

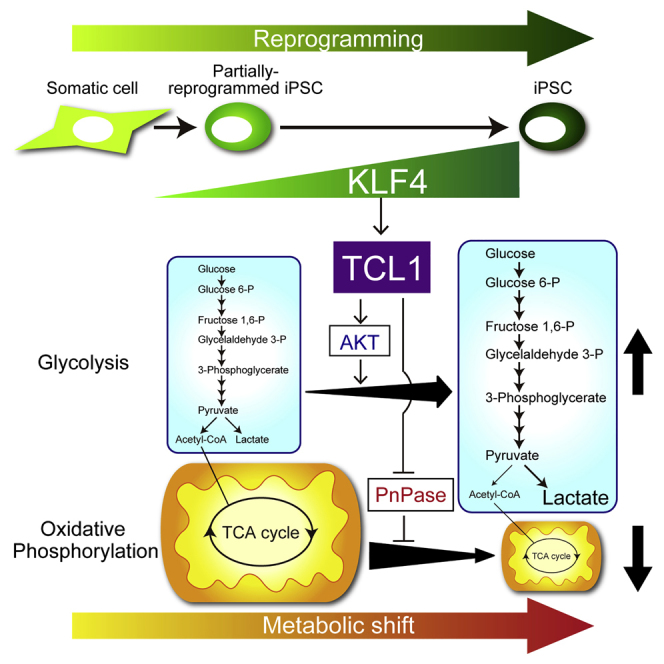
Summary
Reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) is accompanied by morphological, functional, and metabolic alterations before acquisition of full pluripotency. Although the genome-wide effects of the reprogramming factors on gene expression are well documented, precise mechanisms by which gene expression changes evoke phenotypic responses remain to be determined. We used a Sendai virus-based system that permits reprogramming to progress in a strictly KLF4-dependent manner to screen for KLF4 target genes that are critical for the progression of reprogramming. The screening identified Tcl1 as a critical target gene that directs the metabolic shift from oxidative phosphorylation to glycolysis. KLF4-induced TCL1 employs a two-pronged mechanism, whereby TCL1 activates AKT to enhance glycolysis and counteracts PnPase to diminish oxidative phosphorylation. These regulatory mechanisms described here highlight a central role for a reprogramming factor in orchestrating the metabolic shift toward the acquisition of pluripotency during iPSC generation.
Keywords: iPS cells, reprogramming, KLF4, TCL1, metabolic shift, glycolysis, oxidative phosphorylation
Graphical Abstract
Highlights
-
•
KLF4 upregulates Tcl1 expression by directly binding to its enhancer and promoter
-
•
TCL1 enhances glycolysis by activating AKT during reprogramming
-
•
TCL1 reduces oxidative phosphorylation by counteracting PnPase during reprogramming
-
•
TCL1 promotes the metabolic shift to facilitate reprogramming
In this article, Nishimura, Hisatake, and colleagues screened for genes regulated by KLF4 and identified Tcl1 as its major target gene, which promotes the metabolic shift from oxidative phosphorylation to glycolysis. KLF4-induced TCL1 activates AKT to enhance glycolysis and counteracts PnPase to diminish oxidative phosphorylation, thereby facilitating the progression of reprogramming toward full pluripotency.
Introduction
Forced expression of OCT4, SOX2, KLF4, and c-MYC or other combinations of reprogramming factors reprogram somatic cells into induced pluripotent stem cells (iPSCs), which attract much attention for their potential applications in regenerative medicine and drug development, as well as for understanding how cells specify their fate during reprogramming and normal development (Stadtfeld and Hochedlinger, 2010, Takahashi and Yamanaka, 2016). Cells that undergo reprogramming progress through distinct stages, which can be distinguished by the expression of THY1, alkaline phosphatase (AP), and SSEA1 (Polo et al., 2012), initially losing somatic cell characteristics before acquiring full pluripotency. Reprogramming factors, in general, are transcription factors that alter gene expression and epigenetic status, which ultimately specify the cell fate (Theunissen and Jaenisch, 2014). Therefore, genome-wide analyses of gene expression and epigenetic status provide comprehensive views on the mechanism of reprogramming.
In addition to alterations in gene expression and epigenetic status, cells undergo metabolic changes during reprogramming (Folmes et al., 2011, Samavarchi-Tehrani et al., 2010). Embryonic stem cells (ESCs), which originally comprise the inner cell mass, reside in the low-oxygen environment (Fischer and Bavister, 1993) and have few small mitochondria (Cho et al., 2006, St John et al., 2005), utilizing glycolysis as a main source of ATP production (Xu et al., 2013). By contrast, differentiated somatic cells largely depend on oxidative phosphorylation in mitochondria for efficient ATP production (DeBerardinis et al., 2008). Thus, somatic cell reprogramming by necessity entails a metabolic shift from oxidative phosphorylation to glycolysis, which has been corroborated by recent studies (Folmes et al., 2011, Prigione et al., 2010). Indeed, deliberate acceleration of glycolysis or inhibition of oxidative phosphorylation increases the reprogramming efficiency (Prigione et al., 2014). Furthermore, enhanced glycolysis produces higher amounts of metabolic intermediates that are used as cofactors by chromatin-modifying enzymes (Moussaieff et al., 2015). Thus, the metabolic shift and epigenetic regulation, both initiated by the reprogramming factors, are likely to concur during the reprogramming process. However, the molecular mechanisms by which the reprogramming factors initiate and execute the metabolic shift remains less well known.
We developed a unique gene transfer system termed SeVdp (Sendai virus defective and persistent) vector, which stably expresses multiple genes from a single vector with a relatively constant stoichiometry without integrating into the host cell genome (Nishimura et al., 2011). An SeVdp-based vector, SeVdp(KOSM), which harbors the four Yamanaka factors, generates iPSCs efficiently from various sources of somatic cells (Kyttala et al., 2016, Matsumoto et al., 2016). Modifying SeVdp(KOSM), we devised SeVdp(fK-OSM) that expresses KLF4 tagged with a destabilizing domain (DD) (Banaszynski et al., 2006), the expression level of which can be manipulated by a small molecule, Shield1. In the SeVdp(fK-OSM)-infected cells, the KLF4 level, reduced to ∼30% by the DD, is readily restored to its original level by the addition of 100 nM Shield1. Reprogramming with SeVdp(fK-OSM) at a defined KLF4 level generates partially reprogrammed iPSCs, termed paused iPSCs, which have stalled at a specific intermediate stage but nonetheless resume reprogramming toward full pluripotency once the KLF4 level is restored by increased Shield1 (Nishimura et al., 2014). Because the SeVdp(fK-OSM)-based system, named the SeVdp-based stage-specific reprogramming system (3S reprogramming system), allows reprogramming to progress strictly in a KLF4-dependent manner, the system enables us to analyze the role for KLF4 in a gene regulatory network, which may occur only transiently during the complex process of reprogramming.
In this article, we searched for genes that are induced by KLF4 in a dose-dependent manner and identified Tcl1 as its direct target. KLF4 induces Tcl1 transcription during reprogramming via direct binding to its enhancer and promoter regions, concurrently changing repressive histone marks into active ones at the Tcl1 promoter. TCL1 induced by KLF4 activates AKT to enhance glycolysis and inhibits mitochondrial polynucleotide phosphorylase (PnPase) to suppress oxidative phosphorylation. Knockdown of Tcl1 expression not only eliminates the metabolic shift but severely compromises the expression of Cdh1 and Rex1 as well. Thus, KLF4-promoted metabolic shift concurs with the acquisition of pluripotency and is essential for generating fully reprogrammed iPSCs.
