, Nisenbaum et al., 1994), suggesting that at least some of the iPSC-derived cells had MSN-like properties. The remaining cells fired APs with much shorter delays (113 ± 7 ms). Some cells from each group also generated spontaneous APs (sAPs) (Figure 4E); an example is shown in Figure 4D. However, the frequency of sAP generation was not significantly different across the three different groups of hiPSC-derived cells (Figure 4F; p = 0.74, one-way ANOVA).
The passive membrane properties of hiPSC-derived cells also were very similar across all three groups; there were no significant differences in membrane capacitance (Figure 4G; p = 0.08, one-way ANOVA), input resistance (Figure 4H; p = 0.49), and resting membrane potential (Figure 4I; p = 0.34). In addition, the membrane properties of putative MSNs from each group were very similar (data not shown). Thus, while most hiPSC-derived cells exhibited morphological and electrical properties characteristic of neurons, any differences between the three hiPSC lines were difficult to discern as a result of the heterogeneity in the types of neurons generated.
Reversal of HD-Related Phenotypic Abnormalities in Corrected hiPSCs
Previous studies have shown that HD hiPSCs are impaired in their ability to form neural rosettes (Jeon et al., 2012). Consistent with these studies, we observed marked impairment in neural rosette formation in the CAG180 line compared with the non-isogenic CAG33 control line (Figure 5A). Correction of the HD mutation in the isogenic HD-C#1 and HD-C#2 lines rescued neural rosette formation (Figure 5A), indicating that this impairment is indeed a consequence of the HD mutation and not related to differences in genetic background.
Figure 5.

Phenotypic Abnormalities of HD Cells Are Reversed in the Corrected hiPSCs
(A) Assessment of neural rosette formation following neural induction of CAG33, CAG180, and isogenic HD-C#1 and HD-C#2 control hiPSCs. Staining with Nestin, a marker for neural stem cells, and ZO-1, a luminal neural rosette marker, reveal impaired rosette formation in the CAG180 HD line compared with the control CAG33 lines. Correction of the HD mutation in the HD-C#1 and HD-C#2 rescues the HD-related impairment in neural rosette formation. Results are representative of three independent experiments.
(B) TUNEL immunofluorescence images showing cell death in CAG180 and HD-C#1 neurons following growth factor withdrawal.
(C) Quantification of cell death as measured by TUNEL staining in CAG180 and isogenic control neurons following growth factor withdrawal. n = 3 independent biological replicates; values shown as mean ± SEM, and ns, no significance and ∗∗p < 0.01 for shown comparison, §p < 0.05 and §§§p < 0.001 relative to CAG180-GF withdrawal group, was determined by unpaired t test.
Increased cell death of neurons differentiated from HD hiPSCs following growth factor withdrawal has been reported previously (An et al., 2012, HD iPSC Consortium, 2012). Consistent with these studies, we observed increased cell death following growth factor withdrawal in neurons differentiated from HD hiPSCs, a phenotype rescued in the corrected lines (Figures 5B and 5C). The consistent sensitivity of HD neurons to growth factor withdrawal highlights the likely contribution of trophic support deficits to the pathogenesis of HD.
Analysis of Differential Gene Expression in HD and Isogenic Control hiPSCs and Differentiated NPCs
To identify transcriptional changes resulting from mutant HTT in our established human HD isogenic models, we performed a global differential gene expression analysis in hiPSC and NPC lines (Figure 6A). NPCs were differentiated from the iPSC lines using a previously published protocol (Li et al., 2011) (Figure S5A). For each cell type, all clones were grown in tandem with three biological replicates each. Global principal-component analysis (PCA) showed distinct clustering of sample groups for both hiPSCs and NPCs, with the non-isogenic CAG33 control clustering furthest from the HD CAG180 and corrected isogenic control samples (Figure 6B).
Figure 6.

Genome-wide Gene Expression Analysis of HD and Isogenic Control hiPSCs and NPCs
(A) Workflow for the transcriptional analysis performed on hiPSCs and NPCs. For each cell type, three biological replicates for HD CAG180, its isogenic corrected clones HD-C#1 and HD-C#2, and the non-isogenic CAG33 control were assayed (a total of 24 samples).
(B) 3D principal-component analysis (PCA) scatterplots for microarray data of hiPSCs (top) and NPCs (bottom) shows good clustering within replicates of the different cell lines, signifying high biological reproducibility.
(C and D) hiPSCs (C) and NPCs (D): heatmap representing relative expression levels of the DEGs common to all controls (HD-C#1, HD-C#2, CAG33) versus HD CAG180, with fold change ≥2 and corrected p < 0.05. Samples (in columns) and genes (in rows) are clustered by similarity. Shades of red represent upregulation, shades of green represent downregulation.
