

Abstract
The aims of the present study were to undertake gene expression profiling of the blood of glioma patients to determine key genetic components of signaling pathways and to develop a panel of genes that could be used as a potential blood-based biomarker to differentiate between high and low grade gliomas, non-gliomas and control samples. In this study, blood samples were obtained from glioma patients, non-glioma and control subjects. Ten samples each were obtained from patients with high and low grade tumours, respectively, ten samples from non-glioma patients and twenty samples from control subjects. Total RNA was isolated from each sample after which first and second strand synthesis was performed. The resulting cRNA was then hybridized with the Agilent Whole Human Genome (4×44K) microarray chip according to the manufacturer's instructions. Universal Human Reference RNA and samples were labeled with Cy3 CTP and Cy5 CTP, respectively. Microarray data were analyzed by the Agilent Gene Spring 12.1V software using stringent criteria which included at least a 2-fold difference in gene expression between samples. Statistical analysis was performed using the unpaired Student's t-test with a P<0.01. Pathway enrichment was also performed, with key genes selected for validation using droplet digital polymerase chain reaction (ddPCR). The gene expression profiling indicated that were a substantial number of genes that were differentially expressed with more than a 2-fold change (P<0.01) between each of the four different conditions. We selected key genes within significant pathways that were analyzed through pathway enrichment. These key genes included regulators of cell proliferation, transcription factors, cytokines and tumour suppressor genes. In the present study, we showed that key genes involved in significant and well established pathways, could possibly be used as a potential blood-based biomarker to differentiate between high and low grade gliomas, non-gliomas and control samples.
Keywords: blood, biomarker, gene expression, signaling pathway, glioma
Introduction
Cancer is the leading cause of morbidity and mortality worldwide with the number of cases expected to increase by 70% over the next 2 decades (1). Brain and central nervous system tumors are ranked 17th in incidence among all cancers worldwide, being the 13th and 15th most common tumor in men and women, respectively (2). Cancer is the 2nd leading cause of death in the paediatric age group (3), with brain and other nervous system tumors ranked 2nd in incidence after leukaemias (4).
Brain tumors can be either primary or secondary. Gliomas are the most common of the primary brain tumors consisting mainly of oligodendroglioma and astrocytoma with a small number of mixed oligoastrocytoma. Glioblatoma multiforme (GBM) is the most malignant and aggressive of these tumors (5).
Despite advances in surgery, radiation and chemotherapy together with more recently available therapies such as molecularly targeted therapies, prognosis is generally poor. The median survival for patients with malignant gliomas is less than 15 months with GBM patients having the worst prognosis with less than 5% surviving after 5 years (6).
One of the reasons that cancers are detected at a late stage is because many tumors do not have symptoms until the disease has spread. The current methods available for the detection of gliomas are computed tomography (CT) scan and magnetic resonance imaging (MRI) of the brain. However, the definitive diagnosis is by stereotactic guided biopsy of the tumor sample which is technically demanding and has its risks but is, however, considered acceptable (7–9). Therefore, the development of a simple, non-invasive blood test which involves RNA profiling in whole blood, can be used as an addition to the more traditional methods of cancer screening and detection (10).
The inspiration for whole-blood, transcriptome profiling in the context of gliomas originates from the ‘sentinel’ principle (10). Inherent in this principle is the fact that blood is in intimate contact and interacts with all human tissues including cancerous tissue. Blood is considered a connective tissue and is a transporter for various substances such as oxygen, nutrients, cells of the immune system including B cells, T cells, dendritic and natural killer cells, cytokines, growth factors and hormones (11). In addition, blood cells are affected in many disease processes such as hematological malignancies, solid tumors, asthma, autoimmune diseases such as rheumatoid arthritis to common chronic illnesses such as hypertension, diabetes and cardiovascular disease (12–15).
Peripheral blood cells have the ability to respond to changes that affect the physiology, microenvironment and systems biology of the human body. Perturbations or disturbances in the homeostasis of the system can also be subtly detected by peripheral blood cells (11,16). Thus, blood, being easily accessible could serve as a molecular gene expression profile reflecting changes that occur within tissues of the human body (10). The term ‘bloodomics’ has thus been coined to reflect this function of blood in regulation of gene expression and in the molecular profiling of human diseases (11).
One of the earliest models where the sentinel principle has been studied is colorectal carcinoma where a 5- and 7-gene biomarker panel has been developed to assess the current relative risk of patients developing this cancer in Canada and Malaysia (17–19). Molecular gene profiling of the blood transcriptome has also been studied in other diseases including neurological disorders such as schizophrenia and bipolar disorders, chronic fatigue syndrome, tuberous sclerosis complex 2, neurofibromatosis type 1, Down's syndrome, epilepsy, Tourette syndrome, ischemic stroke, migraine, Huntington's and Alzheimer's diseases (20–27).
In the case of an insiduous development of a tumor, substances are secreted by the tumor into the bloodstream and as a systemic response, there are subtle alterations in the level of expression of genes within peripheral blood cells in order to maintain homeostasis or as a reaction to the disease entity itself (10). In the brain, disruption of the blood-brain barrier is due to loss of substances such as the tight junction proteins claudin-1 and claudin-3, decrease in polarity of glioma cells, loss of the molecule agrin and upregulation of the aqueous channel protein, aquaporin 4 (AQP4) resulting in brain oedema formation (28–34). Since blood-brain barrier disruption occurs in brain tumors (35,36), substances that play a role in both homeostasis and tumorigenesis are likely to be secreted into the bloodstream under such conditions and may give a molecular signature profile.
In this study, we have extrapolated the fascinating theory of the sentinel principle to the development of adult gliomas and to determine if such expression profiling in blood could be used to distinguish between high and low grade gliomas, non-gliomas and control samples. The justification for the present study is that such profiling will help not only in the stratification of gliomas, but also in the early detection of tumors when they are far more amenable to complete surgical resection, thus, improving prognosis and survival of the patient.
Materials and methods
Clinical patient data
Upon admission to the hospital, demographic data and a brief clinical history was elicited from 30 of the 50 patients. The demographic data included the age and gender of the patient and the state in which the patient was domiciled. The 30 patients comprised of 10 high grade glioma (HG), 10 low grade glioma (LG) (Table I) and 10 non-glioma (NG) cases (Table II). The remaining 20 patients were normal, healthy controls (C) (Table III). The incidence of gliomas is 2–3 new cases per 100,000 population per year (37). As such, the number of samples we were able to collect on our own was small.
Table I.
WHO classification, histopathology of tumor samples and demographic data.
| Histopathology | Grade | Age (years) | Gender |
|---|---|---|---|
| Pilocytic astrocytoma | I | 31 | Male |
| Diffuse astrocytoma | II | 17 | Male |
| Diffuse astrocytoma | II | 32 | Male |
| Fibrillary astrocytoma | II | 62 | Female |
| Recurrent astrocytoma | II | 45 | Female |
| Diffuse astrocytoma | II | 36 | Male |
| Low grade astrocytoma | II | 59 | Male |
| Low grade oligodendroglioma | II | 45 | Male |
| Low grade oligodendroglioma | II | 56 | Male |
| Recurrent oligodendroglioma | II | 59 | Male |
| Anaplastic oligoastrocytoma | III | 37 | Female |
| Anaplastic oligoastrocytoma | III | 58 | Male |
| Recurrent anaplastic oligoastrocytoma | III | 66 | Male |
| Anaplastic astrocytoma | III | 29 | Female |
| Anaplastic astrocytoma | III | 43 | Male |
| Glioblastoma multiforme | IV | 24 | Male |
| Glioblastoma multiforme | IV | 54 | Male |
| Glioblatoma multiforme | IV | 24 | Male |
| Glioblastoma multiforme | IV | 34 | Male |
| Glioblastoma multiforme | IV | 56 | Female |
Table II.
Demographics and types of non-glioma samples.
| Patient no. | Age (years) | Gender | Sample type |
|---|---|---|---|
| 1 | 40 | Male | Hemangioblastoma |
| 2 | 77 | Male | Blood clot |
| 3 | 44 | Female | Inflammatory pseudotumour |
| 4 | 27 | Male | Arteriovenous malformation (AVM) |
| 5 | 51 | Female | Ischaemic stroke |
| 6 | 53 | Female | Hemangioblastoma |
| 7 | 61 | Male | Haemorrhagic stroke |
| 8 | 56 | Female | Multiple sclerosis |
| 9 | 34 | Female | Ischaemic stroke |
| 10 | 46 | Female | Haemorrhagic stroke |
Table III.