Results
Identification of Key Target Genes of KLF4 during Reprogramming
To gain insight into the physiological events that KLF4 regulates during reprogramming toward full pluripotency, we selected for genes that are regulated by KLF4 in a dose-dependent manner. We performed genome-wide expression analyses of the paused iPSCs, iPSCs(Low-K) and iPSCs(High-K). iPSCs(Low-K), generated by SeVdp(fK-OSM) without Shield1, express a low level of KLF4 and are THY1−, AP+, SSEA1−, and NANOG−. iPSCs(High-K), generated by SeVdp(fK-OSM) with 100 nM Shield1, express a high level of KLF4 and are THY1−, AP+, SSEA1+, and NANOG+ (Figure 1A) (Nishimura et al., 2014). The transition from iPSCs(Low-K) to iPSCs(High-K), based upon our previous principal component analyses, corresponds to a middle-to-late stage of reprogramming (Nishimura et al., 2014). Comparison of these two datasets identified 152 genes that are highly expressed only in iPSCs(High-K), which were further narrowed down to 115 genes by the presence of direct KLF4 binding around their transcriptional start sites in mouse ESCs (Chen et al., 2008) (Figure 1B).
Figure 1.

Screening of Downstream Targets of KLF4 Using the 3S-reprogramming System
(A) Outline of generating iPSCs(Low-K) and iPSCs(High-K) in the 3S-reprogramming system and their characteristics.
(B) Flowchart of the screening. Genome-wide gene expression datasets (Nishimura et al., 2014) were used to select 152 genes that are expressed highly in iPSCs(High-K), but not in iPSCs(Low-K). Other two datasets (Chen et al., 2008, Polo et al., 2012) were also used to enrich genes whose expression is regulated by KLF4 and induced in a late stage of reprogramming.
(C) Changes in the mRNA expression level of seven candidate genes as well as Cdh1 and Nanog genes during iPSC generation. MEFs were reprogrammed by SeVdp(KOSM) and their mRNA levels were determined at days 0, 2, 6, 13, and 25. Data represent means ± SEM of three independent experiments.
(D) KLF4 dose-dependent mRNA expression of six candidate genes as well as Cdh1 and Nanog genes. MEFs were reprogrammed for 27 days by SeVdp(fK-OSM) with 0, 10, 30, or 100 nM of Shield1, and their mRNA levels were determined. Data represent means ± SEM of three independent experiments.
The reprogramming pathway toward pluripotency may vary among different reprogramming systems (O'Malley et al., 2013, Parchem et al., 2014). To enrich for genes whose late-stage upregulation is shared among different reprogramming systems and is therefore universally important, we referred to yet another database to select for genes that are induced at a late stage (>9 days) of reprogramming in a lentivirus-mediated secondary reprogramming system (Polo et al., 2012) (Figure 1B). Among the selected ten genes (Figure S1A), qRT-PCR showed that seven of them (Apoc1, Dmtn, Foxh1, Mapt, Tcl1, Tmem8, and Rex1) exhibited a late-stage upregulation during reprogramming in our SeVdp(KOSM)-based system (Figures 1C and S1B). Six of the genes (Apoc1, Dmtn, Foxh1, Tcl1, Tmem8, and Rex1) were upregulated more or less proportionately to the KLF4 level (Figures 1D and S1C).
To examine the relative importance of the six candidate genes, we tested whether each gene could complement the low level of KLF4 expressed from SeVdp(fK-OSM) for reprogramming. Each candidate gene was transduced into mouse embryonic fibroblasts (MEFs) by the MCsΔYY1 retrovirus vector, which is refractory to silencing during reprogramming due to mutations in the YY1 binding site and primer binding site (Iba et al., 2003, Kitamura et al., 2003). The MEFs expressing each candidate gene were subsequently infected by SeVdp(fK-OSM), which, without Shield1, generates only paused iPSCs(Low-K). Retrovirus-mediated expression of only FOXH1 and TCL1 increased the mRNA levels of early and late pluripotency markers (Cdh1 and Rex1, respectively) (Figure 2A) and the number of SSEA1+ colonies (Figure S2A) similarly to those by the retrovirus-mediated expression of KLF4 or in iPSCs(High-K). TCL1 increased the mRNA levels of other pluripotency markers (Fbxo15, Oct4, Esrrb, and Nanog) to similar degrees by KLF4 (Figure S2B). Next, we tested if each candidate gene, when expressed from the retrovirus, resumes reprogramming of paused iPSC(Low-K) clones. Paused iPSC(Low-K) clones were initially generated by 20 days of infection with SeVdp(fK-OSM) without Shield1 and subsequently transduced with the MCsΔYY1 retrovirus vector expressing each candidate gene. The expression levels of Cdh1 and Rex1 were increased significantly only when TCL1 was expressed in paused iPSC(Low-K) clones (Figure 2B). TCL1 also increased the number of SSEA1+ colonies (Figure S2C) and the mRNA levels of Fbxo15, Oct4, Esrrb, and Nanog (Figure S2D). These results indicate that Tcl1 is upregulated by KLF4 in a dose-dependent manner and may be its key target critical for bypassing or resuming the paused reprogramming caused by the low KLF4 level. We then asked if Tcl1 was a major downstream KLF4 target gene that mediated the effects of KLF4 to promote reprogramming. The expression of TCL1 was first knocked down by transducing a retrovirus that expresses small hairpin RNA (shRNA) against the Tcl1 gene into MEFs, which were then reprogrammed by SeVdp(fK-OSM) with or without 100 nM Shield1. The shRNA reduced the expression levels of both Cdh1 and Rex1 in iPSCs(High-K) almost to those observed in iPSCs(Low-K) (Figure 2C), indicating that Tcl1 is a major mediator of the KLF4 effects during reprogramming.
Figure 2.

TCL1 Enhances Reprogramming as a Downstream Target of KLF4
(A) Cdh1 and Rex1 mRNA levels in MEFs reprogrammed for ∼30 days by SeVdp(fK-OSM). Two days prior to the reprogramming, MEFs were infected with a retrovirus expressing each candidate gene and selected with G418. Data represent means ± SEM of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005 versus iPSCs(Low-K).
(B) Cdh1 and Rex1 mRNA levels in paused iPSC(Low-K) clones that express each candidate gene after ∼30 days of retrovirus infection. MEFs were first reprogrammed by SeVdp(fK-OSM) without Shield1 to generate paused iPSC(Low-K) clones, which were then infected with an empty retrovirus or a retrovirus expressing each candidate gene and selected with G418. Control paused iPSC(Low-K) clones were not infected with retrovirus but treated with 100 nM Shield1 to allow them to undergo reprogramming for ∼30 days. Data represent means ± SEM of three independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.005 versus paused iPSC(Low-K) clones infected with an empty retrovirus (Low-K + vector).
(C) Cdh1 and Rex1 mRNA levels in MEFs reprogrammed by SeVdp(fK-OSM) with or without the retrovirus-transduced shRNA against Tcl1 at day 30. Data represent means ± SEM of three independent experiments. ∗p < 0.05.
