

Abstract
ABCB1 (P-glycoprotein, ABCB1/MDR1) is one of the major members of the ABC transporters linked to MDR in cancer cells. In this study, we observed that pristimerin, a natural triterpenoid, potently decreased P-gp in a dose-dependent manner in both drug-resistant KBv200 and stable transfected HEK293/ABCB1 cell lines. Moreover, pristimerin also inhibited cell proliferation and induced apoptosis in both cell lines. Intriguingly, reverse transcription-PCR, real-time PCR and protein turn-over assay revealed that the decrease of P-gp was independent of mRNA level but primarily owing to its protein stability. Furthermore, immunofluorescence study with anti-P-gp antibody showed that pristimerin disturbed the subcellular distribution of P-gp with decreased location in the plasma membrane. Taken together, these data suggest that subcellular distribution of P-gp and subsequent downregulation by pristimerin contribute to overcoming ABCB1-mediated chemotherapeutic drug resistance. Our findings suggested inducing the decrease of P-gp membrane protein could be a new promising alternative therapeutic strategy in ABCB1-mediated MDR.
Keywords: pristimerin, ABCB1, P-glycoprotein, multidrug resistance, protein degradation
Introduction
It is generally agreed that MDR is one of the most important causes of failure in cancer chemotherapy. MDR is a phenomenon in which cancer cells are resistant to the cytotoxic effects of various structurally and mechanistically unrelated chemotherapeutic agents. The result of MDR is often caused by overexpression of cell membrane-bound ATP-binding cassette (ABC) transporters (1–4). Forty-nine ABC transporters have been identified in the human genome and are divided into seven subfamilies (termed A-G) on the basis of sequence similarities (5), among which, ABCB1 (P-glycoprotein, ABCB1/MDR1), ABCCs (multidrug resistance-associated proteins), and ABCG2 (breast cancer resistance protein/mitoxantrone resistance-associated transporter/ABCP) play major roles in producing MDR in tumor cells. Central to the mechanism of resistance to most chemotherapeutic regimens is the overexpression of mdr1 gene, which encodes a 170-kDa transmembrane glycoprotein named as P-glycoprotein (P-gp). Novel strategies targeting ABCB1, including the downregulation of ABCB1 expression and/or function, may effectively circumvent drug resistance and eliminate MDR cells to achieve better chemotherapeutic effect (4). In this regard, we have previously demonstrated that apatinib (6,7), axitinib (8), crizotinib (9) and vandetanib (10) inhibit various ABC transporters and reverse MDR in leukemia and solid tumors.
Pristimerin is a quininemethide triterpenoid compound which has been found in various species belonging to Celastraceae and Hippocrateaceae families and has long been used as anti-inflammatory, antioxidant, antimalarial, and insecticidal agents (11,12). It has been reported that pristimerin, as a new proteasome inhibitor, has promising clinical potential as both a therapeutic and chemopreventive agent for cancer (13). Indeed, pristimerin induces apoptotic cell death in certain human cancer cells, including breast and lung cancer (14) and human acute myeloid leukemia (15). Our previous data showed that triterpenoid pristimerin induced HepG2 cells apoptosis through ROS-mediated mitochondrial dysfunction (16) which revealed that pristimerin might be a promising compound offering better anticancer treatment options. In this study, we further investigated the effect of this compound overcoming ABCB1-mediated chemotherapeutic drug resistance in vitro and related molecular mechanisms.
Materials and methods
Chemicals and reagents
Pristimerin with a purity of >98% was purchased from the PI & PI Technology Inc. (Guangzhou, China) and the molecular structure is shown in Fig. 1A. Monoclonal antibodies against ABCB1 for western blotting and immunofluorescence assay, and for flow cytometry were from Santa Cruz Biotechnology, respectively. Antibodies against Bax, Bcl-2, caspase-3 and PARP were obtained from Cell Signaling Technology Inc. (Danvers, MA, USA). Antibodies against Akt, ERK1/2, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), anti-mouse and anti-rabbit IgG-horseradish peroxidase were purchased from Kangchen Biotechnology (Shanghai, China). DMEM and RPMI-1640 were products of Gibco BRL. Platinum® SYBR® Green qPCR SuperMix-UDG with ROX was obtained from Invitrogen Co. Protein synthesis inhibitor cycloheximide and 3-(4,5)-dimethylthiazol-2-y1)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma (St. Louis, MO, USA). Other routine laboratory reagents of analytical or high-performance liquid chromatography grade were obtained from Whiga Biotechnology (Guangzhou, China).
Figure 1.

(A) Chemical structure of pristimerin. (B) Expression of ABCB1 in HEK293/pcDNA3.1 and HEK293/ABCB1 cells. (C and D) Pristimerin showed equally potent anticancer effect on parental and ABCB1-mediated MDR cell lines. The cytotoxicity of pristimerin (C) for KB and KBv200 cells; and (D) for HEK293/pcDNA3.1 and HEK293/ABCB1 cells. Cytotoxicity was measured by MTT assay. The cells having grown for 24 h were exposed to a full range of concentrations of pristimerin for 72 h. Cell viability was assessed by model 550 microplate reader after staining with MTT for 4 h. Data are shown as means ± SD of at least triplicate determinations. Each experiment was performed in three replicate wells.