(E) KEGG and Pathway Commons (PC) pathway enrichment analysis of the 131 DEGs common to all controls (HD-C#1, HD-C#2, CAG33) versus HD CAG180 identified for NPCs (corrected p < 0.05). See also Figure S5B.
(F) Top: Venn diagram comparison of 240 DEGs between non-isogenic control CAG33 versus HD CAG180, and the 169 DEGs between corrected isogenic controls HD-C#1 and HD-C#2 versus HD CAG180; bottom: Venn diagram comparison of 241 DEGs between HD-C#1 versus HD CAG180, and the 241 DEGs between HD-C#2 versus HD CAG180.
(G and H) qRT-PCR validation of a subset of genes showing differential expression in CAG33 versus CAG180 that are rescued in HD-C#1 and HD-C#2 NPCs (G), and those that are not (H). n = 3 per clone; values for three independent biological replicates shown as mean ± SEM; ns, no significance; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 was determined by unpaired t test.
To identify differentially expressed genes (DEGs) in hiPSCs and NPCs, we assessed the effect of the HD mutation (HD CAG180 versus DEGs common to all control CAG33 and corrected isogenic lines; ANOVA, false discovery rate using Benjamini-Hochberg correction: 5%). We identified 159 DEGs for hiPSCs (98 upregulated and 61 downregulated) and 131 DEGs for NPCs (72 upregulated and 59 downregulated) in the controls compared with CAG180. Hierarchical clustering of DEGs resulting from the presence of the HD mutation was performed for both hiPSCs (Figure 6C) and NPCs (Figure 6D). Functional annotation of the DEGs in the differentiated NPCs using WebGestalt (Wang et al., 2013) revealed ten significantly enriched gene ontology (GO) categories, including nervous system development (corrected p value = 0.012) (Figure S5B). Pathway enrichment analyses on DEGs in NPCs (Figure 6E) revealed significantly enriched KEGG pathways including fatty acid metabolism (corrected p = 0.004), a transforming growth factor β (TGF-β) signaling pathway (corrected p = 0.011), and a peroxisome proliferator-activated receptor (PPAR) signaling pathway (corrected p = 0.030), as well as Pathway Commons (PC) pathways such as BMP receptor signaling (corrected p = 0.025). In contrast, functional analysis of DEGs in hiPSCs revealed only one significantly enriched GO category, central element (corrected p = 0.038).
To compare the effect of genetic background versus mutation status on differential gene expression, we performed a Venn analysis on the list of genes identified as being differentially expressed relative to the CAG180 HD line when using the non-isogenic (CAG33) versus isogenic (HD-C#1 and HD-C#2) lines as control (Figure 6F, top). Strikingly, almost half (109) of the 240 NPC DEGs for CAG33 versus CAG180 were not common to the 169 DEGs for corrected isogenic controls versus CAG180. Similarly, 38 of the 169 DEGs found in the corrected isogenic controls versus CAG180 comparison were not identified when comparing the CAG33 and HD CAG180 lines. We validated by qRT-PCR a subset of the DEGs that were common to the CAG33 and corrected control lines (Figure 6G) or not (Figure 6H). It should be noted that while gene correction may reduce the phenotypic variability related to genetic background, variability due to clonal differences remains. This, for example, is reflected in the incomplete overlap in the DEGs between HD-C#1 and HD-C#2 (Figure 6F, bottom). Indeed, of the 12 DEGs assessed by qRT-PCR, 3 showed discordance between HD-C#1 and C#2 (Figures 6G and 6H). This highlights the need to assess multiple clones, even when using isogenic controls.
To further validate and investigate the possible biological relevance of the DEGs identified, we focused on CHCHD2, a gene recently identified as a genetic risk factor for a number of neurodegenerative disorders (Zhou et al., 2016). We examined whether the changes in CHCHD2 mRNA levels observed in NPCs are also present in hiPSCs. Using qRT-PCR, we find that, indeed, CHCHD2 levels are significantly elevated in CAG180 HD hiPSCs relative to the CAG33 and isogenic control lines (Figure 7A). Using immunoblotting, we further find that the changes in CHCHD2 mRNA levels are paralleled by similar changes on the protein level (Figure 7B). Our findings in HD hiPSCs and NPCs are consistent with a previous study showing dysregulated CHCHD2 levels in HD human embryonic stem cells (hESCs) (Feyeux et al., 2012).
Figure 7.

Validation of CHCHD2 Dysregulation and Mitochondrial Respiratory Deficits in CAG180 HD Cells
(A) CHCHD2 qRT-PCR in CAG180 and isogenic control hiPSCs; n = 3 per clone; values for three independent biological replicates shown as mean ± SEM; ns, no significance; and ∗∗∗p < 0.001 was determined by unpaired t test.
(B) CHCHD2 protein levels in CAG180 and isogenic control hiPSCs and NPCs; representative blot shown; n = 3 per clone; values for three independent biological replicates shown as mean ± SEM; ∗p < 0.05 was determined by unpaired t test.