To further confirm the effect of TCL1 on reprogramming, we constructed a new SeVdp vector that expresses TCL1 in addition to the four Yamanaka factors (SeVdp(fKiT-OSM)) (Figure 3A). Without Shield1, SeVdp(fKiT-OSM) expressed KLF4 only at a low level, as in the case of SeVdp(fK-OSM) (Figure 3B); however, SeVdp(fKiT-OSM) induced expression of pluripotency markers, Cdh1, Fbxo15, Oct4, and Esrrb, at levels comparable with those by SeVdp(fK-OSM) with 100 nM Shield1 (iPSCs(High-K)) or by SeVdp(KOSM) (Figure 3C). Notably, SeVdp(fKiT-OSM) markedly augmented the expression of the late reprogramming markers, Rex1 and Nanog, to the levels even higher than those by SeVdp(KOSM), a representative Sendai virus vector that establishes fully reprogrammed iPSCs from various somatic cells (Kyttala et al., 2016, Matsumoto et al., 2016) (Figure 3C). Moreover, SeVdp(fKiT-OSM)-infected cells showed earlier induction of NANOG protein than SeVdp(KOSM)-infected ones (Figures 3D and 3E). The effect of TCL1, however, was diminished when the KLF4 level was increased by the addition of Shield1 (Figure S3A). TCL1 also facilitated the reprogramming of MEFs by retrovirus-mediated gene transfer of the four Yamanaka factors (Figures S3B and S3C). When transplanted into mice, the derived iPSC clone, from which the SeVdp(fKiT-OSM) vector was removed by small interfering RNA against the SeV L gene (Nishimura et al., 2011), formed teratomas consisting of three germ layers (Figure 3F), indicating that SeVdp(fKiT-OSM) generates fully reprogrammed iPSCs. Thus, when expressed from the start of reprogramming, TCL1 not only complements the low level of KLF4 for full pluripotency, but also appears to expedite the reprogramming process, suggesting that TCL1 controls important physiological events that may otherwise delay or block reprogramming.
Figure 3.

Expression of TCL1 with the Reprogramming Factors Expedites the Reprogramming Process
(A) Structures of SeVdp(fK-OSM) and its derivative, SeVdp(fKiT-OSM), which expresses TCL1 in addition to the four reprogramming factors.
(B) The KLF4 protein levels from SeVdp(fKiT-OSM). Left panel, western blots to determine the relative KLF4 protein levels. Cell extracts were prepared from MEFs infected for 2 days by either SeVdp(fKiT-OSM) or SeVdp(fK-OSM). High-K and Low-K indicate MEFs cultured with or without Shield1, respectively. An anti-α-tubulin was used to normalize the sample amounts. Right panel, the relative KLF4 protein levels were calculated from the intensity of each band in the blots. Data represent means ± SEM of three independent experiments. ∗∗∗p < 0.005. n.s., not significant.
(C) Expression of pluripotency markers in MEFs infected with the indicated vector and then cultured with or without Shield1 (High-K or Low-K, respectively) for 21 days. Data represent means ± SEM of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005.
(D) Induction of Nanog-GFP by co-expression of TCL1 with the four reprogramming factors. Nanog-GFP MEFs were reprogrammed with the indicated vector for 11 days, and GFP fluorescence was used to assess the expression of the Nanog-GFP reporter transgene. Scale bars, 100 μm.
(E) Time course analyses of Nanog-GFP+ colony induction. MEFs were reprogrammed as in (D) and the ratio of the number of Nanog-GFP+ colonies to that of total colonies was calculated at each indicated day. Data represent means ± SEM of three independent experiments. ∗∗∗p < 0.005 versus SeVdp(KOSM) vector.
(F) Histology of teratomas derived from iPSCs generated with SeVdp(fKiT-OSM). Vector removal from iPSC clones was confirmed by immunostaining of SeV NP protein (a). A vector-free clone (clone no. 1) was injected subcutaneously into severe combined immunodeficiency mice to form teratomas. Teratoma sections stained with H&E showed all three germ layers. Scale bars, 100 μm (b).
KLF4 Directly Regulates Tcl1 Expression in a Dose-Dependent Manner
When iPSCs(Low-K) transition into iPSCs(High-K), KLF4 upregulates transcription of Tcl1 (Figure 1D). The chromatin immunoprecipitation (ChIP) sequencing analyses in mouse ESCs (Chen et al., 2008) indicate the presence of two peaks of KLF4 binding, presumably corresponding to an enhancer (−2.6 kb) and the promoter (−0.2 kb) upstream of the Tcl1 transcription start site (Figure 4A). To confirm whether KLF4 regulates Tcl1 expression through the putative enhancer and promoter, we constructed two reporter plasmids in which the luciferase gene is driven by both the Tcl1 enhancer and promoter regions (pL-Tcl1-Luc) or by the promoter region alone (pS-Tcl1-Luc) (Figure 4A). We used OCT4 as a positive control for activation of the Tcl1 promoter because OCT4 binds and directly upregulates the Tcl1 promoter (Matoba et al., 2006). As shown in Figure 4B, co-expressed KLF4 increased pL-Tcl1-Luc by 2.4-fold and pS-Tcl1-Luc by 1.5-fold, indicating that the KLF4 binding sites in both the enhancer and promoter regions are functional and display additive effects. In these assays using transiently transfected plasmids, however, even overexpressed KLF4 elicited a mere 2.4-fold increase of the Tcl1 reporter gene activity, a response that is far below the upregulation of the endogenous Tcl1 gene (over 60-fold) when iPSCs(Low-K) transition into iPSCs(High-K) (Figure 4D). These results raise the possibility that additional layers of regulatory mechanisms such as epigenetic changes may operate on the Tcl1 chromatin when KLF4 upregulates the endogenous Tcl1 gene.
Figure 4.

KLF4 Directly Regulates Tcl1 Expression by Binding to Its Regulatory Regions
(A) KLF4 binding on the Tcl1 gene in mouse ESCs is shown by the red graph created by GeneProf (http://www.geneprof.org), based upon the published ChIP data (Chen et al., 2008). Red lines along the Tcl1 gene indicate the positions of KLF4 consensus sequences. Gray bars indicate the regions amplified in ChIP-qPCR, which overlap with the putative enhancer, promoter, and gene body. The regulatory regions included in the reporter plasmids, pL-Tcl1-Luc and pS-Tcl1-Luc, are also shown at the bottom.
(B) Luciferase reporter gene assays for the putative regulatory regions of the Tcl1 gene. MEFs were collected 2 days after co-transfection with the indicated reporter plasmid (pL-Tcl1-Luc or pS-Tcl1-Luc) and an expression plasmid for either KLF4 or OCT4. Data represent means ± SEM of six independent experiments. ∗p < 0.05, ∗∗p < 0.01 versus MEFs transfected only with the reporter plasmid (control).
(C) Structure of SeVdp(fKg-OSM) that expresses FLAG-tagged KLF4 fused with the destabilizing domain (DD). SeVdp(fKg-OSM) reprograms MEFs into iPSCs(Low-K) or iPSCs(High-K) similarly to SeVdp(fK-OSM).