Demographics of control samples.
| Patient no. | Age (years) | Gender |
|---|---|---|
| 1 | 30 | Female |
| 2 | 38 | Female |
| 3 | 41 | Male |
| 4 | 57 | Male |
| 5 | 25 | Male |
| 6 | 57 | Male |
| 7 | 33 | Male |
| 8 | 51 | Male |
| 9 | 28 | Male |
| 10 | 25 | Male |
| 11 | 56 | Male |
| 12 | 32 | Male |
| 13 | 22 | Male |
| 14 | 59 | Female |
| 15 | 42 | Female |
| 16 | 55 | Male |
| 17 | 58 | Male |
| 18 | 48 | Male |
| 19 | 33 | Male |
| 20 | 55 | Male |
Informed consent was obtained prior to blood taking and brain tumor removal from the patient during surgery. After obtaining consent, blood was immediately drawn from the patient on the day before surgery. Surgery was performed the next day, typically within 12–24 h of obtaining consent and drawing blood from the patient. In addition, this study received ethics approval from the Medical Research Ethics Committee (MREC) of the Ministry of Health, Malaysia.
Histopathological examination
Brain tumor tissue was sectioned onto glass slides and stained with hematoxylin and eosin (H&E). The slides were read by neuropathologists at the respective hospital. The diagnosis was made based on the World Health Organization (WHO) classification of tumors of the central nervous system (2007) (5). Of the 20 tumor samples, 10 each were high and low grade gliomas, respectively. Grade I and II tumors were classified as low grade while grade III and IV tumors were classified as high grade.
Non-glioma and control samples
In addition to the 20 tumor samples, 10 non-glioma and 20 control samples were also obtained. The 10 non-glioma cases constituted patients with an inflammatory, non-malignant condition of the brain and included cases of hemangioblastoma, haemorrhagic and ischaemic stroke, inflammatory pseudotumor, arteriovenous malformation and multiple sclerosis (Table II). The 20 control subjects were healthy with no known medical illness (Table III).
Blood sample collection
A total of 2.5 ml of venous blood was drawn from each patient using the BD Vacutainer (Becton-Dickinson, Franklin Lakes, NJ, USA) with attached 21G × 3/4′′ × 12′′ butterfly needle directly into the PreAnalytiX PAXgene Blood RNA Tube (BRT) (Qiagen, Hilden, Germany). The samples were kept at room temperature for 2 h to allow for complete lysis of cell components after which they were stored at −20°C.
RNA extraction
RNA was extracted from each blood sample using the PreAnalytiX PAXgene™ Total RNA Blood Extraction kit (Qiagen). After collection, the blood sample in the PAXgene Blood RNA Tube (BRT) was incubated at a minimum of 2 h at room temperature to ensure complete lysis of blood cells. The BRT was then spun for 10 min at 3,000–5,000 × g. The supernatant was removed and the pellet containing the blood cells vortexed until dissolved in 4 ml of RNase-free water. The BRT was centrifuged again and the supernatant removed. A total of 350 µl of resuspension buffer was added and the pellet vortexed until dissolved. The sample was transferred into a 1.5-ml microcentrifuge tube where 300 µl of binding buffer was added to bind the RNA which was predominantly derived from leukocytes; 40 µl of proteinase was also added to dissolve any protein present in the sample. The lysate was transferred directly into a PAXgene Shredder spin column and centrifuged to remove cell debris. The flow through supernatant containing the total RNA was mixed with 350 µl of 96–100% ethanol and vortexed. Sample (700 µl) was pipetted into the PAXgene RNA spin column to which DNase I was added to remove any contaminating DNA. The PAXgene RNA spin column was washed several times with wash buffers 1 and 2 after which 40 µl of elution buffer was added directly onto the PAXgene RNA spin column membrane. This was centrifuged for 1 min at 8,000–20,000 × g to elute the RNA. The eluate containing the total RNA was incubated at 65°C for 5 min and then chilled immediately on ice.
The concentration and purity of the RNA was analyzed using the Spectrophotometer NanoDrop ND-1000 (Thermo Fisher Scientific, Tewksbury, MA, USA). The integrity of the RNA was analyzed using the Agilent 2100 BioAnalyzer RNA 6000 Nano Chip platform (Agilent Technologies, Santa Clara, CA, USA). The concentration of RNA obtained ranged from 37 to 442 ng/µl. The average value for the RNA integrity number (RIN) for the samples was 7.4 with a standard deviation of 0.87. The samples were stored at −80°C until further use.
Microarray processing
Two-colour microarray-based gene expression utilizing the Agilent 4×44K Whole Human Genome microarray, was performed on RNA isolated from the 50 blood samples. Standard protocols were followed for sample preparation, probe labeling and hybridization according to the Two-Colour Microarray-Based Gene Expression Analysis Protocol (Agilent Technologies).
For sample preparation, the Two-Colour RNA Spike-In kit (Agilent Technologies) was used. Spike A and Spike B Mix were thawed, mixed vigorously on a vortex mixer and then heated at 37°C in a water bath for 5 min. Three serial dilutions of 1:20, 1:40 and 1:4 were performed for each spike mix. For the labeling reactions, the Low Input Quick Amp Labeling kit (Agilent Technologies) was used. Total RNA (150 ng) to a volume of 1.5 µl was labeled; 2 µl of the Spike A Mix/Cy3-CTP was used to label the Universal Human Reference RNA (Stratagene, La Jolla, CA, USA) while 2 µl of the Spike B/Cy5-CTP was used to label the HG, LG, NG and C samples, respectively. A total of 1.8 µl of T7 Promoter Primer Mix (consisting of 0.8 µl T7 promoter primer and 1 µl nuclease-free water) was added to the reaction containing 3.5 µl of total RNA and diluted RNA Spike-in Mix. The primer and template were denatured by incubating the reaction in a water bath at 65°C for 10 min. The reactions were then placed on ice for 5 min. cDNA Master mix (4.7 µl) (2 µl 5X first strand buffer, 1 µl 0.1 M DTT, 0.5 µl 10 mM dNTP and 1.2 µl AffinityScript RNase block mix) was added to each sample tube to a total volume of 10 µl. Samples were incubated at 40°C in a water bath for 2 h after which they were moved to a 70°C water bath and incubated for a further 15 min. The samples were then incubated on ice for 5 min. Finally, 6 µl of transcription master mix (0.75 µl nuclease-free water, 3.2 µl 5X transcription buffer, 0.6 µl 0.1 M DTT, 1 µl NTP mix, 0.21 µl T7 RNA polymerase blend and 0.24 µl Cy3-CTP/Cy5-CTP) was added to each sample tube for a total volume of 16 µl and incubated at 40°C in a water bath for 2 h.
The resulting labeled/amplified cRNA was purified as per protocol using the RNeasy mini spin columns (Qiagen). The cleaned cRNA sample was eluted by transferring the RNeasy column to a new 1.5 ml collection tube. RNase-free water (30 µl) was added directly onto the RNeasy filter membrane and allowed to stand for 60 sec. The RNeasy column in the collection tube was then centrifuged at 4°C for 30 sec at 13,000 rpm. The flow-through containing the cRNA sample was maintained on ice. If not used immediately, the samples were stored at −80°C.
The cRNA was quantified using the NanoDrop spectrophotometer as previously described. The yield and specific activity of each reaction was determined respectively as follows:
For the 4-pack microarray format, almost all yields obtained were ≥0.825 µg and had specific activity (pmol Cy3 or Cy5 per µg cRNA) ≥6.
The initial step for the hybridization reactions involved the fragmentation of RNA. For the 4-pack microarray format, 825 ng each of Cy3- and Cy5-labeled, linearly amplified cRNA, 11 µl of 10X blocking agent were made up to a volume of 52.8 µl with nuclease-free water, after which 2.2 µl of 25X fragmentation buffer was added to a total volume of 55 µl. The samples were incubated at 60°C for exactly 30 min to fragment the RNA and then immediately cooled on ice for 1 min. Fragmentation mix (55 µl) containing cRNA was mixed with an equal volume of 2X GE Hybridization buffer HI-RPM. The samples were spun in a microcentrifuge at 13,000 rpm for 1 min at room temperature to drive any residual sample from the walls and lid of the tubes and to help with bubble reduction. The samples were then placed on ice and loaded onto the array immediately. Sample (100 µl) was pipetted into the gasket slide well of the Agilent SureHyb chamber and the ‘active side’ of the array placed directly on top of the gasket slide to form a sandwich pair. The SureHyb chamber cover was placed on the sandwich slides and the clamp assembly tightened onto the chamber. The assembled slide chamber was then placed in a rotisserie hybridization oven at 20 rpm and the samples allowed to hybridize at 65°C for 17 h. The slides were then washed with Gene Expression Wash Buffer 1 followed by Prewarm Gene Expression Wash Buffer 2. In addition, 0.0005% Triton X-102 was added to both buffers which reduced the possibility of array wash artifacts. The microarray slides were scanned using the DNA microarray scanner (Agilent Technologies).