(C–F) Measures of mitochondrial respiration in CAG180 and isogenic control NPCs. (C) Mitochondrial oxidative phosphorylation; (D) mitochondrial basal respiration; (E) mitochondrial ATP production; and (F) mitochondrial maximal respiration; for (D) (E), and (F) n = 5 independent biological replicates; values shown as mean ± SEM; ns, no significance; ∗p < 0.05 and ∗∗p < 0.01, was determined by paired t test.
Given that CHCHD2 has been implicated in mitochondrial oxidative phosphorylation (Baughman et al., 2009), we investigated mitochondrial respiration in CAG180 and isogenic control NPCs (Figure 7C). We find that genetic correction results in improved basal respiration and maximal respiration (significant improvement in two of three isogenic lines) (Figures 7D and 7F), and significant improvement in ATP production (three of three isogenic lines) (Figure 7E).
Discussion
Using a CRISPR/Cas9 nickase- and piggyBac transposon-based HR approach, we demonstrate that the expanded trinucleotide repeat in HTT can be efficiently corrected in hiPSCs. We show that corrected isogenic hiPSC lines retain pluripotency and normal karyotypes, and can be differentiated into excitable and synaptically active neurons. We further show that a number of phenotypic abnormalities in HD hiPSC-derived neural cells, including impaired neural rosette formation, increased susceptibility to growth factor withdrawal, and deficits in mitochondrial respiration, are rescued in isogenic controls. Importantly, using genome-wide expression analysis, we reveal that a number of apparent differences in gene expression seen when comparing HD with non-isogenic control lines are not seen when comparing HD with isogenic corrected lines, indicating that these differences are likely related to genetic background and are not HD-specific effects. We also show that while gene correction may reduce the phenotypic variability related to genetic background, variability due to clonal differences remains, highlighting the importance of assessing multiple clones, even when using isogenic controls.
Genetic correction of HD hiPSCs by HR was reported previously using CRISPR/Cas9-assisted methods (An et al., 2014). However, removal of a selection cassette from the genomic locus of the HTT gene after targeting was not reported. As the presence of a selection cassette may affect HTT expression and regulation, its removal is important to ensure optimal modeling of disease effects.
A number of differentiation protocols for the derivation of MSNs have been published (Arber et al., 2015, Delli Carri et al., 2013). In this study, using a modified version of the protocol of Delli Carri et al. (2013), we derived neurons from CAG180 hiPSCs capable of survival for over 3 weeks, the majority (88%) of which generated sAPs. This is in contrast to previous work using CAG180 hiPSCs, which reported no sAP firing by cells derived following 2 weeks of neuronal differentiation. Moreover, no cells survived after 3 weeks of differentiation (HD iPSC Consortium, 2012). Thus, our study demonstrates that improvements in differentiation protocols can lead to enhanced viability and maturity of hiPSC-derived neurons.
While we show that hiPSC-derived neurons can generate APs, differences in the electrophysiological properties of human HD and control neurons remain to be determined. Indeed, that no discernable differences in the electrophysiological characteristics of HD and control neurons were observed likely reflects the heterogeneity in the type of neurons generated by the differentiation protocol employed and not a lack of genotypic difference per se. As such, future studies aiming to tease apart the electrophysiological properties of disease versus control neurons should be coupled with careful definition of not only the neurons being examined but also the presence of other cell types, most notably glia, that can influence neuronal properties.
There are conflicting reports on the impact of mutant HTT on neural rosette formation by human pluripotent stem cells. An early study using HD hESC lines with 37 and 51 CAG repeats reported formation of “characteristic” neural rosette structures (Niclis et al., 2009). Subsequent studies using HD iPSCs with 72 CAG repeats reported impaired rosette formation, including a significantly decreased number of rosettes per colony and a reduced area of rosette as a percentage of colony area (Jeon et al., 2012). Consistent with these latter studies, we observed a marked impairment in neural rosette formation in the CAG180 HD line, which was rescued in corrected isogenic control lines. Our findings are reminiscent of the phenotype observed with Hdh-deficient ESCs, where a role for ADAM10 and N-cadherin has been identified (Lo Sardo et al., 2012). It is interesting to speculate whether the differences in neural rosette formation among the HD pluripotent stem cell lines reflect a loss of this aspect of normal HTT function in the lines with high CAG repeat lengths (72 CAGs in Jeon et al., 2012 and 180 CAGs in our study) but not low CAG lengths (37 and 51 CAGs in Niclis et al., 2009).