(D) Tcl1 mRNA expression in MEFs infected with vectors expressing DD-fused KLF4 with or without the FLAG tag; namely, SeVdp(fKg-OSM) or SeVdp(fK-OSM), respectively. SeVdp vector-infected MEFs were reprogrammed with or without Shield1 for 14 days. Data represent means ± SEM of three independent experiments. ∗∗∗p < 0.005.
(E) KLF4 or OCT4 binding to the enhancer, promoter, and gene body at the Tcl1 locus. ChIP assays were performed for iPSCs(Low-K) or iPSCs(High-K) at day 50 using an anti-FLAG or anti-OCT4 antibody. Data represent means ± SEM of three independent experiments. ∗p < 0.05.
(F and G) KLF4-dependent alterations in active (H3K4me1, H3K4me3, and H3K27Ac) and repressive (H3K27me3) histone modifications at the Tcl1 enhancer (F) or promoter region (G). ChIP assays were performed using antibodies against indicated histone modifications for MEFs, iPSCs(Low-K), iPSCs(High-K), or mouse ESCs. Data represent means ± SEM of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005.
To explore the KLF4-mediated regulation of the endogenous Tcl1 gene, we constructed a Sendai virus vector, SeVdp(fKg-OSM), which expresses FLAG-tagged KLF4 fused with the DD (Figure 4C). This vector permits regulation of the KLF4 level by Shield1 and detection of the chromatin-bound exogenous KLF4 with an anti-FLAG antibody. SeVdp(fKg-OSM) was similar to SeVdp(fK-OSM) with respect to both Shield1-mediated regulation of the KLF4 level (Figures S4A and S4B) and the reprogramming efficiency of MEFs (Figures 4D and S4C). ChIP analyses of iPSCs(Low-K) and iPSCs(High-K), both of which were generated with SeVdp(fKg-OSM), revealed that iPSCs(Low-K) showed little KLF4 binding on both the Tcl1 enhancer and promoter regions; by contrast, iPSCs(High-K) showed substantial KLF4 binding at the enhancer region and, more markedly, on the promoter region (Figure 4E). Thus, the binding of KLF4 to the Tcl1 promoter region increases significantly during the transition from iPSCs(Low-K) into iPSCs(High-K). Despite the invariable level of OCT4 between iPSCs(Low-K) and iPSCs(High-K) (Nishimura et al., 2014), the binding of OCT4 to the Tcl1 promoter region parallels that of KLF4 (Figure 4E), indicating the cooperative binding of KLF4 and OCT4 on the Tcl1 gene.
To analyze if KLF4 and OCT4 bindings alter the epigenetic status of the Tcl1 promoter region, we performed ChIP assays for H3K27me3 and H3K4me3, which are important histone marks that undergo reciprocal changes during reprogramming (Zhang et al., 2015). The Tcl1 enhancer region had a reduced level of a repressive mark, H3K27me3, in iPSCs(Low-K) as compared with MEFs (Figure 4F), while enhancer marks (H3K4me1 and H3K27Ac) (Creyghton et al., 2010) remained relatively constant. The Tcl1 promoter region also had a markedly reduced level of H3K27me3, which appear to have been replaced by H3K27Ac in iPSCs(Low-K) as compared with MEFs (Figure 4G, middle and right panels). The levels of H3K4me3, a mark of active transcription (Zhang et al., 2015), in the Tcl1 promoter region were similarly low in both MEFs and iPSCs(Low-K). However, iPSCs(High-K) showed increased H3K4me3 around the Tcl1 transcription start site (Figure 4G, left panel), probably because of the bindings of KLF4 to the enhancer region (Figure 4E, upper panel) as well as of KLF4 and OCT4 to the promoter region (Figure 4E, upper and lower panels). Together, these results suggest that, during iPSC generation, the Tcl1 gene may be regulated by two steps; first by OCT4 binding to the enhancer region, which removes the repressive histone mark both at the enhancer and promoter regions, and then by KLF4 binding to the enhancer region, followed by OCT4 and KLF4 bindings to the promoter, which deposit active histone marks around the transcription start site.
KLF4-Induced TCL1 Activates the AKT Pathway to Enhance Glycolysis during Reprogramming
A well-recognized function of TCL1 is to bind the AKT kinase and enhance its activity as a coactivator (Laine et al., 2000). To dissect the molecular mechanism by which TCL1 promotes reprogramming, we first analyzed the roles for AKT during the transition from iPSCs(Low-K) to iPSCs(High-K). As shown in Figures 5A and 5B, we found that an active form of AKT, phosphorylated at Ser473, is increased in iPSCs(High-K) or when exogenous TCL1 was expressed in iPSCs(Low-K), suggesting that KLF4-induced TCL1 enhances the AKT activity. A highly selective inhibitor of AKT, MK2206 (Hirai et al., 2010), markedly attenuated the induction of Cdh1 and Rex1 by the increased KLF4 level or exogenously expressed TCL1 (Figures 5C and 5D). The attenuated Cdh1 and Rex1 levels were similar to those observed when the endogenous TCL1 expression was knocked down by shRNA (Figure 2C). Moreover, an AKT activator, SC79 (Jo et al., 2012), upregulated Cdh1 but not Rex1, partly recapitulating the effects of the increased KLF4 level (Figure 5C). Taken together, these data indicate that KLF4-induced TCL1 activates the AKT pathway, which in turn promotes reprogramming toward pluripotency.
Figure 5.

TCL1 Upregulates Pluripotency Markers through AKT Activation
(A) Activation of AKT by KLF4 or TCL1. Western blotting was performed using anti-phosphorylated AKT (p-AKT (S473)), anti-AKT, or anti-α-tubulin antibody for the whole-cell extracts prepared from MEFs, iPSCs(Low-K), iPSCs(High-K), or iPSCs(Low-K) expressing TCL1 at day 30.
(B) The intensity of each band in (A) was quantified using LAS4000. Data represent means ± SEM of three independent experiments. ∗p < 0.05.
(C and D) Cdh1 and Rex1 mRNA levels in MEFs reprogrammed by SeVdp(fK-OSM) with or without Shield1 for 30 days. The cells were treated only with an inhibitor (1 μM MK2206) or activator (4 μg/mL SC79) of AKT (C), or infected with a TCL1-expressing retrovirus and then treated with MK2206 (D). Data represent means ± SEM of three independent experiments. ∗p < 0.05, ∗∗∗p < 0.005.
See also Figures S5, S6, and S7 and Table S1.
Although the AKT pathway is involved in cell proliferation and survival (Manning and Cantley, 2007), iPSCs(Low-K) and iPSCs(High-K), which differ in AKT activity (Figures 5A and 5B), showed unexpectedly similar rates of cell proliferation (Figure 6A). We observed, however, that the media of iPSCs(High-K) turned yellow faster than that of iPSCs(Low-K) (Figure 6B). This often indicates lactic acidosis resulting from enhanced glycolysis, a metabolic state that is reminiscent of the Warburg effect in cancer. Indeed, both glucose uptake and lactate production were higher in iPSCs(High-K) than in iPSCs(Low-K) (Figures 6C and 6D). Moreover, when expressed in iPSCs(Low-K), exogenous TCL1 elevated glucose uptake and lactate production to similar levels observed in iPSCs(High-K) (Figures 6C and 6D). Importantly, an AKT inhibitor, MK2206, significantly diminished the enhancement of glucose uptake and almost completely counteracted the enhancement of lactate production (Figures 6C and 6D). Retrovirus-mediated expression of TCL1 in paused iPSC(Low-K) clones (Figure S5A) also increased the phosphorylation of AKT (Figure S5B), which is followed by the increased glucose uptake and lactate production (Figure S5C). These metabolic changes concurred with the increase in Cdh1 expression but preceded the increases in Fbxo15, Rex1, and Nanog expression (Figure S5E). Similar changes in metabolism and marker gene expression were observed when Shield1-stabilized KLF4 induced TCL1 in paused iPSC(Low-K) clones (Figures S5C and S5E). Furthermore, inhibition of AKT by MK2206 in iPSC(High-K) clones (Figure S6A) diminished glucose uptake and lactate production (Figure S6B), but did not change the pluripotency marker expression (Figure S6D). These results demonstrate that KLF4 enhances glycolysis during the transition from iPSCs(Low-K) to iPSCs(High-K), in large part through the TCL1-AKT pathway.