Data extraction
Data were extracted using Agilent feature extraction software analyzed with Gene Spring version GX 12.5V (Agilent Technologies). The data files were extracted in text (.txt) format after Lowess normalization. The sequence of events involved in processing of the data files were as follows: thresholding, summarization, dye swap, ratio computation, log transformation and baseline transformation.
Thresholding involved a substitution step where all expression values below a certain specified value were made constant. Thresholding was done to remove very small expression values or negative values in the dataset. This was to ensure that there were no very large negative numbers when the data was log transformed. Summarization was done by calculating the geometric mean of the expression values. Raw signal values were then generated which essentially were linear data that had undergone thresholding and summarization for the individual channels (Cy3 and Cy5). Normalized signal values refer to the data after it has undergone ratio computation, log2 transformation and baseline transformation. Normalization was also done using the Human Reference RNA.
Dye-swapping accounts for dye related bias as different dyes (Cy3 and Cy5) bind DNA with different affinities. This dye related bias cannot be removed by standard normalization methods. In GeneSpring, samples that have been marked as dye-swapped were treated as follows: Cy3 was designated as ‘signal’ and Cy5 as ‘control’ and the signal was computed as Cy3/Cy5. For samples that have not undergone dye-swapping, GeneSpring treats Cy5 as ‘signal’ and Cy3 as ‘control’ and the signal is computed as Cy5/Cy3.
In baseline transformation, the baseline to median of control samples was performed. In the Agilent 4×44K Human Array, there are a set of samples designated as controls that can be used for all samples. In this baseline transformation, for each probe, the median of the log summarized values from the control samples was first computed, after which, this value was subtracted from the sample.
As mentioned previously, Lowess normalization was performed before the raw data were extracted. Lowess normalization is critical for reducing intra-array (within slide) variation. In 2 colour experiments, 2 fluorescent dyes, red and green, are used. The intensity-dependent variation in dye bias may introduce spurious variations in the dataset. Lowess normalization is performed which merges the 2 colour data and applies a smoothing adjustment which removes such variations.
Investigating the effects of tumor status, age, gender and experimental array batch on gene expression
A linear mixed regression analysis was performed using the ‘R’ statistical package, to investigate the effects of tumor pathology, age, gender and experimental batch effects on gene expression. The tumor status was defined by the four groups of samples, HG, LG, NG and C. The age and gender of patients are represented in Tables I–III, respectively. Samples were run on arrays in 4 experimental batches as follows: Batch 1, 6 HG and 6 LG samples; Batch 2, 4 HG, 4 LG and 4C samples; Batch 3, 8C and 4 NG samples; Batch 4, 8C and 6 NG samples. The explanatory power of each factor was assessed in a stepwise manner by examining the increase in the variation explained when a new covariate or set of covariates was added to the existing model. This resulted in the investigation of the following four models: i) model 1, gene expression as a function of tumor status; ii) model 2, gene expression as a function of tumor status and age; iii) model 3, gene expression as a function of tumor status, age and gender; and iv) model 4, gene expression as a function of tumor status, age, gender and array batch.
In total there were 50 microarray samples each with 29,092 gene expression values from 44,000 probe sets. In preparation of input data for multiple regression analysis, a table of 50 microarray samples (50 rows) × 29,092 gene expression values (29,092 columns) was generated. The metadata for each sample that included tumor status, age, gender and array batch were combined as columns in the prepared table. The input data were then read into the ‘R’ software. For each gene, a linear model was fitted using the Im function to its respective gene expression values vs. variable(s) of interest as per the four models. For each model, there were 29,092 r2 (coefficient of determination) values that were generated. Each r2 value was then modified to generate an adjusted r2 value to account for the number of variables and the sample size. A median, mean and range for r2 was then calculated for each model as shown in Fig. 1 and Table IV.
Figure 1.
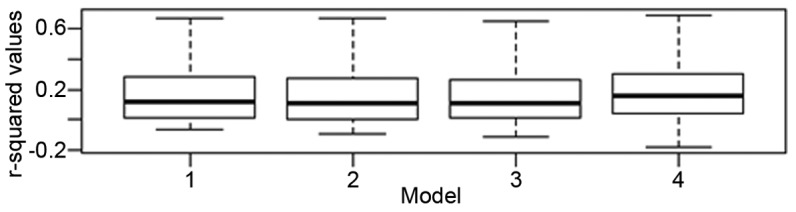
Box plot of adjusted r2 values for the 4 models. The range for the error bars for models 1–4 are as follows: model 1, −0.06508 (min) to 0.91450 (max); model 2, −0.088070 (min) to 0.915400 (max); model 3, −0.11050 (min) to 0.91510 (max) and model 4, −0.1779 (min) to 0.9123 (max).
Table IV.
Median and mean adjusted r2 values for models 1–4.
| Model | Median | Mean |
|---|---|---|
| 1 | 0.11830 | 0.17220 |
| 2 | 0.110700 | 0.163200 |
| 3 | 0.11180 | 0.16360 |
| 4 | 0.1540 | 0.1882 |
Unsupervised hierarchical clustering
Unsupervised hierarchical clustering using the Euclidean distance method and Ward's linkage was performed on each of the 4 different pairs of conditions and all 4 conditions. One of the limitations in unsupervised hierarchical clustering is that this form of analysis could be influenced by noise and outliers particularly when sample sizes are small.
Principal component analysis (PCA)
Principal component analysis was performed on the complete data set. The first step in PCA was to subtract the mean from each of the data dimensions. Then, the covariance matrix and the eigenvectors and eigenvalues of the covariance matrix were calculated. Data compression and reduced dimensionality was performed when converting the data into components and to form feature vectors in 3 dimensions along the x-, y- and z-axis.
Identification of significant differences in gene expression between the 4 different conditions
The moderated t-test, a modification of the Students t-test, was used to identify significant differences in gene expression between the 4 sets of conditions (HG vs. C, LG vs. C, HG vs. LG and NG vs. C). While the Students t-test calculates variance from the data that is available for each gene, the moderated t-test uses information from all of the genes to calculate variance. This is particularly useful when a small number of samples is available in each group (as in this case) making the variance estimates unstable.
When testing was performed across these different conditions, each gene was considered independently from the other as a moderated t-test was performed on each gene separately. Given that in this microarray experiment, the expression levels of 44,000 probes was measured simultaneously across each condition, multiple testing correction (MTC) was required. With this in mind, the Benjamini and Hochberg (B-H) false discovery rate was used to control for the large number of tests performed. This procedure is one of the less stringent methods of MTC but it provides a good balance between identification of many genes that are statistically significant and protection against false positives (type I error).
Pathway analysis
For each group, genes were selected based on at least a 2-fold difference in expression and a B-H corrected P<0.01. Pathway analysis was performed using the ingenuity pathway analysis (IPA) programme (Johns Hopkins University, Baltimore, MD, USA).
IPA is based on the Ingenuity Knowledge Base. In IPA, canonical pathways are well characterized pathways that have been curated and hand-drawn by PhD level scientists and the information comes from specific journal articles, review articles, text books and HumanCyc, an encyclopaedia of human genes and metabolism (http://humancyc.org). Gene selection for the canonical pathways is based on this analysis.
cDNA synthesis
RNA from each sample was converted to cDNA using the high capacity RNA-to-cDNA kit (Applied Biosystems). Optimal blend priming was performed with a mixture of random octamers and oligo dT primers.
Total RNA (200 ng) was mixed with 10.0 µl of 2X reverse transcriptase (RT) buffer, 1.0 µl 20X enzyme mix and nuclease-free water to a total volume of 20.0 µl. The tube containing the reaction mix was then incubated in the Professional basic thermocycler (Biometra, Gottingen, Germany) at 37°C for 60 min after which the reaction was terminated by heating to 95°C for 5 min. The reactions were then used for droplet digital PCR (ddPCR) or stored long-term at −80°C.