Enrichment analyses of the DEGs common to the non-isogenic and isogenic control lines identified a number of pathways that have been previously implicated in HD. For example, TGF-β signaling was previously shown to be altered in HD (Kandasamy et al., 2010, Ring et al., 2015), drawing interesting links between its role in temporal neurogenesis (Dias et al., 2014) and the known impairment of mutant HTT in striatal and cortical neuronal development (Molina-Calavita et al., 2014). Pathway enrichment of fatty acid metabolism and PPAR signaling in this study is particularly interesting given their known dysregulation in HD (Block et al., 2010, Dickey et al., 2016). PPARs are ligand-activated transcription factors activated by lipids and fatty acid derivatives, and they perform essential regulatory roles in various processes including cellular differentiation, metabolism (lipid, carbohydrate, protein) and energy production (Grygiel-Górniak, 2014). Indeed, mitochondrial dysfunction and metabolic deficits in HD have been attributed to mutant HTT interference with PPAR-γ coactivator-1 α, a key transcriptional regulator of mitochondrial biogenesis and metabolism (Weydt et al., 2006). With further relevance to mitochondrial dysfunction in HD is the gene CHCHD2, the expression of which we found to be dysregulated in the CAG180 HD NPCs relative to isogenic and non-isogenic control lines. CHCHD2 was first identified in a computational screen as a regulator of mitochondrial respiration (Baughman et al., 2009), and subsequently validated in knockdown and overexpression studies (Aras et al., 2015, Baughman et al., 2009). More recently, mutations in CHCHD2 have been identified as risk factors for a number of neurodegenerative disorders (Zhou et al., 2016). The marked upregulation of CHCHD2 in CAG180 HD NPCs that we observed, which is consistent with a previously published report (Feyeux et al., 2012), is paralleled by deficits in mitochondrial respiration. Given its described role in oxidative phosphorylation, the upregulation in CHCHD2 may represent a compensatory adaptation to cellular energy deficits in HD. However, whether such upregulation in CHCHD2 levels does indeed moderate the energetic status or other phenotypic abnormalities of HD cells remains to be determined.
There is widespread recognition of the complementary value of hiPSCs in disease modeling. Indeed, a number of studies have revealed mutant HTT-related molecular and cellular abnormalities using human pluripotent stem cells and differentiated cells, e.g., alterations in transcriptomes, proteomes, ATM-p53 and TGF-β signaling, and monoamine oxidase activity (Mattis and Svendsen, 2015, Ooi et al., 2015). Cellular abnormalities include enhanced lysosomal activity in HD hiPSCs, altered neuronal glutamate signaling and calcium homeostasis, reduced mitochondrial length and function, reduced neurite length in GABAergic and MSNs, impaired neuronal brain-derived neurotrophic factor (BDNF)-vesicular transport, and reduced viability in response to a number of cellular stress paradigms including BDNF withdrawal, H2O2 treatment, and inhibition of autophagy (Mattis and Svendsen, 2015). While many of these pathological features had been previously identified in animal models of HD, some human-specific discoveries are starting to emerge from studies in hiPSCs (Ruzo et al., 2015). Ultimately the use of isogenic control lines will facilitate such efforts, and will help authenticate mutant HTT-specific effects.
Experimental Procedures
Cloning of Constructs
Synthesized oligos for sgRNA-a (Addgene, no. 87201) and sgRNA-b (Addgene, no. 87200) expression were cloned into the Cas9 nickase expression vector pX335. A 1.7-kb 5′ homology arm containing human HTT exon 1 with 18 “CAG” repeats and a 2.4-kb 3′ homology arm were cloned into the MCS1 and MCS2 sites of the PB HR targeting donor vector (pJOP-HTT-HR18Q, Addgene, no. 87228). The oligo sequences for the sgRNAs are listed in Table S1.
Cell Culture
HEK293 cells for testing CRISPR-Cas9 activity were cultured in DMEM supplemented with 10% fetal bovine serum. Human CAG33 hiPSCs (ND36997) and CAG180 hiPSCs (ND36999) were obtained from the NINDS iPSC Repository at Coriell Institute and were cultured on Matrigel-coated plates in mTeSR-1 medium (STEMCELL Technologies, no. 05850).
Nucleofection of hiPSCs
hiPSCs were dissociated with Accutase, and 1 × 106 cells were electroporated using the Neon Transfection System (Life Technologies) with 1 μg sgRNA-a, 1 μg sgRNA-b, and 4 μg PB donor plasmids at 1,400 V for 3 pulses of 10 ms. Targeted hiPSCs were selected by 1 μg/mL puromycin treatment for 48 hr from day 3 and another 48 hr from day 10 post transfection. Surviving colonies at 2 weeks post transfection were manually picked and expanded for culture and PCR screening.
Selection Cassette Excision
To remove the PB cassette, corrected hiPSCs were transfected with PB Excision-Only Transposase vector (System Biosciences). Seventy-two hours after transfection, 0.2 μM fialuridine (Sigma) was used to eliminate PB cassette-containing clones for 5 days. Resistant colonies were picked and expanded, and further screened by junction PCR.