Cell lines and culture
The following cell lines were cultured in Dulbecco's modified Eagle's medium or RPMI-1640 medium supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere of 5% CO2. The human oral epidermoid carcinoma cell line KB and its vincristine-selected, ABCB1-overexpressing derivative KBv200 were gifts from Dr Xu-Yi Liu (Cancer Hospital of Beijing, Beijing, China). The human primary embryonic kidney cell line HEK293 and its stably pcDNA3.1- and ABCB1-transfected cell lines HEK293/pcDNA3.1 and HEK293/ABCB1 (Fig. 1B) were obtained from Dr S.E. Bates (National Cancer Institute, National Institutes of Health, Bethesda, MD, USA). All of the transfected cells were cultured in medium with 2 mg/ml G418 (Geneticin) (17). All resistant cells were authenticated through comparison of their fold resistance with that of the parental, drug-sensitive cells and examination of the expression levels of ABC transporters. All cells were grown in drug-free culture medium for >2 weeks before assays.
Cell viability assay
Cells harvested during logarithmic growth phase were seeded in 96-well plates in a volume of 190 µl/well. After 24 h of incubation, 10 µl of pristimerin full-range concentration was added to the 96-well plates. After 68 h of treatment, 10 µl MTT (10 mg/ml stock solution of saline) was added to each well for 4 h. Subsequently, the supernatant was removed, and MTT crystals were solubilized with 100 µl anhydrous DMSO each well. Thereafter, cell viability was measured by model 550 microplate reader (Bio-Rad) at 540 nm with 655 nm as reference filter. The 50% inhibitory concentration (IC50) was determined as the anticancer drug concentration causing 50% reduction in cell viability and calculated from the cytotoxicity curves (Bliss's software). Cell percent survival rate was calculated using the following formula: survival (%) = (mean experimental absorbance/mean control absorbance) × 100%.
Assessment of apoptosis morphology by Hoechst 33258 staining
After KB and KBv200 or HEK293/pcDNA3.1 and HEK293/ABCB1 cells grown on coverslips were treated with 2.0 µmol/l pristimerin for 48 h, both floating and trypsinized adherent cells were collected, washed once with ice-cold phosphate-buffered saline (PBS), fixed with 1 ml 4% paraformaldehyde for 20 min, and washed once with ice-cold PBS. Then the cells were incubated in 1 ml PBS containing 10 µmol/l Hoechst 33258 at 37°C for 30 min, washed twice, and observed using fluorescence microscopy with standard excitation filters (Leica Dmirb, Germany) in random microscopic fields at ×400 magnification (18).
Annexin V/PI double-staining assay
Annexin V and PI staining was performed using ApopNexin™ FITC Apoptosis Detection kit. Cells (6×105) were seeded in 25-cm2 flasks and allowed to attach for 24 h. After HEK293/pcDNA3.1 and HEK293/ABCB1 were treated with 0.5–2.0 µmol/l pristimerin for 48 h, both floating and attached cells were collected, washed with ice-cold PBS twice and resuspended in 200 µl 1X binding buffer containing Annexin V (1:50 according to the manufacturer's instructions) and 40 ng/sample of PI for 15 min at 37°C in the dark. Then the number of viable, apoptotic and necrotic cells were quantified by flow cytometer (Becton-Dickinson, USA) and analysed by CellQuest software. Cells were excited at 488 nm and the emissions of Annexin V at 525 nm and PI were collected through 610 nm band pass filters, respectively. At least 10,000 cells were analyzed for each sample. The percent apoptosis (%) = (the number of apoptotic cells/the number of total cells observed) × 100%.
Expression of ABCB1 analyzed by flow cytometry
Expression of ABCB1 in the cell lines KB, KBv200, HEK293/pcDNA3.1, and HEK293/ABCB1 were assessed through flow cytometry. After KBv200 cells were treated with 2.0 µmol/l pristimerin for indicated time, and HEK293/ABCB1 were treated with 0.5–4.0 µmol/l pristimerin for 48 h, single-cell suspensions were prepared and washed three times with isotonic PBS (supplemented with 0.5% bovine serum albumin). Then, 10 µl of phycoerythrin-conjugated, mouse anti-human ABCB1 antibody was mixed with 25 µl of cells (4×106 cells per ml). After incubation for 45 min at 4°C in the dark, the cells were washed twice with PBS (supplemented with 0.5% bovine serum albumin) and were resuspended in 400 µl of PBS for flow cytometric analysis. Isotype control samples were treated in an identical manner with phycoerythrin-conjugated mouse IgG2a (19).