Droplet digital polymerase chain reaction (ddPCR)
Selected genes (Table V) from each of the 4 group pairs as previously mentioned, were verified using ddPCR. Reactions for each sample were done either singly or in duplicate. Beta-glucuronidase (GUSB) was used as the reference gene as it showed the least variation with gene expression amongst the other housekeeping genes used, namely TATA binding protein (TBP) and human acidic ribosomal protein (HuPO). All reagents and equipment used for ddPCR were from Bio-Rad Laboratories (Hercules, CA, USA). cDNA (10 ng) was mixed with 10 µl of 2X ddPCR Supermix for probes (No dUTP), 1 µl 20X target primers/probe mix (FAM) or 20x reference primers/probe (HEX) and nuclease-free water to a total reaction volume of 20 µl. The entire reaction mix of 20 µl was then loaded into a sample well of a DG8 Cartridge for the QX200/QX100 droplet generator. This was then followed by adding 70 µl of droplet generation oil for probes into the oil wells of the cartridge, according to the QX200/QX100 Droplet Generator Instruction Manual. The cartridge was then inserted into the Automated Droplet Generator. After droplet generation, the droplets were transferred to a 96-well plate and then sealed with foil using the PX1 PCR plate sealer.
Table V.
The significant genes after Bonferroni correction.
| Condition | Gene | Fold change from GeneSpring | Fold change from ddPCR | P-value | Bonferroni correction: new 0.05 threshold, 10 tests | Result (P-value) |
|---|---|---|---|---|---|---|
| NG vs. C | MMP9 | +2.35 | +6.49 | 0.0068 | 0.005 | False |
| LG vs. C | MAP3K8 | +2.46 | +1.61 | 0.00003 | 0.005 | True |
| TP53 | −2.81 | +2.00 | 0.00007 | 0.005 | True | |
| SOS1 | −2.62 | −1.69 | 0.00362 | 0.005 | True | |
| HG vs. C | FOS | +2.28 | +3.55 | 0.00853 | 0.005 | False |
| IL6 | +4.06 | +3.05 | 0.00001 | 0.005 | True | |
| TNF | −2.90 | +1.60 | 0.00620 | 0.005 | False | |
| HG vs. LG | EGFR | +2.44 | −1.25 | 0.43 | 0.005 | False |
| VEGFA | +2.13 | +1.36 | 0.24 | 0.005 | False | |
| MAPK12 | −4.09 | +1.19 | 0.27 | 0.005 | False |
NG, non-glioma; C, control; LG, low grade glioma; HG, high grade glioma; MMP9, matrix metallopeptidase 9; MAP3K8, mitogen-activated protein kinase 8; TP53, tumor protein p53; SOS1, son of sevenless homolog 1; FOS, FBJ murine osteosarcoma viral oncogene homolog; IL6, interleukin 6; TNF, tumor necrosis factor; EGFR, epidermal growth factor receptor; VEGFA, vascular endothelial growth factor A; MAPK12, mitogen-activated protein kinase 12.
Thermal cycling was then performed on the droplets using the C1000 Touch Thermal Cycler with 96-deep well reaction module according to the following protocol: enzyme activation at 95°C for 10 min (1 cycle), denaturation at 94°C for 30 sec followed by annealing/extension at 55°C for 1 min (40 cycles), enzyme deactivation at 98°C for 10 min (1 cycle) followed by hold at 4°C. The ramp rate was set at 2°C/sec, the heated lid to 105°C and the sample volume at 40 µl. After thermal cycling, the sealed plate was placed in a QX200/QX100 droplet reader and the absolute gene expression level per well for the probes and reference genes were quantitated using QuantaSoft software.
For analysis of the gene expression data, we assumed a normal distribution. Each gene was evaluated for its expression in a minimum of 3 to a maximum of 6 samples under each pair of conditions. The gene expression values for each sample were normalized to the housekeeping gene. The values for the absolute level of gene expression as obtained by ddPCR were then subjected to the t-test for the genes selected under the 4 sets of conditions, with a resulting fold change and P-value. Statistical outliers were removed using the box and whisker plot.
In summary, ddPCR is a method for performing digital PCR that utilizes a water-oil emulsion droplet system using a combination of microfluidics and proprietary surfactant chemistries. Droplets are formed in a water-oil emulsion that partitions the nucleic acid samples into 20,000 nanoliter sized droplets, with background and target DNA randomly distributed among the droplets. Sample partitioning is key to ddPCR. PCR amplification is then carried out on each droplet thus enabling the measurement of thousands of independent amplification events within a single sample. Nucleic acids are quantified by counting the regions that contain the PCR end product. Thus, ddPCR is not dependent on the number of amplification cycles to determine the initial sample amount, hence eliminating the reliance on uncertain exponential data to quantify target nucleic acids. This allows clonal amplification of nucleic acids with direct and absolute quantification. Therefore, the main benefits of ddPCR technology are simplified and absolute quantification of nuclei acids with superior partitioning, unparalleled precision, increased signal-to-noise ratio and removal of PCR bias.
Results
Modeling the effects of tumor status, age, gender and experimental array batch effects on gene expression
Using a linear mixed regression analysis, the effect of tumor pathology, age, gender and experimental batch on gene expression was investigated. For models 1, 2, 3 and 4, the median and mean adjusted r2 values did not vary significantly (Table IV and Fig. 1). The change in median values for models 2 and 3 were 0.0077 and 0.0065%, respectively as compared to model 1, suggesting that age and gender had a minimal impact on gene expression globally. For model 4, the change in the median adjusted r2 value when compared to tumor status alone was 3.57%, indicating that array batch had some impact on gene expression globally, but that this was still small. The mean adjusted r2 values showed a similar trend. Based on these findings, all subsequent analysis focused on the impact of tumor pathology alone on gene expression.
Microarray analysis of samples
(a) Unsupervised hierarchical clustering was performed on each of the 4 different pairs of conditions (HG vs. C, LG vs. C, HG vs. LG and NG vs. C) and all 4 conditions together, with the total gene input list, using the Euclidean distance method and Ward's linkage. The gene input list consisted of genes which were found to be differentially expressed with a corrected P<0.01 and a fold change of at least 2. The results are depicted in Fig. 2 showing 3 clusters of samples: one cluster for the high grade tumors, a second cluster for the low grade tumors and a third cluster for the non-glioma and control samples. The non-glioma and controls clustered together and were distinct from the glioma samples, with the low grade glioma being furthest from the control and non-glioma samples. Therefore, not only were we able to show a distinction between the glioma and control samples but were also able to distinguish between high and low grade gliomas.
Figure 2.

Unsupervised hierarchical clustering of all 4 groups by use of the Euclidean similarity measure and Wards linkage to visualize the expression level of genes between the groups. The heat map shows the gene expression for the different groups in columns, with a dendogram representing their similarity. The clustering was performed on a filtered gene list of normalized signal intensity values (averaged over replicates) for all the 4 groups.
(b) Principal component analysis (PCA) of samples
The PCA plot (Fig. 3) of the first 3 axes, showed results that were very similar to that of the microarray analysis, demonstrating clear separation into the 3 clusters of samples as mentioned in (a). Of specific note, is that the 2 sample types which were closest to each other were the control and non-glioma sets.
Figure 3.

Principal component analysis (PCA) plot for the 4 different conditions. The axes corresponds to principal component 1 (PC1, x-axis), PC2 (y-axis) and PC3 (z-axis). The ellipses (2 standard deviation coverage; see colour key for the different conditions) shows a distinct directionality in the different groups based on similarities in gene expression.
Volcano plots
Multiple testing correction using the Benjamini-Hochberg (B-H) analysis with a corrected P<0.01, and a 2-fold change cut-off, for each of the four conditions were as follows: HG vs. C: total number of genes, 1055, with 479 upregulated and 576 downregulated; LG vs. C: total number of genes, 2708, with 713 upregulated and 1995 downregulated; HG vs. LG: total number of genes, 1629, with 1287 upregulated and 342 downregulated; and NG vs. C: total number of genes, 82, with 56 upregulated and 26 downregulated. The results were represented on volcano plots (Fig. 4).
Figure 4.

Volcano plots to determine differentially expressed genes for the individual pairs of conditions: (A) HG vs. C; (B) LG vs. C; (C) HG vs. LG; and (D) NG vs. C. The x-axis represents the log2 fold change of genes for the different condition pairs, while the y-axis represents the - log10 of the corrected P-values for the different pairs of conditions. Each dot represents a gene and the red coloured area represents the differentially expressed genes that met the selection criteria of a fold change (FC) of at least 2 (FC≥2 or ≤2) and a P<0.01.
The results showed that there were relatively few genes which were differentially expressed between control and non-glioma samples. In comparing the glioma samples to the controls, the predominant effect was the downregulation of genes in the glioma samples. When comparing the high and low grade samples, there was in general an upregulation occurring in the high grade samples.