Analysis of piggyBac TTAA Site Post-excision
The genomic region flanking the TTAA site (PB excision site) from pre- and post-excision clones were PCR amplified and sequenced. Amplicons were analyzed by Sanger sequencing.
Surveyor Assay for OT Analysis
To predict potential OT effects, guide sequences sgRNA-a and sgRNA-b (Table S2) were analyzed using the CasOT script (Xiao et al., 2014) and the top-ranked hits were selected for screening. Selected genomic regions were amplified and amplicons were then mixed in a 1:1 ratio of parental (CAG180):corrected (HD-C#1, #2 or #3) to a final volume of 20 μL for Surveyor assay following the manufacturer's instructions (IDT, no. 706020).
Whole-Exome Sequencing
Whole-exome sequencing of the parental CAG180 hiPSCs and three isogenic control lines was performed. See Supplemental Experimental Procedures for details of the protocol.
Mitochondrial Respiration Analysis
Mitochondrial respiration of CAG180 and corrected control NPCs was carried out using a Seahorse XF96 Extracellular Flux Analyzer followed the instructions of Seahorse XF Cell Mito Stress Test Kit (Seahorse Bioscience). Readings were normalized to total protein measured by Bradford assay, and data were analyzed using the Seahorse Wave software.
Immunoblotting
Cells were lysed with RIPA buffer (Sigma-Aldrich) containing complete protease inhibitor Cocktail Tablets (Roche). A total of 30 μg of protein per sample was separated on NuPAGE 3%–8% Tris-acetate (for HTT/calnexin) or NuPAGE 4%–12% bis-Tris (for CHCHD2/calnexin) gradient gels followed by transfer to nitrocellulose membrane. Membranes were imaged using the LI-COR Odyssey infrared imaging system and quantified by ImageJ software.
Immunofluorescence Staining
Cells grown on coverslips were fixed with 4% paraformaldehyde (PFA) for 15 min, permeabilized using 0.3% Triton X-100, then blocked with 3% normal donkey serum containing 0.1% Triton X-100 in DPBS, before being stained with primary antibodies at 4°C overnight, followed by the appropriate secondary antibodies for 1 hr at room temperature and 1 μg/mL DAPI (Sigma) for 10 min. Images were acquired using an Olympus FV1000 inverted confocal microscope.
RNA Isolation, cDNA Synthesis, and qPCR
RNA was purified using the RNeasy Plus Mini Kit (QIAGEN) and cDNA was generated using PrimeScript RT reagent kit (TAKARA BIO). Quantitative real-time PCR was performed on the StepOnePlus or QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems) using the primers listed in Table S3. Relative gene expression levels were analyzed using the Comparative CT Method (ΔΔCT Method).
Three Germ Layers Differentiation
To form embryonic bodies (EBs), hiPSCs were dissociated into clumps using Dispase, and cultured on low-attachment tissue culture plates in knockout serum replacement (KSR) medium (DMEM/F12 with 20% knockout serum replacer, 1% GlutaMAX, 1% non-essential amino acids, and 0.1 mM 2-mercaptoethanol). On day 7, EBs were transferred onto Matrigel-coated coverslips and left to spontaneously differentiate for 9 days in KSR medium before fixation and staining.
Differentiation of hiPSCs
NPC differentiation: hiPSCs were induced into NPCs according to a previously published protocol (Li et al., 2011). Cells between passage 3 and passage 6 were used for experiments.
Forebrain neuronal differentiation: hiPSCs were differentiated into forebrain neurons using an established protocol (Delli Carri et al., 2013) incorporating some modifications from other published protocols (Maroof et al., 2013, Xu et al., 2013). NPCs were induced in N2B27 medium supplemented with certain small molecules and growth factors for 15 days. For final neuronal differentiation, the cells were cultured in N2B27 medium supplemented with BDNF (20 ng/mL), glial cell-derived neurotrophic factor (20 ng/mL), cAMP (N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate; Sigma, 0.5 mM), and ascorbic acid (0.2 mM).
MEA Recordings
Neurons on day 46 were dissociated and re-plated on 0.1% polyethylenimine (Sigma)-coated 12-well MEA plates (Axion BioSystems) and spontaneous neuronal activity was observed and recorded at 37°C for 5 min every other day using the Maestro MEA system (Axion BioSystems).
Whole-Cell Patch-Clamp Recordings
Whole-cell patch-clamp recordings were performed as described previously (Ma et al., 2015). See Supplemental Experimental Procedures for details of the protocol.
Neural Rosette Formation Assay
hiPSCs were dissociated into single cells using Accutase, and 4.5 × 106 cells were seeded into AggreWell800 plates to form neural aggregates in STEMdiff Neural Induction Medium. On day 5, neural aggregates were harvested and transferred into poly-L-ornithine/laminin-coated plates. On day 10, cells were fixed using 4% PFA and stained with antibodies against ZO-1 and Nestin.