Immunofluorescence assay
After KBv200 or HEK293/ABCB1 cells grown on coverslips were treated with 2.0 µmol/l pristimerin for 48 h, cells were fixed with 4% polyoxymethylene for 20 min, and Triton X-100 was added for 10 min at room temperature. Then, the cells were rinsed with PBS three times, and the non-specific binding sites were blocked in PBS with 1% bovine serum albumin for 1 h. The cells were incubated overnight at 4°C with ABCB1 antibody followed by FITC-conjugated secondary antibody at room temperature for 1 h. Subcellular distribution of P-gp protein was observed under fluorescence microscopy with standard excitation filters (Leica Dmirb) in random microscopic field at ×400 magnification (20).
RT-PCR and real-time RT-PCR
ABCB1 expression was assayed as described previously (21). After pristimerin treatment for indicated time, total cellular RNA was isolated with a TRIzol reagent RNA extraction kit (Molecular Research Center, Cincinnati, OH, USA), by following the manufacturer's instructions. The first-strand cDNA was synthesized with oligo(dT) primers with reverse transcriptase (Promega, Madison, WI, USA). The PCR primers were 5′-cccatcattgcaatagcagg-3′ (forward) and 5′-gttcaaacttctgctcctga-3′ (reverse) for ABCB1 and 5′-ctttggtatcgtggaagga-3′ (forward) and 5′-caccctgttgctgtagcc-3′ (reverse) for GAPDH. With the use of a GeneAmp 9700 PCR system (Applied Biosystems, Foster City, CA, USA), reactions were performed at 94°C for 2 min for initial denaturation and then at 94°C for 30 sec, 58°C for 30 sec and 72°C for 1 min. After 32 cycles of amplification, additional extension was performed at 72°C for 10 min. Products were resolved and examined through 1.5% agarose gel electrophoresis. Expected PCR products were 157 bp for ABCB1 and 475 bp for GAPDH.
Real-time RT-PCR was performed with a Bio-Rad CFX96 real-time system (Applied Biosystems). The geometric mean of GAPDH levels was used as an internal control, to normalize the variability in expression levels. The forward primer for GAPDH was 5′-gagtcaacggatttggtcgt-3′, and the reverse primer was 5′-gatctcgctcctggaagatg-3′. The forward primer for ABCB1 was 5′-gtggggcaagtcagttcatt-3′, and the reverse primer was 5′-tcttcacctccaggctcagt-3′. PCR was performed at 50°C for 2 min, at 95°C for 5 min, and then at 95°C for 15 sec and 60°C for 30 sec for 40 cycles. Relative quantification of ABCB1 was performed by using the threshold cycle difference method (22). To ensure reproducibility of the results, all genes were tested in triplicate in three independent experiments (19).
Protein turnover assay
After KBv200 cells were treated with 2.0 µmol/l pristimerin or DMSO for 12 h, the protein synthesis inhibitor cycloheximide (CHX, 10 µg/ml) was added to the cells. After treated for additional indicated time, the whole cells were harvested at different time-points and whole cell lysates were analyzed by western blotting (20).
Western blot analysis
Cells were harvested and washed twice with ice-cold PBS, and the pellets were collected in 1X lysis buffer [50 mmol/l Tris-HCl (pH 6.8), 10% glycerol, 2% SDS, 0.25‰ bromophenol blue, and 0.1 mol/l DTT] was added for 100 µl/5×106 cells. After heated at 95°C for 20 min, the lysates were centrifuged at 12,000 rpm for 10 min and the supernatant was collected. The protein concentration was determined by nucleic acid-protein analyzer (Beckman). Equal amount of lysate protein was separated on 8–12% SDS-PAGE and transferred onto polyvinylidene difluoride membrane (Pall). The non-specific binding sites were blocked with TBST buffer containing 5% non-fat dry milk for 2 h at room temperature. The membranes were incubated overnight at 4°C with specific primary antibodies, and the membranes were then washed thrice with TBST buffer and incubated at room temperature for 1 h with horseradish peroxidase-conjugated secondary antibody. After three washes with TBST buffer, the immuno-blots were visualized by the enhanced Phototope-Horseradish Peroxidase Detection kit purchased from Cell Signaling Technology and exposed to Kodak medical X-ray processor (Kodak, Rochester, NY, USA) (23).
Statistical analysis
For each protocol, three independent experiments were performed. Results were expressed as the mean ± standard error of the mean (SEM). Statistical calculations were performed by using SPSS16.0 software. Differences in measured variables between experimental and control groups were assessed by the Student's t-test. P<0.05 was indicative of significant difference and P<0.01 was indicative of very significant difference.
Results
Pristimerin shows equally potent anticancer effect on parental and ABCB1-mediated MDR cell lines
The cytotoxicity of pristimerin in different cell lines was determined with the MTT assay. As shown in Fig. 1, the IC50 values were 0.54±0.01 and 0.52±0.01 µmol/l for KB and KBv200 cell lines, respectively (P>0.05) (Fig. 1C). Moreover, the IC50 values were 0.69±0.04 and 0.93±0.20 µmol/l for HEK293/pcDNA3.1 and HEK293/ABCB1 cells, respectively (P>0.05) (Fig. 1D). The data suggested that pristimerin exerted potent and similar cytotoxicity to both parental and ABCB1-mediated MDR cell lines.