Venn diagram of differentially expressed genes
The Venn Diagram represented the genes with at least a 2-fold difference in expression and a P<0.01, that were unique to each condition and also those that overlapped between the various conditions (Fig. 5A). There were 104 genes common to both the HG vs. C and the HG vs. LG pairs. These included genes belonging to the zinc finger transcription factor, ZNF 649 and ZNF 205, homeobox genes such as HOXB2 and SOX8, a transcription factor involved in embryonic development and determination of cell fate. For the HG vs. LG pair, there were a total of 1629 genes, of which 644 were unique to this pair and included EGFR, TGFβ1 and VEGFA. There were 573 genes common to both the HG vs. C and LG vs. C pairs. These common genes included IL12RB1, FOS, TP53 and TNF. One important gene common to the HG vs. C, LG vs. C and HG vs. LG pairs was IL6.
Figure 5.

(A) Venn diagram of differentially expressed genes for the different condition pairs. The Venn diagram summarizes the number of distinct and overlapping differentially expressed genes found in the four condition pairs: HG vs. C (gene list 1), LG vs. C (gene list 2), NG vs. C (gene list 3) and HG vs. LG (gene list 4). (B) Venn diagram of canonical pathways. The Venn diagram summarizes the number of distinct and overlapping pathways found in the 4 condition pairs: HG vs. C (pathway list 1), LG vs. C (pathway list 2), NG vs. C (pathway list 3) and HG vs. LG (pathway list 4).
For the NG vs. C pair, there were 46 unique genes, 19 that overlapped with the HG vs. C pair, 7 that overlapped with the LG vs. C pair and another 7 that were common to the HG vs. LG and LG vs. C pairs. There were no genes common to all 4 conditions (Fig. 5A).
Canonical pathways
The significance of association between differentially expressed genes and the canonical pathways (as annotated by the HumanCyc Pathway database) were assumed to follow a normal distribution and assessed using the B-H multiple testing correction to calculate a P-value. Only those pathways with a corrected P<0.05 were selected. This determined the probability that the association between the genes and the pathways, relative to all functionally characterized human genes, were not explained by chance alone (data not shown). The IPA also determines whether the pathways are activated or inhibited by assigning a z score. The ratio defined the proportion of differentially expressed genes from a pathway to the total number of genes that make up that particular pathway. For the HG vs. C pair, 4 significant pathways were identified (ratios ranging from 0.084 to 0.136) with no evidence for significant activation or inhibition as shown by z scores close to zero (data not shown). The 4 significant pathways included those involved in innate and adaptive immunity. For the LG vs. C pair, the IPA predicted a mixed pattern of activity for the 46 significant pathways with 23 pathways having no activity pattern available, 5 pathways having a positive z-score (predicted activation), 16 pathways having a negative z-score (predicted inhibition) and 2 pathways having a z-score of zero (data not shown). The z-score of zero corresponded to the standard mean of the normal distribution curve. Pathways having no activity pattern available meant that a z-score could not be calculated. The significant pathways with a positive z-score included those involved in LXR/RXR activation, RhoG, Ephrin B, IL-8 and cholecystokinin/gastrin-mediated signaling. The z-score of zero included pathways involved in NF-κB activation by viruses and glioma invasiveness signaling. The significant pathways with a negative z-score were signaling by the Rho family of GTPases, TEC kinase signaling, HGF, eicosanoid, integrin, acute phase response, PEDF and thrombin signaling. It also included pathways involved in PKC, actin nucleation and immune system signaling.
For the HG vs. LG glioma pair, 9 significant pathways were predicted with 6 having no activity pattern and 1 each with a positive, negative and zero z-score respectively (data not shown). The activity pattern referred to the differential expression of genes that made up the pathway. The 6 pathways with no activity pattern were those involved in FXR/RXR activation, superoxide radical degeneration, hepatic fibrosis/hepatic stellate cell activation, role of tissue factor in cancer, clathrin-mediated endocytosis and atherosclerosis signaling. The pathways that had a positive z-score, a z-score of zero and a negative z-score were pathways involved in LXR/RXR activation, coagulation system and acute phase response signaling respectively. For the NG vs. C pair, there was only one significant pathway, hepatic fibrosis/hepatic stellate cell activation that had no activity pattern available (data not shown).
Venn diagram of significant pathways
The Venn diagram for the pathways showed pathways that were unique to each pair of conditions and also pathways that overlapped between the 4 different groups (Fig. 5B). For the HG vs. C pair, there was 1 unique pathway and 3 pathways that overlapped with the LG vs. C pair. The LG vs. C pair had 39 unique pathways. The pathways that overlapped between the HG vs. C and LG vs. C pairs were pathways involved in the innate and adaptive immune response. The HG vs. LG pair had a total of 9 significant pathways, with 4 unique pathways, 4 overlapping with the LG vs. C pair and 1 overlapping with the NG vs. C pair. The 4 unique pathways were the superoxide radicals degradation, clathrin-mediated endocytosis, coagulation system and role of tissue factor in cancer pathways. The 4 pathways overlapping with the LG vs. C pair were the acute phase response, FXR/RXR activation, LXR/RXR activation and atherosclerosis signaling pathways. The 1 pathway overlapping with the NG vs. C pair was the hepatic fibrosis/hepatic stellate cell activation pathway.
Heat map
A heat map (Fig. 6) with genes commonly involved in tumor signaling pathways especially in high and low grade brain tumors was generated with the four types of samples, namely C, NG, LG and HG glioma, respectively. The results showed a unique differential pattern of expression for each of the 4 sample types. In addition, genes commonly upregulated in high grade tumors such as EGFR and VEGFC, are also highly expressed in blood. On the other hand, these genes are downregulated in the low grade tumor heat map. Specific isoforms of Bcl2 such as Bcl2L11 and Bcl2A1 are upregulated in the low grade but not high grade samples. None of the genes involved in tumorigenesis are significantly upregulated in the non-glioma and control samples.
Figure 6.

Heat map of selected differentially expressed genes for the 4 different conditions.
Genes chosen for validation by ddPCR
Ten genes were selected for statistical validation by ddPCR (Table V). These genes were selected from the list of differentially expressed genes that were significant from the 4 pairs of conditions. These genes were selected because they were known to be common genes involved in pathways related to tumorigenesis including the pathogenesis of brain tumors. Only the NG vs. C had no significant genes that were downregulated. The other 3 conditions had significant genes that were both upregulated and downregulated.
Each gene was evaluated for its expression in a minimum of 3 to a maximum of 6 samples under each pair of conditions. The values for the absolute level of gene expression as obtained by ddPCR was then subjected to statistical analysis. A normal distribution of the values was assumed and the t-test applied to each gene with a resulting P-value. Seven of the 10 genes had P<0.05 and 3 genes had P>0.05. The genes with a P<0.05 were MMP, MAP3K8, TP53, SOS1, FOS, IL6 and TNF. The genes with a P>0.05 were EGFR, VEGFA and MAPK12 (Table I). Multiple testing correction of the P-value using the Bonferroni correction with a threshold P-value of 0.05 and 10 test samples, resulted in only 4 genes that were highly significant. The genes were MAP3K8, TP53, SOS1 and IL6 (Table V).
Discussion
This study has advanced the idea of using blood-based gene expression studies as an indicator of neoplastic changes occurring in brain tissue. This idea was based upon the sentinel principle and extrapolated to the study of brain tumors. In this study, we have used the sentinel principle not only to identify patients with a glioma but also to differentiate between high grade, low grade, non-glioma and control subjects.
The unsupervised hierarchical clustering and principal component analysis clearly showed that the four groups of subjects clustered into 3 statistically significant groups as represented by the ellipses, which showed a distinct directionality in the different groups based on similarities in gene expression (Fig. 3). The fact that the non-glioma and control subjects clustered together and were distinct from the high and low grade tumor patients, indicated that the changes in gene expression in blood in these 2 groups were clearly different from that of the glioma patients indicating specificity of expression. This lends further credence to the sentinel principle that substances are released from the tumor into the bloodstream (10,11) and may be distinct for each tumor subtype. Although the blood samples in this study were taken from patients after presentation to the hospital with neurological symptoms, it is highly likely that these substances were released during the early stages of tumor formation (10) and continued to persist in blood even as the tumor enlarged based upon the theory and evidence from the sentinel principle (10,11).