Growth Factor Withdrawal Assay
hiPSC-derived neurons on day 40 were switched to N2B27 medium supplemented with 50 ng/mL BDNF or N2B27 medium only for 48 hr. Cells were then fixed with 4% PFA and used for TUNEL staining (In Situ Cell Death Detection Kit; Roche) following the manufacturer's instructions.
Microarray Analysis
Total RNA from hiPSC and NPC samples was analyzed on Illumina HumanHT-12-v4 Expression BeadChip. See Supplemental Experimental Procedures for details of the protocol.
The detailed methods for immunoblotting, immunofluorescence staining, qPCR, fragment sizing analysis, TTAA site analysis, OT Surveyor analysis, whole-exome sequencing, three germ layer differentiation, differentiation of hiPSCs, MEA recordings, whole-cell patch-clamp recordings, neural rosette formation, growth factor withdrawal, and microarray analysis are found in the Supplemental Experimental Procedures.
Author Contributions
M.A.P. conceived and was responsible for the overall supervision of the study. X.X. and M.A.P. designed the research. X.X., Y.T., Y.H., S.-I.Y., B.S., J.O., K.H.U., J.H.N., A.Z., A.N., and D.L. carried out the experiments. X.X., Y.T., Y.H., S.-I.Y., B.S., C.R., B.V., M.L., F.G., G.J.A., and M.A.P. performed analysis of the data. X.X., Y.T., and M.A.P. wrote the paper with feedback and input from the other authors.
Acknowledgments
We thank Qiyu Chen for technical assistance, Sumanty Tohari for whole-exome sequencing, and Kerry McLaughlin for editorial assistance. The work was partly funded by a Strategic Positioning Fund for Genetic Orphan Diseases (SPF2012/005) and a Joint Council Office Project grant (1431AFG122) from the Agency for Science, Technology and Research (Singapore), and a Tier 1 grant R-172-000-297-112 from the Ministry of Education (Singapore) to M.A.P. F.G. and D.L. are supported by the Singapore Immunology Network (SIgN) core funding. C.R. is supported by the A∗STAR Research Attachment Program (ARAP) and A.Z. is supported by the A∗STAR Singapore International Graduate Award (SINGA). We thank the NINDS iPSC Repository for the HD and control iPSC lines (ND36997 and ND36999).
Published: February 23, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and six tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.01.022.
Accession Numbers
The microarray data reported in this paper have been deposited in the GEO under accession number GEO: GSE93767.
Supplemental Information
References
- An M.C., Zhang N., Scott G., Montoro D., Wittkop T., Mooney S., Melov S., Ellerby L.M. Genetic correction of Huntington's disease phenotypes in induced pluripotent stem cells. Cell Stem Cell. 2012;11:253–263. doi: 10.1016/j.stem.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An M.C., O'Brien R.N., Zhang N., Patra B.N., Cruz La, De M., Ray A., Ellerby L.M. Polyglutamine disease modeling: epitope based screen for homologous recombination using CRISPR/Cas9 system. PLoS Curr. 2014;6 doi: 10.1371/currents.hd.0242d2e7ad72225efa72f6964589369a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aras S., Bai M., Lee I., Springett R., Hüttemann M., Grossman L.I. MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion. 2015;20:43–51. doi: 10.1016/j.mito.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Arber C., Precious S.V., Cambray S., Risner-Janiczek J.R., Kelly C., Noakes Z., Fjodorova M., Heuer A., Ungless M.A., Rodríguez T.A. Activin A directs striatal projection neuron differentiation of human pluripotent stem cells. Development. 2015;142:1375–1386. doi: 10.1242/dev.117093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardy C., van den Hurk M., Eames T., Marchand C., Hernandez R.V., Kellogg M., Gorris M., Galet B., Palomares V., Brown J. Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proc. Natl. Acad. Sci. USA. 2015;112:E2725–E2734. doi: 10.1073/pnas.1504393112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman J.M., Nilsson R., Gohil V.M., Arlow D.H., Gauhar Z., Mootha V.K. A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet. 2009;5:e1000590. doi: 10.1371/journal.pgen.1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block R.C., Dorsey E.R., Beck C.A., Brenna J.T., Shoulson I. Altered cholesterol and fatty acid metabolism in Huntington disease. J. Clin. Lipidol. 2010;4:17–23. doi: 10.1016/j.jacl.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delli Carri A., Onorati M., Castiglioni V., Faedo A., Camnasio S., Toselli M., Biella G., Cattaneo E. Human pluripotent stem cell differentiation into authentic striatal projection neurons. Stem Cell Rev. 2013;9:461–474. doi: 10.1007/s12015-013-9441-8. [DOI] [PubMed] [Google Scholar]