Pristimerin induces apoptosis in pairs of parental and ABCB1-mediated MDR cell lines
To observe the morphological characteristics of apoptosis, cells were stained with Hoechst 33258 after KB, KBv200, HEK293/pcDNA3.1 and HEK293/ABCB1 cells were exposed to 2.0 µmol/l pristimerin for 48 h respectively, and detected by fluorescence microscopy. Control cells showed even distribution of the stain and round homogeneous nuclei feature. Apoptotic cells displayed typical changes including reduction of cellular volume, staining bright and condensed or fragmented nucleus (Fig. 2A). For a further assessment of apoptosis induced by pristimerin, we examined the exposure of phosphatidylserine on the cell surface by using Annexin V and PI double staining. Flow cytometric analysis revealed that the percentage of apoptotic cells with Annexin V-positive but PI-negative cells increased gradually with concentration in pristimerin treated cells (Fig. 2B). The early percent apoptosis was 1.30±1.20, 1.50±1.85, 20.20±2.45 and 39.17±3.15% in HEK293/pcDNA3.1 cells and 1.60±1.06, 4.57±1.11, 19.47±0.93 and 36.33±2.12% in HEK293/ABCB1 cells, respectively (Fig. 2C). Intriguingly, we found that the early apoptosis rate was approximately equal between HEK293/pcDNA3.1 and HEK293/ABCB1 cells treated by the same concentration of pristimeirin (P>0.05).
Figure 2.

(A) Cell apoptosis induced by pristimerin was examined by Hoechst 33258 staining and observed under fluorescence microscope at ×400 magnification. KB and KBv200 or HEK293/pcDNA3.1 and HEK293/ABCB1 cells grown on coverslips were treated with 2.0 µmol/l pristimerin for 48 h. The apoptotic cells detected by the fluorescence microscopy displayed condensed and fragmented nuclei, shrinkage of cell volume, showed the morphological changes of the above cells after exposure to pristimerin. (B and C) Apoptosis analysis in HEK293/pcDNA3.1 and HEK293/ABCB1 cells were assessed by Annexin V/PI double staining. After cells were exposed to 0.5–2.0 µmol/l pristimerin for 48 h, respectively, the attached and detached cells were collected. Following staining with Annexin V and PI, cells were subjected to flow cytometer analysis. (B) Bottom right quadrant, cells stained mainly by Annexin V (early apoptotic cells); top right quadrant, cells stained by both PI and Annexin V (late apoptotic/necrotic secondary necrosis); top left quadrant, cells stained mainly by PI viable cells; bottom left quadrant, cells negative for both Annexin V and PI. (C) Percent apoptosis. Early apoptotic cell population with Annexin V-positive but PI-negative cells increased gradually from 1.30 to 39.17% and from 1.60 to 36.33% in HEK293/pcDNA3.1 and HEK293/ABCB1 cells, respectively. Means ± SD of three assays. *P<0.05; **P<0.01 versus control.
Pristimerin inhibits cell proliferation and induces cell apoptosis in pairs of MDR and corresponding parental cell lines
To further explore the mechanisms of pristimerin inducing apoptosis of ABCB1-mediated MDR cell lines, western blotting was done to detected the related protein expression. The MAPK (mitogen-activated protein kinase) and PI3K (phosphatidylinositol 3-kinase)/Akt signaling pathway are the two important signalling pathway controlling cancer cell proliferation. As shown in Fig. 3A, the expression of total Akt and ERK1/2 proteins were inhibited in a dose-dependent manner in KB and KBv200 cells. The results conformed that pristimerin induced apoptosis similarly in both parental and its corresponding ABCB1-mediated MDR cell lines. Western blotting also revealed pristimerin induced growth inhibition and apoptosis in stable transfected HEK293/ABCB1 cell lines, showing inhibition of Akt and ERK1/2 proteins, and downregulation of Bcl-2/Bax in a dose-dependent manner after pristimerin exposure in HEK293/ABCB1 cells (Fig. 3B), which suggested that intrinsic pathways might take part in pristimerin-induced apoptosis in ABCB1-overexpressing cell lines.
Figure 3.

Pristimerin induced growth inhibition and apoptosis in ABCB1-mediated MDR cell lines. KB, KBv200 (A) and HEK293/ABCB1 cells (B) were treated with various concentrations of pristimerin for 48 h, equal amounts of total cell lysates were loaded, then related proteins of cell proliferation and apoptosis were detected by western blotting.