Figure 6.

TCL1 Enhances Glycolysis through AKT Activation
(A) Cell growth of iPSCs(Low-K) and iPSCs(High-K). One hundred cells of iPSCs(Low-K) or iPSCs(High-K) were passaged to 24-well plate and cell number was counted every day from day 1. Data represent means ± SEM of three independent experiments.
(B) Different colors of the cell culture media of iPSCs(Low-K) and iPSCs(High-K). 1 × 103 cells of iPSCs(Low-K) and iPSCs(High-K) were cultured for 2 days.
(C and D) Glucose uptake (C) or lactate production (D) in iPSCs(Low-K), iPSCs(High-K), or iPSCs(High-K) with an AKT inhibitor (High-K + MK), iPSCs(Low-K) expressing TCL1 (Low-K + TCL1), or Low-K + TCL1 with an AKT inhibitor (Low-K + TCL1+MK) at day 30. Data represent means ± SEM of three independent experiments. ∗p < 0.05, ∗∗∗p < 0.005.
(E) Key proteins in the glycolytic pathway.
(F) The mRNA levels of glycolysis-related genes in the reprogrammed cells at day 30. Data represent means ± SEM of three independent experiments. ∗p < 0.05 versus iPSCs(Low-K).
See also Figures S5, S6, and S7 and Table S1.
Time course analyses of reprogramming of MEFs with SeVdp(fK-OSM) show that iPSCs(Low-K) display early increases in glucose uptake and lactate production by day 4, which remain relatively constant thereafter (Figure S7A). In addition to these metabolic changes by day 4, iPSCs(High-K) show further increases in glucose uptake and lactate production at days 10 and 20, above those of iPSCs(Low-K) (Figure S7A). These additional metabolic changes are preceded by the increase in Tcl1 expression, concurrent with the increase in Cdh1 expression, and are followed by increases in Fbxo15, Rex1, and Nanog expression (Figure S7C).
To identify the genes that the TCL1-AKT pathway regulates to enhance glycolysis, we analyzed five genes that are critical for the Warburg effect in the glucose metabolism pathway (Figure 6E) (Ito and Suda, 2014). qRT-PCR showed that the high level of KLF4 or expression of exogenous TCL1 in iPSCs(Low-K) is accompanied by increased mRNA expression levels of glucose transporter (Glut1), pyruvate kinase M2 (Pkm2), and lactate dehydrogenase A (Ldha) (Figure 6F). The increases in the expression of the three genes were eliminated almost completely by an AKT inhibitor, MK2206 (Figure 6F). These results show that KLF4 upregulates Tcl1 expression to enhance the activity of AKT, which in turn increases expression of key glycolytic proteins for robust glycolysis.
KLF4-Induced TCL1 Counteracts PnPase to Diminish Mitochondrial Function
In addition to robust glycolysis, ESCs and iPSCs display diminished oxidative phosphorylation because they only possess a small number of immature mitochondria (Bukowiecki et al., 2014). We therefore asked if the mitochondria undergo numerical and morphological changes during the transition from iPSCs(Low-K) to iPSCs(High-K). As shown in Figures 7A and 7B, MitoTracker Green staining of iPSCs(Low-K) and iPSCs(High-K), quantified by AxioVision, revealed that the mitochondrial content in iPSCs(High-K) is reduced by more than 60% compared with that in iPSCs(Low-K). The effect of KLF4 in reducing the mitochondrial content is mediated by TCL1 because the mitochondrial reduction was recapitulated by expressing exogenous TCL1 in iPSCs(Low-K) (Figures 7A and 7B). However, MK2206 failed to block the effect of high KLF4 or expression of exogenous TCL1 (Figures 7A and 7B), showing that KLF4-induced TCL1 reduces the mitochondrial content independent of the AKT signaling pathway.
Figure 7.

TCL1 Suppresses Mitochondrial Function through PnPase
(A) Mitochondrial contents were visualized by staining cells with MitoTracker Green FM at day 30. The stained cells were iPSCs(Low-K), iPSCs(High-K), or iPSCs(Low-K) expressing TCL1 with or without an AKT inhibitor (MK). Scale bars, 10 μm.
(B) Fluorescence intensities in (A) were quantified using AxioVision software. Data represent means ± SEM of at least three independent experiments. ∗∗∗p < 0.005 versus iPSCs(Low-K).
(C) Mitochondrial contents were visualized as in (A), but retrovirus-mediated expression of PNPT1 was performed instead of treating with MK. Scale bars, 10 μm.
(D) Fluorescence intensities in (C) were quantified as in (B). ∗∗p < 0.01, ∗∗∗p < 0.005.
(E) Oxygen consumption in the same set of reprogrammed cells as in (C). Data represent means ± SEM of three independent experiments. ∗∗∗p < 0.005.
(F) Cdh1 and Rex1 mRNA levels in the same set of reprogrammed cells as in (C). Data represent means ± SEM of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005.
(G) Schematic model summarizing the role for KLF4 in promoting the metabolic shift through TCL1.
See also Figures S5, S6, and S7 and Table S1.
Recent studies show that, in addition to functioning as a coactivator of AKT, TCL1 interacts directly with mitochondrial PnPase to inhibit mitochondrial biogenesis and oxidative phosphorylation (Khaw et al., 2015). PnPase, encoded by the Pnpt1 gene, is localized in the mitochondrial inner membrane space and facilitates RNA import into mitochondria, thereby maintaining the mitochondrial content and their homeostasis by an as-yet-unknown mechanism (Wang et al., 2010). To find if the KLF4-TCL1 pathway directly reduces the mitochondrial content by counteracting PnPase, we investigated the role for PnPase in KLF4-induced mitochondrial reduction. Expression of exogenous PnPase in iPSCs(High-K) or TCL1-expressing iPSCs(Low-K) almost completely prevented the reduction of the mitochondrial content by high KLF4 or exogenous TCL1 (Figures 7C and 7D). Thus, PnPase antagonizes the effect of high KLF4 and exogenous TCL1, in congruent with the direct physical association and functional antagonism between TCL1 and PnPase (Khaw et al., 2015). Analyses of oxygen consumption, which reflects the mitochondrial respiration, also confirmed the antagonism between the KLF4-TCL1 pathway and PnPase, indicating that TCL1 reduces mitochondrial content and function by counteracting PnPase (Figure 7E), presumably through direct physical interaction with PnPase. Moreover, the expression of exogenous PnPase in iPSCs(High-K) or TCL1-expressing iPSCs(Low-K) decreased the expression of Cdh1 and Rex1 (Figure 7F), suggesting that the increased mitochondrial content by itself may negatively impact the KLF4-induced progression of reprogramming.