- Dias J.M., Alekseenko Z., Applequist J.M., Ericson J. Tgfβ signaling regulates temporal neurogenesis and potency of neural stem cells in the CNS. Neuron. 2014;84:927–939. doi: 10.1016/j.neuron.2014.10.033. [DOI] [PubMed] [Google Scholar]
- Dickey A.S., Pineda V.V., Tsunemi T., Liu P.P., Miranda H.C., Gilmore-Hall S.K., Lomas N., Sampat K.R., Buttgereit A., Torres PPAR-δ is repressed in Huntington's disease, is required for normal neuronal function and can be targeted therapeutically. Nat. Med. 2016;22:37–45. doi: 10.1038/nm.4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyeux M., Bourgois-Rocha F., Redfern A., Giles P., Lefort N., Aubert S., Bonnefond C., Bugi A., Ruiz M., Dιglon N. Early transcriptional changes linked to naturally occurring Huntington's disease mutations in neural derivatives of human embryonic stem cells. Hum. Mol. Genet. 2012;21:3883–3895. doi: 10.1093/hmg/dds216. [DOI] [PubMed] [Google Scholar]
- GeM-HD Consortium Identification of genetic factors that modify clinical onset of Huntington's disease. Cell. 2015;162:516–526. doi: 10.1016/j.cell.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Group T.H.D.C.R. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Grygiel-Górniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications–a review. Nutr. J. 2014;13:17. doi: 10.1186/1475-2891-13-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HD iPSC Consortium Induced pluripotent stem cells from patients with Huntington's disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell. 2012;11:264–278. doi: 10.1016/j.stem.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichida J.K., Kiskinis E. Probing disorders of the nervous system using reprogramming approaches. EMBO J. 2015;34:1456–1477. doi: 10.15252/embj.201591267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon I., Lee N., Li J.-Y., Park I.-H., Park K.S., Moon J., Shim S.H., Choi C., Chang D.-J., Kwon J. Neuronal properties, in vivo effects, and pathology of a Huntington's disease patient-derived induced pluripotent stem cells. Stem Cells. 2012;30:2054–2062. doi: 10.1002/stem.1135. [DOI] [PubMed] [Google Scholar]
- Kajiwara M., Aoi T., Okita K., Takahashi R., Inoue H., Takayama N., Endo H., Eto K., Toguchida J., Uemoto S., Yamanaka S. Donor-dependent variations in hepatic differentiation from human-induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA. 2012;109:12538–12543. doi: 10.1073/pnas.1209979109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandasamy M., Couillard-Despres S., Raber K.A., Stephan M., Lehner B., Winner B., Kohl Z., Rivera F.J., Nguyen H.P., Riess O. Stem cell quiescence in the hippocampal neurogenic niche is associated with elevated transforming growth factor-beta signaling in an animal model of Huntington disease. J. Neuropathol. Exp. Neurol. 2010;69:717–728. doi: 10.1097/NEN.0b013e3181e4f733. [DOI] [PubMed] [Google Scholar]
- Klapstein G.J., Fisher R.S., Zanjani H., Cepeda C., Jokel E.S., Chesselet M.F., Levine M.S. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J. Neurophysiol. 2001;86:2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- Li W., Sun W., Zhang Y., Wei W., Ambasudhan R., Xia P., Talantova M., Lin T., Kim J., Wang X. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc. Natl. Acad. Sci. USA. 2011;108:8299–8304. doi: 10.1073/pnas.1014041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Sardo V., Zuccato C., Gaudenzi G., Vitali B., Ramos C., Tartari M., Myre M.A., Walker J.A., Pistocchi A., Conti L. An evolutionary recent neuroepithelial cell adhesion function of Huntingtin implicates ADAM10-Ncadherin. Nat. Neurosci. 2012;15:713–721. doi: 10.1038/nn.3080. [DOI] [PubMed] [Google Scholar]
- Ma D., Yoon S.-I., Yang C.-H., Marcy G., Zhao N., Leong W.-Y., Ganapathy V., Han J., Van Dongen A.M.J., Hsu K.-S. Rescue of methyl-CpG binding protein 2 dysfunction-induced defects in newborn neurons by pentobarbital. Neurotherapeutics. 2015;12:477–490. doi: 10.1007/s13311-015-0343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald V., Halliday G. Pyramidal cell loss in motor cortices in Huntington's disease. Neurobiol. Dis. 2002;10:378–386. doi: 10.1006/nbdi.2002.0528. [DOI] [PubMed] [Google Scholar]
- Maroof A.M., Keros S., Tyson J.A., Ying S.-W., Ganat Y.M., Merkle F.T., Liu B., Goulburn A., Stanley E.G., Elefanty A.G. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattis V.B., Svendsen C.N. Modeling Huntington's disease with patient-derived neurons. Brain Res. 2015 doi: 10.1016/j.brainres.2015.10.001. [DOI] [PubMed] [Google Scholar]