Downregulation of P-gp by pristimerin is primarily dependent of P-gp protein stability
Our results show that pristimerin significantly downregulated P-gp expression in a dose-dependent manner in both drug-resistant KBv200 and stable transfected HEK293/ABCB1 cell lines (Fig. 3A and B). Moreover, after KBv200 cells were treated with 2.0 µmol/l pristimerin for the indicated time, and HEK293/ABCB1 cells were treated with 0.5–4.0 µmol/l pristimerin for 48 h, pristimerin induced decrease of P-gp membrane protein in a dose- and time-dependent manner detected by flow cytometry (Fig. 4). The expression of P-gp membrane protein was 0.17±0.15% for KB cells, 97.37±1.60, 93.23±1.00, 75.00±0.78, 55.17±1.27 and 27.17±1.96% for KBv200 with 2.0 µmol/l prestimerin treatment for 0, 8, 24, 36 and 48 h, respectively (Fig. 4A and C). The expression of P-gp membrane protein was 0.37±0.21% for HEK293/pcDNA3.1 cells, 95.23±1.91, 92.53±1.14, 86.10±3.50, 44.70±0.56 and 17.97±1.65% for HEK293/ABCB1 cells with 0, 0.5, 1.0, 2.0 and 4.0 µmol/l prestimerin treatment for 48 h, respectively (Fig. 4B and D).
Figure 4.

Pristimerin induces decrease of P-gp membrane protein in ABCB1-overexpressing cancer cells. KBv200 cells were treated with 2.0 µmol/l pristimerin for indicated time, and HEK293/ABCB1 were treated with 0.5–4.0 µmol/l pristimerin for 48 h. (A and B) Determination of the expression of P-gp of cell surface by flow cytometry, isotype control samples were treated in an identical manner with phycoerythrin-conjugated mouse IgG2a antibody. (C and D) Statistical analysis of expression of P-gp. Means ± SD of three assays. *P<0.05; **P<0.01, compared with 0 h and DMSO control, respectively.
To examine whether P-gp was downregulated by pristimerin at the mRNA level, we analyzed ABCB1 mRNA expression. As shown in Fig. 5A, after KBv200 and HEK293/ABCB1 cells were exposed to pristimerin, no significant change in ABCB1 mRNA expression was observed by semiquantitative RT-PCR. This result comfirmed that only a slight change (P>0.05) was detected by quantitative real-time RT-PCR analysis (Fig. 5B and C). These results indicated that the decrease of P-gp protein caused by pristimerin was independent of its mRNA level.
Figure 5.
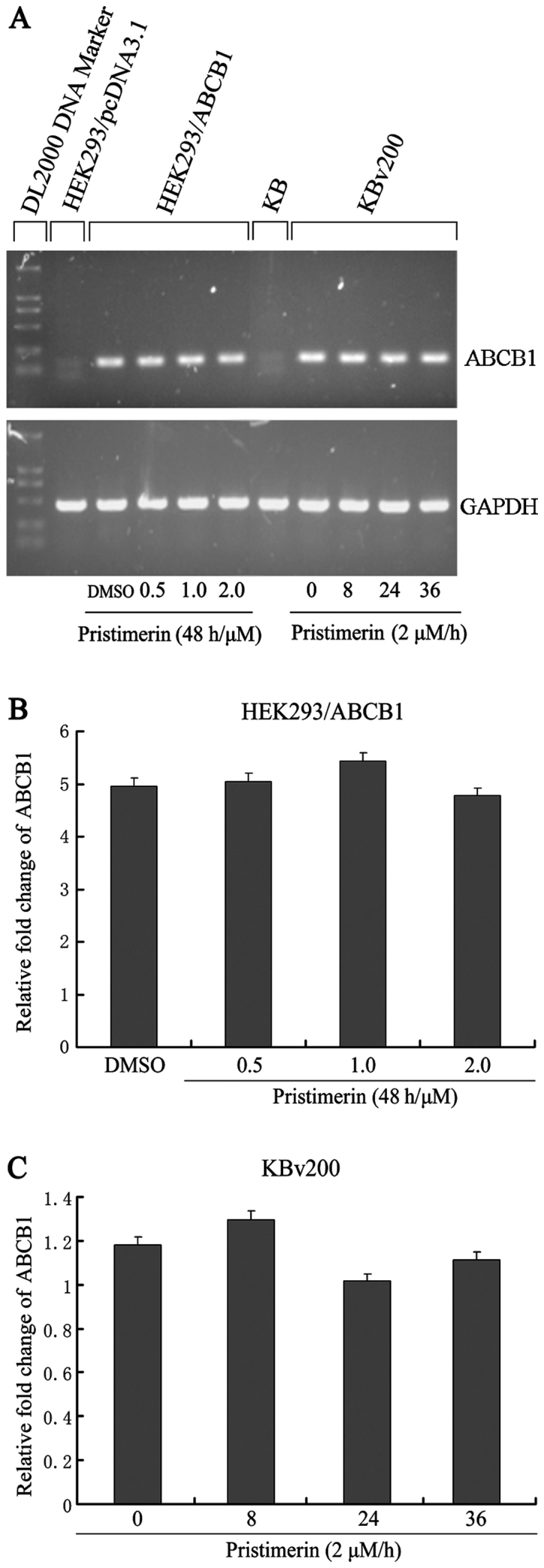
Pristimerin does not affect ABCB1 mRNA expression. HEK293/ABCB1 cells were treated with pristimerin at various concentrations for 48 h, and KBv200 exposed to 2.0 µmol/l pristimerin for various times, respectively. Semiquantitative RT-PCR (A) and quantitative real-time PCR (B and C) assays were performed for the analysis of ABCB1 mRNA expression, respectively. Values represent the mean ± SD for three independent experiments.