To further confirm the above findings, we tested the effect of increased KLF4 and exogenous TCL1 in paused iPSC(Low-K) clones (Figure S5A). Restoration of the KLF4 level and retrovirus-mediated expression of TCL1 in paused iPSC(Low-K) clones diminished the mitochondrial content and oxygen consumption (Figure S5D) and increased Cdh1 expression, which was followed by the increased expression of Fbxo15, Rex1, and Nanog (Figure S5E). Furthermore, expression of PnPase, but not inhibition of AKT by MK2206, in iPSC(High-K) clones (Figure S6A) increased the mitochondrial content and oxygen consumption (Figure S6C). However, the expression of PnPase did not affect glucose uptake and lactate production (Figure S6B). Time course analyses of reprogramming (Figure S7) also indicate that reduction of the mitochondrial content and oxygen consumption (Figure S7B) parallel the increases in glucose uptake and lactate production (Figure S7A). Taken together, our results indicate that, besides enhancing glycolysis via AKT activation, KLF4-induced TCL1 diminishes oxidative phosphorylation through antagonizing PnPase to promote the metabolic shift during reprogramming toward full pluripotency (Figure 7G).
Discussion
We found here that Tcl1 is one of the important direct targets of KLF4 and KLF4-induced TCL1 directs a metabolic shift during reprogramming by both enhancing glycolysis and diminishing oxidative phosphorylation. Our analyses revealed that KLF4 regulates TCL1 expression at the level of transcription by directly binding the Tcl1 enhancer and promoter when cells transition from iPSCs(Low-K) to iPSCs(High-K), simultaneously recruiting OCT4 to the Tcl1 promoter. The increased binding of KLF4 and OCT4 is accompanied by deposition of H3K4me3 at the Tcl1 promoter, which has already lost H3K27me3 and acquired H3K27Ac before the transition (Figures 4E and 4G). Given that the 3-fold increase of the KLF4 permits the deposition of H3K4me3, there may be a critical threshold for deposition of each histone mark. Moreover, because subsets of genes increase expression at a distinct stage of reprogramming (Nishimura et al., 2014), the critical threshold during reprogramming may differ for individual genes.
Rapidly proliferating cells have a high demand for essential cell components, which is met by increased glycolysis that shunts the metabolic intermediates toward synthesis of building blocks for nucleic acids, lipids, and proteins (Vander Heiden et al., 2009). This is achieved by aerobic glycolysis, also known as the Warburg effect, which has recently been acknowledged as an important metabolic state in cancer cells (Courtnay et al., 2015), adult stem cells as well as ESCs (Ito and Suda, 2014). In cancer cells, central to aerobic glycolysis is the phosphatidylinositol 3-kinase-AKT-mammalian target of the rapamycin signaling pathway and the hypoxia-inducible factor 1α (HIF1α) pathway, which regulate cell proliferation, survival, and metabolism (Courtnay et al., 2015). In adult stem cells, the same pathways also play an essential role for maintaining aerobic glycolysis in hypoxic stem cell niches (Ito and Suda, 2014). ESCs also display aerobic glycolysis and reduced oxidative phosphorylation, and the AKT pathway is an important downstream target of leukemia inhibitory factor (LIF), which is essential for self-renewal and pluripotency of ESCs (Niwa et al., 2009). However, the role for the AKT pathway in the metabolic state of ESCs is less clear.
Our results indicate that Tcl1 regulates AKT to enhance glycolysis and inhibits PnPase to diminish oxidative phosphorylation in fully reprogrammed iPSCs, and perhaps in ESCs as well. Although ESCs express a high level of Tcl1 (Miyazaki et al., 2013), its role as a metabolic regulator in ESCs remains undefined. Further contributing to this uncertainty is the fact that only a slight decrease in cell proliferation is observed in Tcl1(−/−) ESCs, without any change in the phosphorylated form of AKT (Miyazaki et al., 2013). Tcl1(−/−) embryos, however, show a more unambiguous phenotype, displaying a defect in early embryonic development (Narducci et al., 2002). Given that TCL1 is not the mainstream pathway that connects to AKT but merely modulates its activities, augmentation of the AKT activities via LIF in cell culture medium, for instance, may compensate for the lack of Tcl1 in ESCs cultured in vitro. Its requirement, however, may be more stringent in the developing embryo, where such signals could be less variable.
Regardless of the detailed mechanisms of metabolic regulation in ESCs, recent studies indicate that reprogramming MEFs into iPSCs involves dramatic changes in cell metabolism from oxidative phosphorylation to glycolysis (Folmes et al., 2011). Somewhat paradoxically, however, the early phase of the metabolic shift involves a burst in oxidative phosphorylation (Hawkins et al., 2016, Kida et al., 2015), which is mediated by estrogen-related receptors, when cell proliferation increases dramatically (Kida et al., 2015). This burst in oxidative phosphorylation generates reactive oxygen species (ROS), which in turn leads to the increased expression of nuclear factor erythroid-derived 2-like 2, nuclear factor κB, and AP-1, eventually increasing the level of HIF1α. Sequential activation of these transcription factors gradually diminishes oxidative phosphorylation and enhances glycolysis (Hawkins et al., 2016).
The metabolic shift elicited by KLF4-induced TCL1 during the transition from iPSCs(Low-K) to iPSCs(High-K) is probably distinct from, and preceded by, the ROS-induced changes in metabolism (Hawkins et al., 2016, Kida et al., 2015). Indeed, no difference in the cell proliferation rate was observed between iPSCs(Low-K) and iPSCs(High-K). Moreover, the protein level of HIF1α, which is regulated by proteolysis (Courtnay et al., 2015), and the mRNA levels of Pdk1 and Hk2, which are regulated by HIF1α (Semenza, 2010), remain essentially unchanged during this transition (Figure 6F and data not shown). Thus, the metabolic shift may involve a series of events, consisting of at least two phases, and cells respond appropriately to the ongoing metabolic demand in each phase of reprogramming. Notably, the latter metabolic shift probably initiates slightly earlier but largely concurs with the acquisition of full pluripotency when the metabolism in reprogrammed cells becomes indistinguishable from that of ESCs.
In this article, we revealed that one of the key functions of KLF4-induced TCL1 during reprogramming is to promote the metabolic shift from oxidative phosphorylation to glycolysis. Despite its role in a late stage of reprogramming, expression of TCL1 with the four reprogramming factors from the beginning of reprogramming results in faster and higher induction of pluripotency markers (Figures 3C–3E). Indeed, TCL1b1, another member of the TCL family (Hallas et al., 1999), also improves the efficiency of reprogramming (Khaw et al., 2015). Given that the metabolic shift in the earlier stage occurs via ROS production (Hawkins et al., 2016), it is tempting to hypothesize that expression of TCL1 throughout reprogramming may permit cells with lower ROS production to undergo the metabolic shift. This might minimize the exposure of cells to ROS during reprogramming, potentially reducing damage to the genome in the generated iPSCs.