The brain, as an immunologically privileged site, is protected by the blood-brain barrier which restricts the movement of water soluble molecules by tight junctions (38) and a low level of transcytosis (39). The breakdown of the blood-brain barrier in brain tumors can be visualized by either freeze fracture electron microscopy (40) or contrast enhanced magnetic resonance imaging (MRI) using gadolinium (41). The normal blood-brain barrier is impermeable to contrast medium but there is a gradual increase in the degree of disruption of the blood-brain barrier corresponding to the grade of the tumor. WHO grade II tumors show little or no contrast medium enhancement, WHO grade III tumors enrich more contrast medium than grade II tumors while WHO grade IV tumors (GBM) show the greatest gadolinium enhancement (35). This observation fits well with our postulation that substances from the brain are able to cross the blood-brain barrier and enter the circulation due to the varying degrees of disruption of the blood-brain barrier during glioma formation.
In addition, cells may dislodge from the tumor and enter the peripheral circulation as circulating tumor cells (CTCs). These CTCs then colonize a distant tissue or organ and begin to form a new tumor mass. Although most CTCs do not survive in the circulation, a subset of cells known as disseminated tumor cells (DTCs) that have cancer stem cell properties are able to survive. They then invade a distant tissue or organ site and form tumor cell clusters known as micrometastasis (42). Since CTCs are found in extremely low levels in the circulation (<5 cells/10 ml of blood) (43), identification and detection of these cells require analytical methods that are highly sensitive and specific combined with enrichment procedures. We did not perform the isolation of CTCs in this study as the contribution of these CTCs is extremely small compared to the contribution of leucocytes to the gene expression patterns seen in the peripheral blood transcriptome through signaling mechanisms via the sentinel principle.
In gliomas, CTCs are mainly detected in patients with high grade glioma such as GBM. Unlike tumors of epithelial origin which express epithelial cell adhesion molecule (EpCAM), glioma cells instead express Nestin, both in, in vitro and in vivo studies. This suggests that Nestin could be used as a suitable marker for the detection of circulating glioma cells. In addition, glioma cells also express high levels of human telomerase (hTERT) which co-localizes with Nestin in vivo (44).
Athough CTCs may have limited use in studying gene expression patterns in the peripheral blood transcriptome, they may have clinical utility in distinguishing between a persistent signal on MRI which may be due to either true disease progression or pseudoprogression. Thus, the identification of glioma-derived CTCs in the circulation of such patients posttreatment (after chemoradiation therapy) is prognostic, with a reduction in CTCs indicating treatment response and an increase in CTCs indicating disease progression (44,45).
Besides CTCs, circulating tumor-associated nucleic acids (CNAs) can also be used as possible biomarkers. CNAs are particularly promising as biomarkers as this allows the tumor to be sampled at the transcriptomal and genomic level from blood. Nucleic acids can be found in body fluids including blood as a result of tumor apoptosis, necrosis or active secretion into the peripheral circulation (46). Circulating tumor-associated DNA (ctDNA) may harbor the same genetic aberrations found in the tumor. ctDNA in glioma patients have been shown to have similar genetic alterations as found in the parent tumor including LOH for 1p and 19q (47), IDH1 mutation (48) and abnormal methylation of the promoters of certain genes including MGMT (49) and p16 (50). Using circulating tumor-associated RNA (ctRNA) as a biomarker is more challenging, as RNA is easily degraded by RNases which are present in the peripheral blood of cancer patients. However, microRNAs (miRNAs) have shown more promise as biomarkers in glioma patients. These include RNU6-1, miR-320 and miR-574 which are associated with GBM (51) and miR-29 with differential expression in low grade vs. high grade gliomas (52). However, we chose not to include these types of investigations in the present study as we were focusing on gene expression of the peripheral blood transcriptome via the sentinel principle.
The differentially expressed genes for the four different conditions were unique, but also had some commonality. Most of the unique and common genes in the HG and LG tumor samples were transcription factors, cytokines, proto-oncogenes, oncogenes, growth factors and tumor suppressor genes. These genes are involved in inflammation, tumor signaling pathways, glioma formation, tissue necrosis, apoptosis, homeostasis, cytoskeletal architecture, maintenance of the extracellular matrix and determination of cell fate. Notably, there were also a substantial number of genes involved in the innate and adaptive immune system suggesting that modulation of the immune system plays a critical role in tumor response. In addition, genes known to be involved in the pathogenesis of GBM were also upregulated in blood. These genes included EGFR, VEGF and IL-6. This evidence implied that some of the changes occurring in the tumor tissue may be reflected in blood, suggesting that these substances may be released into the circulation through disruption of the blood-brain barrier or through complex signaling mechanisms.
The canonical pathways for the 4 sets of conditions mirrored the differential gene expression pattern. These included pathways involved in the innate and adaptive immune response, interleukin, acute phase response, glioma invasiveness, NF-κB activation and TGF-β signaling. The latter 3 pathways are also involved in the pathogenesis of gliomas. Again, we see much commonality between the signaling pathways in tissue and blood taken from glioma patients. One of the reasons for this could be the fact that peripheral blood cells share more than 80% of the transcriptome with 9 different tissue types including brain (10). More important is the fact that blood cells express organ specific genes and also genes that are responsive to physiological changes and stimuli that were previously thought to be exclusive to certain tissue types (10). In the pathogenesis and formation of gliomas, these interactions between blood and tissue, together with disruption of the blood-brain barrier, could possibly explain some of the similarity observed in gene expression between gliomas and peripheral blood cells.
The validation of selected genes was done by ddPCR. As previously mentioned, these genes were selected because they were known to be involved in signaling pathways that played an important role in tumorigenesis including the pathogenesis and formation of gliomas. In the selection of 10 genes for validation, 4 of the 10 genes, namely TP53, TNF, MAPK12 and EGFR showed fold changes that were reversed to that seen in the microarray experiment. TP53, TNF and MAPK12 were downregulated in the microarray experiment but upregulated by validation and EGFR was upregulated in the microarray experiment but minimally downregulated by validation. The reason for this could be multifactorial. Firstly and most importantly, the probes used for the microarray experiment are different from the primers used in ddPCR. As genes very commonly have isoforms, it is likely that the primers in ddPCR may be amplifying an isoform of the gene resulting in alternative transcripts (26). These transcripts may have expression levels that are different from the parent gene. In addition, there may be a negative feedback loop where one transcript inhibits the expression of the alternative transcript of the same gene or vice versa. This could result in reversal of expression as seen during ddPCR validation. Therefore, great importance should be placed on careful primer design when using qRT-PCR and ddPCR. For these validation assays, the primers should be designed to be on the same exon as the microarray probes. By doing this, variations in gene expression between microarrays and validation assays including ddPCR will be minimized. Secondly, we selected GUSB as the housekeeping gene to normalize our ddPCR data. Although GUSB showed the least variation with samples compared to TBP and HuPO, it might still have shown some variation in gene expression in the tumor samples. This could result in reversal of gene expression after validation. Thirdly, microarray analysis is generally used to screen large numbers of genes and the possibility arises that there may be false positives. In addition, microarray experiments are often performed with a small number of biological replicates, resulting in low statistical power for detecting differentially expressed genes and concomitant high false positive rates. Studies have shown that microarray results were in agreement with qRT-PCR and ddPCR for genes with medium and high expression but there was very little agreement for genes with lower or variable expression (53–55). In this study, the genes generally varied in expression from 2–4 fold which is considered a low fold change. As such, we would expect some differences between the gene expression in microarrays compared to ddPCR including reversal of expression. Fourthly, human samples have huge technical and biological variability and it is likely that the presence of substances such as activators or inhibitors within the samples could be contributing to the differences observed. This is because ddPCR, being far more sensitive and quantitative and having a higher dynamic range, is able to detect the expression of genes affected by either inhibitors or activators, that may not be detected by microarray analysis. Also, the scanning software for microarrays has low sensitivity which can limit the precision of detection of the image, thus, contributing to a lower fold change of the differentially expressed genes. Fifthly, not all samples were used for validation by ddPCR. Only 3–6 samples were used for each set of conditions and this may have affected the level and pattern of gene expression as well.
The initial P-values obtained showed that 7 of the 10 genes chosen for validation had statistically significant P<0.05. The genes with initial P>0.05 were EGFR, VEGFA and MAPK12. After applying the Bonferroni correction for the P-value, only 4 of the 10 genes passed this stringent statistical test. The 4 genes were MAP3K8, IL6, SOS1 and TP53. Although the other genes were not considered to be statistically significant, they could be clinically significant. In addition, P-values are dependent on many factors including sample size, with a larger sample size giving rise to a more reliable P-value (56). In our case with a limited sample size, the P-value could vary by adding or removing even one value. Thus, a larger sample size would definitely add more confidence to the P-values that were obtained in our experiments.