Like several other membrane proteins, the level of P-gp seems to be regulated by protein stability (20). To investigate whether pristimerin could downregulate P-gp through this mechanism, P-gp protein stability was evaluated by using cycloheximide to stop protein synthesis and measuring the amount of remaining P-gp protein at various time-points after 2.0 µmol/l pristimerin treatment by western blot analysis (Fig. 6A). The quantitative analyses shown that a half-life of 744.63 h for P-gp was significantly reduced to 22.09 h by pristimerin in KBv200 cells (Fig. 6B). Immunofluorescence study with anti-P-gp antibody was also performed. As shown in Fig. 7, the control cells had strong immunofluorescence at the plasma membrane. After 2.0 µmol/l pristimerin treatment for 48 h, the immunofluorescence at the plasma membrane was attenuated and replaced with diffuse cytoplasmic and nuclear punctate staining in drug-resistant KBv200 cells (Fig. 7B and C). Similar findings were observed in stable transfected HEK293/ABCB1 cells (Fig. 7E and F).
Figure 6.
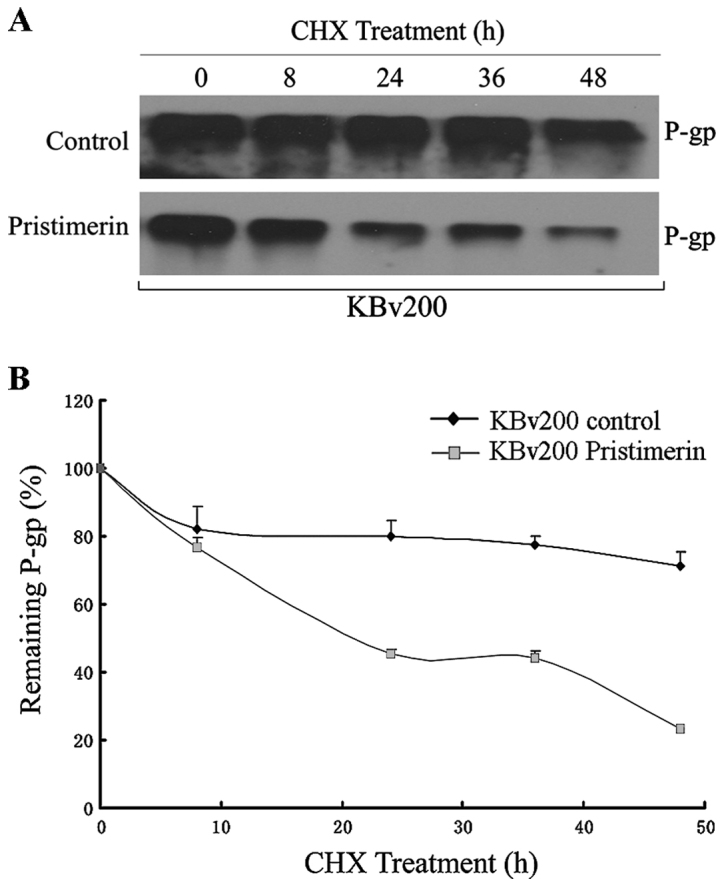
The downregulation of P-gp by pristimerin was primarily dependent of P-gp protein stability. Pristimerin decreased P-gp over time in KBv200 cells. After preincubation with 2.0 mmol/l pristimerin or control (DMSO) for 12 h, cells were treated with the protein synthesis inhibitor cycloheximide (CHX, 10 µg/ml) for the indicated time. The protein level of P-gp was detected by western blotting (A) and quantification of remaining P-gp at different time-points is presented as percent of initial P-gp at 0 h of CHX treatment (B).
Figure 7.

Pristimerin altered the subcellular distribution of P-gp. After KBv200 or HEK293/ABCB1 cells grown on coverslips were treated with 2.0 µmol/l pristimerin or DMSO control for 48 h, immunofluorescence assay was performed according to the experimental section.
Discussion
Cancer has become an increasing public health problem due to its high rates of morbidity and mortality. Conventional cancer chemotherapy is limited by MDR caused by overexpression of integral membrane transporters, such as P-gp, which can efflux intracellular anticancer drugs thus decrease drug accumulation. Developing new anticancer drugs which are efficient to MDR cells is a feasible strategy to overcome MDR. Our previous studies demonstrated that pristimerin induced HepG2 cell apoptosis through ROS-mediated mitochondrial dysfunction (16). This study further showed that pristimerin exhibited equally cytotoxicity and apoptosis in both MDR and parental cell lines (Figs. 1–3). These data seem to beautifully interpret the anti-MDR activities of pristimerin. Pristimerin could not be the substrate of the drug transporter P-gp. Therefore, there could be the same amount of pristimerin inside the cell to induce apoptosis, which will be finally translated into similar biological effects in MDR and corresponding parental cells.