Experimental Procedures
Cell Culture and Generation of iPS Cells
Mouse iPSCs were generated from MEFs isolated from transgenic mice carrying the Nanog-GFP-IRES-Puro reporter construct (Okita et al., 2007) (provided by the RIKEN BioResource Center) (Nanog-GFP MEFs) by infection with an indicated SeVdp vector at 32°C for 14 hr. The infected cells were seeded onto mitomycin C-treated neomycin-resistant SNL76/7 feeder cells and grown in KSR medium (KnockOut DMEM [Thermo Fisher] supplemented with 15% KnockOut Serum Replacement [Thermo Fisher], 2 mM GlutaMAX [Thermo Fisher], 0.1 mM non-essential amino acids [Thermo Fisher], 55 μM 2-mercaptoethanol [Thermo Fisher], 100 U/mL penicillin-streptomycin [Wako], and 1,000 U/mL LIF [Wako]) for 7 days, after which the medium was replaced by mES medium (DMEM [Nacalai Tesque] supplemented with 15% fetal bovine serum [HyClone], 0.1 mM non-essential amino acids, 55 μM 2-mercaptoethanol, 100 U/mL penicillin-streptomycin, and 1,000 U/mL LIF), in the presence or absence of 100 nM Shield1 (Takara Bio).
Nanog-GFP MEFs or iPSCs(Low-K) were infected with the indicated retrovirus for 2 days with 8 μg/mL hexadimethrine bromide (polybrene; Sigma). The cells were then used for iPSC generation or passaged onto feeder cells, respectively, with 800 μg/mL G418 (Nacalai Tesque) and/or 2 μg/mL puromycin (Nakalai Tesque) for selection of retroviral vector-infected cells.
Teratoma Formation
Animal experiments were carried out in accordance with the protocols approved by University of Tsukuba Ethics Committee for Animal Experiments. Teratoma was formed by subcutaneous injection of 1 × 106 iPSCs, generated by SeVdp(fKiT-OSM), into SCID mice. After 4 weeks, tumors were harvested at necropsy, fixed overnight in 4% paraformaldehyde in PBS, and embedded in paraffin wax. The tumor samples were sectioned at 4 μm and stained with H&E. Histological findings were evaluated using a microscope BX50 (Olympus) equipped with a digital camera G11 (Canon).
Determination of Glucose Uptake
Cells (5.0 × 105), cultured in KSR medium without feeder cells, were treated with 1 mM 2-deoxy-D-glucose (2DG) (Nacalai Tesque) for 20 min at 37°C, followed by washing with PBS plus 0.2 mM Phloretin (Tokyo Chemical Industry). 2DG in the cells was determined using a 2DG Uptake Measurement Kit (Cosmo Bio) according to manufacturer's procedures.
Determination of Lactate Production
Cells (2.0 × 105) were cultured in KSR medium with feeder cells for 6 hr, and the cell culture medium was collected and centrifuged to remove cells. The amount of lactate in the medium was determined using a Glycolysis Cell-Based Assay Kit (Cayman Chemical) according to the manufacturer's procedures.
Mitochondrial Staining
Cells were treated with 20 nM MitoTraker Green FM (Molecular Probes) for 30 min at 37°C. To determine the presence of mitochondria, cells were observed under Axio Observer fluorescent microscopy (Zeiss), and the strength of the GFP signal was determined using AxioVision software (Zeiss).
Determination of Oxygen Consumption
Cells (1.0 × 105) were cultured in a 96-well plate with feeder cells for 1 day, and the extracellular oxygen consumption was determined using an Extracellular Oxygen Consumption Assay Kit (Abcam) according to the manufacturer's procedures.
Author Contributions
K.N. and K.H. designed the research. K.N., S.A., F.L.N., E.S., H.T., and A.K. performed the research and analyzed the data. Y.T.H.T., P.L.B., E.B., Y.S., and M.N. prepared the materials. K.N., Y.H., A.F., and K.H. wrote the paper.
Acknowledgments
We thank T. Mizutani for providing pMCsΔYY1-IRES-GFP plasmid. We also thank T. Nishimura for technical assistance. This work was supported by JSPS KAKENHI grant numbers JP26870074, JP16K08610, and JP25460354 (to K.N., A.F., and K.H., respectively), Kato Memorial Bioscience Foundation (to K.N.), Inamori Foundation (to K.N.), and the Program to Disseminate Tenure Tracking System by MEXT (to K.N.).
Published: March 2, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.01.026.
Contributor Information
Ken Nishimura, Email: ken-nishimura@md.tsukuba.ac.jp.
Koji Hisatake, Email: kojihisa@md.tsukuba.ac.jp.
Supplemental Information
References
- Banaszynski L.A., Chen L.C., Maynard-Smith L.A., Ooi A.G., Wandless T.J. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukowiecki R., Adjaye J., Prigione A. Mitochondrial function in pluripotent stem cells and cellular reprogramming. Gerontology. 2014;60:174–182. doi: 10.1159/000355050. [DOI] [PubMed] [Google Scholar]
- Chen X., Xu H., Yuan P., Fang F., Huss M., Vega V.B., Wong E., Orlov Y.L., Zhang W., Jiang J. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Cho Y.M., Kwon S., Pak Y.K., Seol H.W., Choi Y.M., Park D.J., Park K.S., Lee H.K. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem. Biophys. Res. Commun. 2006;348:1472–1478. doi: 10.1016/j.bbrc.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Courtnay R., Ngo D.C., Malik N., Ververis K., Tortorella S.M., Karagiannis T.C. Cancer metabolism and the Warburg effect: the role of HIF-1 and PI3K. Mol. Biol. Rep. 2015;42:841–851. doi: 10.1007/s11033-015-3858-x. [DOI] [PubMed] [Google Scholar]
- Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis R.J., Lum J.J., Hatzivassiliou G., Thompson C.B. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Fischer B., Bavister B.D. Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J. Reprod. Fertil. 1993;99:673–679. doi: 10.1530/jrf.0.0990673. [DOI] [PubMed] [Google Scholar]
- Folmes C.D., Nelson T.J., Martinez-Fernandez A., Arrell D.K., Lindor J.Z., Dzeja P.P., Ikeda Y., Perez-Terzic C., Terzic A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011;14:264–271. doi: 10.1016/j.cmet.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallas C., Pekarsky Y., Itoyama T., Varnum J., Bichi R., Rothstein J.L., Croce C.M. Genomic analysis of human and mouse TCL1 loci reveals a complex of tightly clustered genes. Proc. Natl. Acad. Sci. USA. 1999;96:14418–14423. doi: 10.1073/pnas.96.25.14418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins K.E., Joy S., Delhove J.M., Kotiadis V.N., Fernandez E., Fitzpatrick L.M., Whiteford J.R., King P.J., Bolanos J.P., Duchen M.R. NRF2 orchestrates the metabolic shift during induced pluripotent stem cell reprogramming. Cell Rep. 2016;14:1883–1891. doi: 10.1016/j.celrep.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H., Sootome H., Nakatsuru Y., Miyama K., Taguchi S., Tsujioka K., Ueno Y., Hatch H., Majumder P.K., Pan B.S. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010;9:1956–1967. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- Iba H., Mizutani T., Ito T. SWI/SNF chromatin remodelling complex and retroviral gene silencing. Rev. Med. Virol. 2003;13:99–110. doi: 10.1002/rmv.378. [DOI] [PubMed] [Google Scholar]