There are limitations to the use of the sentinel principle and that of a blood-based biomarker to detect changes in a disease state in another tissue. The main limitation is that the blood transcriptome is susceptible to a vast array of changes such as that due to tobacco smoke, environmental pollutants and toxins, and to diseases such as hypertension, diabetes, cardiovascular disease, ischaemic stroke and asthma (12–15,57,58). Many cancer patients, including the patients in the present study, have these comorbidities and this could have a confounding effect on the differential gene expression pattern observed. In addition, the drawing of blood, temperature and storage conditions can all have an effect on gene expression levels of peripheral blood cells.
This is a preliminary study to assess the possibility of using a blood-based biomarker to differentiate between high grade, low grade, non-glioma and control samples. The main drawback of the present study is the small sample size. In order to take this study forward to a blood-based biomarker panel for gliomas, we would need a much larger sample size to give this study more power and to obtain more reliable P-values for the genes selected. In addition, this study would need to be validated in an independent data set.
Finally, the data in this study will be freely available. As the sample number, n, in this study is small, this will enable those who are interested to verify the results of this study, to use the data as a starting point. They may wish to replicate this study using a similar or larger sample size.
Acknowledgements
We wish to thank the Director General of Health, Malaysia for the permission to publish this study. The present study was financially supported by a grant (NMRR 10-930-7461) from the Ministry of Health, Malaysia, awarded to Dr S.N.P.
References
- 1.Stewart BW, Wild CP, editors. World Cancer Report 2014. International Agency for Research on Cancer (IARC) Publications; Lyon, France: 2014. [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray, F. IARC CancerBase no. 11. IARC Press; Lyon: 2012. Cancer incidence and mortality worldwide. [Google Scholar]
- 3.US Mortality Data, corp-author. National Centre for Health Statistics. Centres for Disease Control and Prevention. 20062009 [Google Scholar]
- 4.Linet MS, Ries LA, Smith MA, Tarone RE, Devesa SS. Cancer surveillance series: Recent trends in childhood cancer incidence and mortality in the United States. J Natl Cancer Inst. 1999;91:1051–1058. doi: 10.1093/jnci/91.12.1051. [DOI] [PubMed] [Google Scholar]
- 5.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group: Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 7.Sawin PD, Hitchon PW, Follett KA, Torner JC. Computed imaging-assisted stereotactic brain biopsy: A risk analysis of 225 consecutive cases. Surg Neurol. 1998;49:640–649. doi: 10.1016/S0090-3019(97)00435-7. [DOI] [PubMed] [Google Scholar]
- 8.Samadani U, Stein S, Moonis G, Sonnad SS, Bonura P, Judy KD. Stereotactic biopsy of brain stem masses: Decision analysis and literature review. Surg Neurol. 2006;66:484–490. doi: 10.1016/j.surneu.2006.05.056. discussion 491. [DOI] [PubMed] [Google Scholar]
- 9.Chen CC, Hsu PW, Erich Wu TW, Lee ST, Chang CN, Wei KC, Chuang CC, Wu CT, Lui TN, Hsu YH, et al. Stereotactic brain biopsy: Single center retrospective analysis of complications. Clin Neurol Neurosurg. 2009;111:835–839. doi: 10.1016/j.clineuro.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 10.Liew CC, Ma J, Tang HC, Zheng R, Dempsey AA. The peripheral blood transcriptome dynamically reflects system wide biology: A potential diagnostic tool. J Lab Clin Med. 2006;147:126–132. doi: 10.1016/j.lab.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Mohr S, Liew CC. The peripheral-blood transcriptome: New insights into disease and risk assessment. Trends Mol Med. 2007;13:422–432. doi: 10.1016/j.molmed.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Gladkevich A, Nelemans SA, Kauffman HF, Korf J. Microarray profiling of lymphocytes in internal diseases with an altered immune response: Potential and methodology. Mediators Inflamm. 2005;2005:317–330. doi: 10.1155/MI.2005.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 14.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 15.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liew CC. Methods for the detection of gene transcripts in blood and uses thereof. United States patent US 20040014059. 2004 Jan 22; [Google Scholar]
- 17.Marshall KW, Mohr S, Khettabi FE, Nossova N, Chao S, Bao W, Ma J, Li XJ, Liew CC. A blood-based biomarker panel for stratifying current risk for colorectal cancer. Int J Cancer. 2010;126:1177–1186. doi: 10.1002/ijc.24910. [DOI] [PubMed] [Google Scholar]
- 18.Han M, Liew CT, Zhang HW, Chao S, Zheng R, Yip KT, Song ZY, Li HM, Geng XP, Zhu LX, et al. Novel blood-based, five-gene biomarker set for the detection of colorectal cancer. Clin Cancer Res. 2008;14:455–460. doi: 10.1158/1078-0432.CCR-07-1801. [DOI] [PubMed] [Google Scholar]
- 19.Yip KT, Das PK, Suria D, Lim CR, Ng GH, Liew CC. A case-controlled validation study of a blood-based seven-gene biomarker panel for colorectal cancer in Malaysia. J Exp Clin Cancer Res. 2010;29:128–134. doi: 10.1186/1756-9966-29-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsuang MT, Nossova N, Yager T, Tsuang MM, Guo SC, Shyu KG, Glatt SJ, Liew CC. Assessing the validity of blood-based gene expression profiles for the classification of schizophrenia and bipolar disorder: A preliminary report. Am J Med Genet B Neuropsychiatr Genet 133B. 2005:1–5. doi: 10.1002/ajmg.b.30161. [DOI] [PubMed] [Google Scholar]
- 21.Glatt SJ, Everall IP, Kremen WS, Corbeil J, Sásik R, Khanlou N, Han M, Liew CC, Tsuang MT. Comparative gene expression analysis of blood and brain provides concurrent validation of SELENBP1 up-regulation in schizophrenia. Proc Natl Acad Sci USA. 2005;102:15533–15538. doi: 10.1073/pnas.0507666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaushik N, Fear D, Richards SC, McDermott CR, Nuwaysir EF, Kellam P, Harrison TJ, Wilkinson RJ, Tyrrell DA, Holgate ST, et al. Gene expression in peripheral blood mononuclear cells from patients with chronic fatigue syndrome. J Clin Pathol. 2005;58:826–832. doi: 10.1136/jcp.2005.025718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang Y, Schapiro MB, Franz DN, Patterson BJ, Hickey FJ, Schorry EK, Hopkin RJ, Wylie M, Narayan T, Glauser TA, et al. Blood expression profiles for tuberous sclerosis complex 2, neurofibromatosis type 1, and Downs syndrome. Ann Neurol. 2004;56:808–814. doi: 10.1002/ana.20291. [DOI] [PubMed] [Google Scholar]
- 24.Tang Y, Gilbert DL, Glauser TA, Hershey AD, Sharp FR. Blood gene expression profiling of neurologic diseases: A pilot microarray study. Arch Neurol. 2005;62:210–215. doi: 10.1001/archneur.62.2.210. [DOI] [PubMed] [Google Scholar]
- 25.Du X, Tang Y, Xu H, Lit L, Walker W, Ashwood P, Gregg JP, Sharp FR. Genomic profiles for human peripheral blood T cells, B cells, natural killer cells, monocytes, and polymorphonuclear cells: Comparisons to ischemic stroke, migraine, and Tourette syndrome. Genomics. 2006;87:693–703. doi: 10.1016/j.ygeno.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Borovecki F, Lovrecic L, Zhou J, Jeong H, Then F, Rosas HD, Hersch SM, Hogarth P, Bouzou B, Jensen RV, et al. Genome-wide expression profiling of human blood reveals biomarkers for Huntington's disease. Proc Natl Acad Sci USA. 2005;102:11023–11028. doi: 10.1073/pnas.0504921102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maes OC, Xu S, Yu B, Chertkow HM, Wang E, Schipper HM. Transcriptional profiling of Alzheimer blood mononuclear cells by microarray. Neurobiol Aging. 2007;28:1795–1809. doi: 10.1016/j.neurobiolaging.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 28.Liebner S, Fischmann A, Rascher G, Duffner F, Grote EH, Kalbacher H, Wolburg H. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 2000;100:323–331. doi: 10.1007/s004010000180. [DOI] [PubMed] [Google Scholar]