- Molina-Calavita M., Barnat M., Elias S., Aparicio E., Piel M., Humbert S. Mutant Huntingtin affects cortical progenitor cell division and development of the mouse neocortex. J. Neurosci. 2014;34:10034–10040. doi: 10.1523/JNEUROSCI.0715-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller F.-J., Schuldt B.M., Williams R., Mason D., Altun G., Papapetrou E.P., Danner S., Goldmann J.E., Herbst A., Schmidt N.O. A bioinformatic assay for pluripotency in human cells. Nat. Methods. 2011;8:315–317. doi: 10.1038/nmeth.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niclis J., Trounson A.O., Dottori M., Ellisdon A., Bottomley S.P., Verlinsky Y., Cram D. Human embryonic stem cell models of Huntington disease. Reprod. Biomed. Online. 2009;19:106–113. doi: 10.1016/s1472-6483(10)60053-3. [DOI] [PubMed] [Google Scholar]
- Nisenbaum E.S., Xu Z.C., Wilson C.J. Contribution of a slowly inactivating potassium current to the transition to firing of neostriatal spiny projection neurons. J. Neurophysiol. 1994;71:1174–1189. doi: 10.1152/jn.1994.71.3.1174. [DOI] [PubMed] [Google Scholar]
- Onorati M., Castiglioni V., Biasci D., Cesana E., Menon R., Vuono R., Talpo F., Goya R.L., Lyons P.A., Bulfamante G.P. Molecular and functional definition of the developing human striatum. Nat. Neurosci. 2014;17:1804–1815. doi: 10.1038/nn.3860. [DOI] [PubMed] [Google Scholar]
- Ooi J., Hayden M.R., Pouladi M.A. Inhibition of excessive monoamine oxidase A/B activity protects against stress-induced neuronal death in Huntington disease. Mol. Neurobiol. 2015;52:1850–1861. doi: 10.1007/s12035-014-8974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishna S., Cho S.W., Kim S., Song M., Gopalappa R., Kim J.-S., Kim H. Surrogate reporter-based enrichment of cells containing RNA-guided Cas9 nuclease-induced mutations. Nat. Commun. 2014;5:3378. doi: 10.1038/ncomms4378. [DOI] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Lin C.-Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y., Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A., Albin R.L., Anderson K.D., D'Amato C.J., Penney J.B., Young A.B. Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. USA. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring K.L., An M.C., Zhang N., O'Brien R.N., Ramos E.M., Gao F., Atwood R., Bailus B.J., Melov S., Mooney S.D. Genomic analysis reveals disruption of striatal neuronal development and therapeutic targets in human Huntington's disease neural stem cells. Stem Cell Rep. 2015;5:1023–1038. doi: 10.1016/j.stemcr.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzo A., Ismailoglu I., Popowski M., Haremaki T., Croft G.F., Deglincerti A., Brivanlou A.H. Discovery of novel isoforms of Huntingtin reveals a new hominid-specific exon. PLoS One. 2015;10:e0127687. doi: 10.1371/journal.pone.0127687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterneckert J.L., Reinhardt P., Schφler H.R. Investigating human disease using stem cell models. Nat. Rev. Genet. 2014;15:625–639. doi: 10.1038/nrg3764. [DOI] [PubMed] [Google Scholar]
- Suzuki K., Yu C., Qu J., Li M., Yao X., Yuan T., Goebl A., Tang S., Ren R., Aizawa E. Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell. 2014;15:31–36. doi: 10.1016/j.stem.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Duncan D., Shi Z., Zhang B. WEB-based GEne SeT AnaLysis toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41:W77–W83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P., Pineda V.V., Torrence A.E., Libby R.T., Satterfield T.F., Lazarowski E.R., Gilbert M.L., Morton G.J., Bammler T.K., Strand A.D. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Xiao A., Cheng Z., Kong L., Zhu Z., Lin S., Gao G., Zhang B. CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics. 2014;30:1180–1182. doi: 10.1093/bioinformatics/btt764. [DOI] [PubMed] [Google Scholar]
- Xu X., Lei Y., Luo J., Wang J., Zhang S., Yang X.-J., Sun M., Nuwaysir E., Fan G., Zhao J. Prevention of β-amyloid induced toxicity in human iPS cell-derived neurons by inhibition of cyclin-dependent kinases and associated cell cycle events. Stem Cell Res. 2013;10:213–227. doi: 10.1016/j.scr.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Yusa K. Seamless genome editing in human pluripotent stem cells using custom endonuclease-based gene targeting and the piggyBac transposon. Nat. Protoc. 2013;8:2061–2078. doi: 10.1038/nprot.2013.126. [DOI] [PubMed] [Google Scholar]
- Zhou Z.-D., Saw W.-T., Tan E.-K. Mitochondrial CHCHD-containing proteins: physiologic functions and link with neurodegenerative diseases. Mol. Neurobiol. 2016 doi: 10.1007/s12035-016-0099-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.