It is noteworthy that we observed that pristimerin achieved effective downregulation of P-gp in a dose-dependent manner (Fig. 3). Moreover, pristimerin induced decrease of P-gp membrane protein in drug-resistant KBv200 cells and stable transfected HEK293/ABCB1 cells detected by flow cytometry (Fig. 4). Further investigation of the signal molecules revealed that components of the proliferation- and survival-associated MAPK and PI3K/Akt pathways were markedly inhibited by pristimerin (Fig. 3). We observed that P-gp downregulation contributed to equal cytotoxicity and apoptosis induced by pristimerin in KB and KBv200 cells (Fig. 3A). Therefore, it is plausible that P-gp interferes with DNA damage signaling to apoptotic machinery or with signal transductions within apoptotic cascades, which is further supported by the findings of the apoptosis inhibition of P-gp (24–26).
How is the expression of P-gp downregulated by pristimerin? Two possible mechanisms may be involved. First, pristimerin downregulated P-gp protein at the mRNA level; and second, ubiquitin-proteasome-, lysosome-, and calpain-mediated protein degradation systems regulated the degradation of P-gp by pristimerin. Interestingly, our data revealed that pristimerin decreased P-gp protein expression independent of its mRNA level (Fig. 5). However, protein turnover assay revealed that the half-life of P-gp protein was significantly reduced by pristimerin (Fig. 6), suggesting that downregulation of P-gp by pristimerin was primarily dependent of P-gp protein stability. We will further explore which protein degradation systems are involved in the degradation of P-gp protein caused by pristimerin.
In conclusion, we presented cell biological evidence that pristimerin inhibited cell proliferation and induced cell apoptosis in ABCB1-overexpressing cancer cells and was a potent cytotoxic agent to ABCB1-mediated MDR cells. Importantly, we demonstrated for the first time that pristimerin treatment led to downregulation of P-gp, and intracellular redistribution, which may represent a novel approach for the targeted therapy of ABCB1-overexpressing cancers.
Acknowledgements
This study was supported by the National Science Foundation for Young Scientists of Shanxi Province (2014021037-6), the Research Foundation of Collaborative Innovation Center for Cancer and the Priming Scientific Research Foundation for Ph.D. in Shanxi Datong University (no. 2010-B-11).
Glossary
Abbreviations
- ABCB1
ATP-binding cassette subfamily B member 1
- P-gp
P-glycoprotein
- MTT
3-(4,5)-dimethylthiazol-2-y1)-2,5-diphenyltetrazolium bromide
- MAPK
mitogen-activated protein kinase
- PI3K
phosphatidylinositol 3-kinase
- ERK
extracellular signal-regulated kinase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- v/v
volume per volume
- RT-PCR
reverse transcription-polymerase chain reaction
- PBS
phosphate-buffered saline
- TBST
Tris-buffered saline/Tween-20
References
- 1.Pérez-Tomás R. Multidrug resistance: Retrospect and prospects in anti-cancer drug treatment. Curr Med Chem. 2006;13:1859–1876. doi: 10.2174/092986706777585077. [DOI] [PubMed] [Google Scholar]
- 2.Eytan GD. Mechanism of multidrug resistance in relation to passive membrane permeation. Biomed Pharmacother. 2005;59:90–97. doi: 10.1016/j.biopha.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Johnson WW. P-glycoprotein-mediated efflux as a major factor in the variance of absorption and distribution of drugs: Modulation of chemotherapy resistance. Methods Find Exp Clin Pharmacol. 2002;24:501–514. doi: 10.1358/mf.2002.24.8.705071. [DOI] [PubMed] [Google Scholar]
- 4.Fojo T, Bates S. Strategies for reversing drug resistance. Oncogene. 2003;22:7512–7523. doi: 10.1038/sj.onc.1206951. [DOI] [PubMed] [Google Scholar]
- 5.Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–1166. doi: 10.1101/gr.GR-1649R. [DOI] [PubMed] [Google Scholar]
- 6.Mi YJ, Liang YJ, Huang HB, Zhao HY, Wu CP, Wang F, Tao LY, Zhang CZ, Dai CL, Tiwari AK, et al. Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters. Cancer Res. 2010;70:7981–7991. doi: 10.1158/0008-5472.CAN-10-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tong XZ, Wang F, Liang S, Zhang X, He JH, Chen XG, Liang YJ, Mi YJ, To KK, Fu LW. Apatinib (YN968D1) enhances the efficacy of conventional chemotherapeutical drugs in side population cells and ABCB1-overexpressing leukemia cells. Biochem Pharmacol. 2012;83:586–597. doi: 10.1016/j.bcp.