- 29.Wolburg H, Wolburg-Buchholz K, Kraus J, Rascher-Eggstein G, Liebner S, Hamm S, Duffner F, Grote EH, Risau W, Engelhardt B. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol. 2003;105:586–592. doi: 10.1007/s00401-003-0688-z. [DOI] [PubMed] [Google Scholar]
- 30.Noell S, Fallier-Becker P, Beyer C, Kröger S, Mack AF, Wolburg H. Effects of agrin on the expression and distribution of the water channel protein aquaporin-4 and volume regulation in cultured astrocytes. Eur J Neurosci. 2007;26:2109–2118. doi: 10.1111/j.1460-9568.2007.05850.x. [DOI] [PubMed] [Google Scholar]
- 31.Wolburg H, Noell S, Wolburg-Buchholz K, Mack A, Fallier-Becker P. Agrin, aquaporin-4, and astrocyte polarity as an important feature of the blood-brain barrier. Neuroscientist. 2009;15:180–193. doi: 10.1177/1073858408329509. [DOI] [PubMed] [Google Scholar]
- 32.Noell S, Fallier-Becker P, Deutsch U, Mack AF, Wolburg H. Agrin defines polarized distribution of orthogonal arrays of particles in astrocytes. Cell Tissue Res. 2009;337:185–195. doi: 10.1007/s00441-009-0812-z. [DOI] [PubMed] [Google Scholar]
- 33.Saadoun S, Papadopoulos MC, Davies DC, Krishna S, Bell BA. Aquaporin-4 expression is increased in oedematous human brain tumors. J Neurol Neurosurg Psychiatry. 2002;72:262–265. doi: 10.1136/jnnp.72.2.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warth A, Kröger S, Wolburg H. Redistribution of aquaporin-4 in human glioblastoma correlates with loss of agrin immunoreactivity from brain capillary basal laminae. Acta Neuropathol. 2004;107:311–318. doi: 10.1007/s00401-003-0812-0. [DOI] [PubMed] [Google Scholar]
- 35.Larsson HB, Stubgaard M, Frederiksen JL, Jensen M, Henriksen O, Paulson OB. Quantitation of blood-brain barrier defect by magnetic resonance imaging and gadolinium-DTPA in patients with multiple sclerosis and brain tumors. Magn Reson Med. 1990;16:117–131. doi: 10.1002/mrm.1910160111. [DOI] [PubMed] [Google Scholar]
- 36.Wolburg H, Noell S, Fallier-Becker P, Mack AF, Wolburg-Buchholz K. The disturbed blood-brain barrier in human glioblastoma. Mol Aspects Med. 2012;33:579–589. doi: 10.1016/j.mam.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 37.Lantos PL, VandenBerg SR, Kleihues P. Tumors of the nervous system. In: Graham DI, Lantos PL, editors. Greenfields neuropathology. Arnold; London: 1996. pp. 583–879. [Google Scholar]
- 38.Brightman MW, Reese TS. Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol. 1969;40:648–677. doi: 10.1083/jcb.40.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peters A, Palay SL, Webster H. The Fine Structure of the Nervous System. 3nd. Oxford University Press; New York: 1991. [Google Scholar]
- 40.Dinda AK, Sarkar C, Roy S, Kharbanda K, Mathur M, Khosla AK, Banerji AK. A transmission and scanning electron microscopic study of tumoral and peritumoral microblood vessels in human gliomas. J Neurooncol. 1993;16:149–158. doi: 10.1007/BF01324702. [DOI] [PubMed] [Google Scholar]
- 41.Sage MR, Wilson AJ. The blood-brain barrier: An important concept in neuroimaging. AJNR Am J Neuroradiol. 1994;15:601–622. [PMC free article] [PubMed] [Google Scholar]
- 42.Pantel K, Alix-Panabières C. Circulating tumour cells in cancer patients: Challenges and perspectives. Trends Mol Med. 2010;16:398–406. doi: 10.1016/j.molmed.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 43.Pantel K, Alix-Panabières C, Riethdorf S. Cancer micrometastases. Nat Rev Clin Oncol. 2009;6:339–351. doi: 10.1038/nrclinonc.2009.44. [DOI] [PubMed] [Google Scholar]
- 44.Macarthur KM, Kao GD, Chandrasekaran S, Alonso-Basanta M, Chapman C, Lustig RA, Wileyto EP, Hahn SM, Dorsey JF. Detection of brain tumor cells in the peripheral blood by a telomerase promoter-based assay. Cancer Res. 2014;74:2152–2159. doi: 10.1158/0008-5472.CAN-13-0813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao F, Cui Y, Jiang H, Sui D, Wang Y, Jiang Z, Zhao J, Lin S. Circulating tumor cell is a common property of brain glioma and promotes the monitoring system. Oncotarget. 2016 Aug 8; doi: 10.18632/oncotarget.11114. (Epub ahead of print). doi: 10.18632/oncotarget.11114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426–437. doi: 10.1038/nrc3066. [DOI] [PubMed] [Google Scholar]
- 47.Lavon I, Refael M, Zelikovitch B, Shalom E, Siegal T. Serum DNA can define tumor-specific genetic and epigenetic markers in gliomas of various grades. Neuro Oncol. 2010;12:173–180. doi: 10.1093/neuonc/nop041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boisselier B, Gállego Pérez-Larraya J, Rossetto M, Labussière M, Ciccarino P, Marie Y, Delattre JY, Sanson M. Detection of IDH1 mutation in the plasma of patients with glioma. Neurology. 2012;79:1693–1698. doi: 10.1212/WNL.0b013e31826e9b0a. [DOI] [PubMed] [Google Scholar]
- 49.Balańa C, Carrato C, Ramírez JL, Cardona AF, Berdiel M, Sánchez JJ, Tarón M, Hostalot C, Musulen E, Ariza A, et al. Tumour and serum MGMT promoter methylation and protein expression in glioblastoma patients. Clin Transl Oncol. 2011;13:677–685. doi: 10.1007/s12094-011-0714-x. [DOI] [PubMed] [Google Scholar]
- 50.Wakabayashi T, Natsume A, Hatano H, Fujii M, Shimato S, Ito M, Ohno M, Ito S, Ogura M, Yoshida J. p16 promoter methylation in the serum as a basis for the molecular diagnosis of gliomas. Neurosurgery. 2009;64:455–461. doi: 10.1227/01.NEU.0000340683.19920.E3. discussion 461–462. [DOI] [PubMed] [Google Scholar]
- 51.Manterola L, Guruceaga E, Gállego Pérez-Larraya J, González-Huarriz M, Jauregui P, Tejada S, Diez-Valle R, Segura V, Samprón N, Barrena C, et al. A small noncoding RNA signature found in exosomes of GBM patient serum as a diagnostic tool. Neuro Oncol. 2014;16:520–527. doi: 10.1093/neuonc/not218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu J, Li L, Jiang C. Identification and evaluation of serum microRNA-29 family for glioma screening. Mol Neurobiol. 2015;52:1540–1546. doi: 10.1007/s12035-014-8937-9. [DOI] [PubMed] [Google Scholar]
- 53.Kuo WP, Liu F, Trimarchi J, Punzo C, Lombardi M, Sarang J, Whipple ME, Maysuria M, Serikawa K, Lee SY, et al. A sequence-oriented comparison of gene expression measurements across different hybridization-based technologies. Nat Biotechnol. 2006;24:832–840. doi: 10.1038/nbt1217. [DOI] [PubMed] [Google Scholar]
- 54.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 55.Huggett JF, Foy CA, Benes V, Emslie K, Garson JA, Haynes R, Hellemans J, Kubista M, Mueller RD, Nolan T, et al. The digital MIQE guidelines: Minimum Information for publication of Quantitative digital PCR experiments. Clin Chem. 2013;59:892–902. doi: 10.1373/clinchem.2013.206375. [DOI] [PubMed] [Google Scholar]
- 56.Sackett DL, Rosenberg WM, Gray JA, Haynes RB, Richardson WS. Evidence based medicine: What it is and what it isnt. 1996. Clin Orthop Relat Res. 2007;455:3–5. [PubMed] [Google Scholar]
- 57.Wang Z, Neuburg D, Li C, Su L, Kim JY, Chen JC, Christiani DC. Global gene expression profiling in whole-blood samples from individuals exposed to metal fumes. Environ Health Perspect. 2005;113:233–241. doi: 10.1289/txg.7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lampe JW, Stepaniants SB, Mao M, Radich JP, Dai H, Linsley PS, Friend SH, Potter JD. Signatures of environmental exposures using peripheral leukocyte gene expression: Tobacco smoke. Cancer Epidemiol Biomarkers Prev. 2004;13:445–453. [PubMed] [Google Scholar]