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 8.Wang F, Mi YJ, Chen XG, Wu XP, Liu Z, Chen SP, Liang YJ, Cheng C, To KK, Fu LW. Axitinib targeted cancer stemlike cells to enhance efficacy of chemotherapeutic drugs via inhibiting the drug transport function of ABCG2. Mol Med. 2012;18:887–898. doi: 10.2119/molmed.2011.00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou WJ, Zhang X, Cheng C, Wang F, Wang XK, Liang YJ, To KK, Zhou W, Huang HB, Fu LW. Crizotinib (PF-02341066) reverses multidrug resistance in cancer cells by inhibiting the function of P-glycoprotein. Br J Pharmacol. 2012;166:1669–1683. doi: 10.1111/j.1476-5381.2012.01849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng LS, Wang F, Li YH, Zhang X, Chen LM, Liang YJ, Dai CL, Yan YY, Tao LY, Mi YJ, et al. Vandetanib (Zactima, ZD6474) antagonizes ABCC1- and ABCG2-mediated multidrug resistance by inhibition of their transport function. PLoS One. 2009;4:e5172. doi: 10.1371/journal.pone.0005172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao JM, Wu WJ, Zhang JW, Konishi Y. The dihydro-beta-agarofuran sesquiterpenoids. Nat Prod Rep. 2007;24:1153–1189. doi: 10.1039/b601473a. [DOI] [PubMed] [Google Scholar]
- 12.Brinker AM, Ma J, Lipsky PE, Raskin I. Medicinal chemistry and pharmacology of genus Tripterygium (Celastraceae) Phytochemistry. 2007;68:732–766. doi: 10.1016/j.phytochem.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salminen A, Lehtonen M, Suuronen T, Kaarniranta K, Huuskonen J. Terpenoids: Natural inhibitors of NF-kappaB signaling with anti-inflammatory and anticancer potential. Cell Mol Life Sci. 2008;65:2979–2999. doi: 10.1007/s00018-008-8103-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu CC, Chan ML, Chen WY, Tsai CY, Chang FR, Wu YC. Pristimerin induces caspase-dependent apoptosis in MDA-MB-231 cells via direct effects on mitochondria. Mol Cancer Ther. 2005;4:1277–1285. doi: 10.1158/1535-7163.MCT-05-0027. [DOI] [PubMed] [Google Scholar]
- 15.Nagase M, Oto J, Sugiyama S, Yube K, Takaishi Y, Sakato N. Apoptosis induction in HL-60 cells and inhibition of topoisomerase II by triterpene celastrol. Biosci Biotechnol Biochem. 2003;67:1883–1887. doi: 10.1271/bbb.67.1883. [DOI] [PubMed] [Google Scholar]
- 16.Guo Y, Zhang W, Yan YY, Ma CG, Wang X, Wang C, Zhao JL. Triterpenoid pristimerin induced HepG2 cells apoptosis through ROS-mediated mitochondrial dysfunction. J BUON. 2013;18:477–485. [PubMed] [Google Scholar]
- 17.Robey RW, Honjo Y, Morisaki K, Nadjem TA, Runge S, Risbood M, Poruchynsky MS, Bates SE. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. 2003;89:1971–1978. doi: 10.1038/sj.bjc.6601370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu QL, Wu XP, Liang YJ, Chen LM, Ding Y, Fu LW. P-glycoprotein is not involved in pathway of anti-Fas/Fas-induced apoptosis in KBv200 cells. World J Gastroenterol. 2005;11:3544–3548. doi: 10.3748/wjg.v11.i23.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao XQ, Xie JD, Chen XG, Sim HM, Zhang X, Liang YJ, Singh S, Talele TT, Sun Y, Ambudkar SV, et al. Neratinib reverses ATP-binding cassette B1-mediated chemotherapeutic drug resistance in vitro, in vivo, and ex vivo. Mol Pharmacol. 2012;82:47–58. doi: 10.1124/mol.111.076299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan YY, Zheng LS, Zhang X, Chen LK, Singh S, Wang F, Zhang JY, Liang YJ, Dai CL, Gu LQ, et al. Blockade of Her2/neu binding to Hsp90 by emodin azide methyl anthraquinone derivative induces proteasomal degradation of Her2/neu. Mol Pharm. 2011;8:1687–1697. doi: 10.1021/mp2000499. [DOI] [PubMed] [Google Scholar]
- 21.Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG, Ashby CR, Jr, Huang Y, Robey RW, Liang YJ, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–7914. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Yan Y, Su X, Liang Y, Zhang J, Shi C, Lu Y, Gu L, Fu L. Emodin azide methyl anthraquinone derivative triggers mitochondrial-dependent cell apoptosis involving in caspase-8-mediated Bid cleavage. Mol Cancer Ther. 2008;7:1688–1697. doi: 10.1158/1535-7163.MCT-07-2362. [DOI] [PubMed] [Google Scholar]
- 24.Johnstone RW, Cretney E, Smyth MJ. P-glycoprotein protects leukemia cells against caspase-dependent, but not caspase-independent, cell death. Blood. 1999;93:1075–1085. [PubMed] [Google Scholar]
- 25.Johnstone RW, Ruefli AA, Tainton KM, Smyth MJ. A role for P-glycoprotein in regulating cell death. Leuk Lymphoma. 2000;38:1–11. doi: 10.3109/10428190009060314. [DOI] [PubMed] [Google Scholar]
- 26.Ruefli AA, Smyth MJ, Johnstone RW. HMBA induces activation of a caspase-independent cell death pathway to overcome P-glycoprotein-mediated multidrug resistance. Blood. 2000;95:2378–2385. [PubMed] [Google Scholar]