- Ito K., Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014;15:243–256. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo H., Mondal S., Tan D., Nagata E., Takizawa S., Sharma A.K., Hou Q., Shanmugasundaram K., Prasad A., Tung J.K. Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc. Natl. Acad. Sci. USA. 2012;109:10581–10586. doi: 10.1073/pnas.1202810109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaw S.L., Min-Wen C., Koh C.G., Lim B., Shyh-Chang N. Oocyte factors suppress mitochondrial polynucleotide phosphorylase to remodel the metabolome and enhance reprogramming. Cell Rep. 2015;12:1080–1088. doi: 10.1016/j.celrep.2015.07.032. [DOI] [PubMed] [Google Scholar]
- Kida Y.S., Kawamura T., Wei Z., Sogo T., Jacinto S., Shigeno A., Kushige H., Yoshihara E., Liddle C., Ecker J.R. ERRs mediate a metabolic switch required for somatic cell reprogramming to pluripotency. Cell Stem Cell. 2015;16:547–555. doi: 10.1016/j.stem.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T., Koshino Y., Shibata F., Oki T., Nakajima H., Nosaka T., Kumagai H. Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp. Hematol. 2003;31:1007–1014. [PubMed] [Google Scholar]
- Kyttala A., Moraghebi R., Valensisi C., Kettunen J., Andrus C., Pasumarthy K.K., Nakanishi M., Nishimura K., Ohtaka M., Weltner J. Genetic variability overrides the impact of parental cell type and determines iPSC differentiation potential. Stem Cell Rep. 2016;6:200–212. doi: 10.1016/j.stemcr.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine J., Kunstle G., Obata T., Sha M., Noguchi M. The protooncogene TCL1 is an Akt kinase coactivator. Mol. Cell. 2000;6:395–407. doi: 10.1016/s1097-2765(00)00039-3. [DOI] [PubMed] [Google Scholar]
- Manning B.D., Cantley L.C. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba R., Niwa H., Masui S., Ohtsuka S., Carter M.G., Sharov A.A., Ko M.S. Dissecting Oct3/4-regulated gene networks in embryonic stem cells by expression profiling. PLoS One. 2006;1:e26. doi: 10.1371/journal.pone.0000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T., Fujimori K., Andoh-Noda T., Ando T., Kuzumaki N., Toyoshima M., Tada H., Imaizumi K., Ishikawa M., Yamaguchi R. Functional neurons generated from T cell-derived induced pluripotent stem cells for neurological disease modeling. Stem Cell Rep. 2016;6:422–435. doi: 10.1016/j.stemcr.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T., Miyazaki S., Ashida M., Tanaka T., Tashiro F., Miyazaki J. Functional analysis of Tcl1 using Tcl1-deficient mouse embryonic stem cells. PLoS One. 2013;8:e71645. doi: 10.1371/journal.pone.0071645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussaieff A., Rouleau M., Kitsberg D., Cohen M., Levy G., Barasch D., Nemirovski A., Shen-Orr S., Laevsky I., Amit M. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015;21:392–402. doi: 10.1016/j.cmet.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Narducci M.G., Fiorenza M.T., Kang S.M., Bevilacqua A., Di Giacomo M., Remotti D., Picchio M.C., Fidanza V., Cooper M.D., Croce C.M. TCL1 participates in early embryonic development and is overexpressed in human seminomas. Proc. Natl. Acad. Sci. USA. 2002;99:11712–11717. doi: 10.1073/pnas.182412399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K., Sano M., Ohtaka M., Furuta B., Umemura Y., Nakajima Y., Ikehara Y., Kobayashi T., Segawa H., Takayasu S. Development of defective and persistent sendai virus vector: a unique gene delivery/expression system ideal for cell reprogramming. J. Biol. Chem. 2011;286:4760–4771. doi: 10.1074/jbc.M110.183780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K., Kato T., Chen C., Oinam L., Shiomitsu E., Ayakawa D., Ohtaka M., Fukuda A., Nakanishi M., Hisatake K. Manipulation of KLF4 expression generates iPSCs paused at successive stages of reprogramming. Stem Cell Rep. 2014;3:915–929. doi: 10.1016/j.stemcr.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H., Ogawa K., Shimosato D., Adachi K. A parallel circuit of LIF signalling pathways maintains pluripotency of mouse ES cells. Nature. 2009;460:118–122. doi: 10.1038/nature08113. [DOI] [PubMed] [Google Scholar]
- O'Malley J., Skylaki S., Iwabuchi K.A., Chantzoura E., Ruetz T., Johnsson A., Tomlinson S.R., Linnarsson S., Kaji K. High-resolution analysis with novel cell-surface markers identifies routes to iPS cells. Nature. 2013;499:88–91. doi: 10.1038/nature12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K., Ichisaka T., Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Parchem R.J., Ye J., Judson R.L., LaRussa M.F., Krishnakumar R., Blelloch A., Oldham M.C., Blelloch R. Two miRNA clusters reveal alternative paths in late-stage reprogramming. Cell Stem Cell. 2014;14:617–631. doi: 10.1016/j.stem.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo J.M., Anderssen E., Walsh R.M., Schwarz B.A., Nefzger C.M., Lim S.M., Borkent M., Apostolou E., Alaei S., Cloutier J. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–1632. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigione A., Fauler B., Lurz R., Lehrach H., Adjaye J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells. 2010;28:721–733. doi: 10.1002/stem.404. [DOI] [PubMed] [Google Scholar]
- Prigione A., Rohwer N., Hoffmann S., Mlody B., Drews K., Bukowiecki R., Blumlein K., Wanker E.E., Ralser M., Cramer T. HIF1alpha modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1-3 and PKM2. Stem Cells. 2014;32:364–376. doi: 10.1002/stem.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samavarchi-Tehrani P., Golipour A., David L., Sung H.K., Beyer T.A., Datti A., Woltjen K., Nagy A., Wrana J.L. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell. 2010;7:64–77. doi: 10.1016/j.stem.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Semenza G.L. HIF-1: upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John J.C., Ramalho-Santos J., Gray H.L., Petrosko P., Rawe V.Y., Navara C.S., Simerly C.R., Schatten G.P. The expression of mitochondrial DNA transcription factors during early cardiomyocyte in vitro differentiation from human embryonic stem cells. Cloning Stem Cells. 2005;7:141–153. doi: 10.1089/clo.2005.7.141. [DOI] [PubMed] [Google Scholar]
- Stadtfeld M., Hochedlinger K. Induced pluripotency: history, mechanisms, and applications. Genes Dev. 2010;24:2239–2263. doi: 10.1101/gad.1963910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016;17:183–193. doi: 10.1038/nrm.2016.8. [DOI] [PubMed] [Google Scholar]
- Theunissen T.W., Jaenisch R. Molecular control of induced pluripotency. Cell Stem Cell. 2014;14:720–734. doi: 10.1016/j.stem.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Chen H.W., Oktay Y., Zhang J., Allen E.L., Smith G.M., Fan K.C., Hong J.S., French S.W., McCaffery J.M. PNPASE regulates RNA import into mitochondria. Cell. 2010;142:456–467. doi: 10.1016/j.cell.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Duan S., Yi F., Ocampo A., Liu G.H., Izpisua Belmonte J.C. Mitochondrial regulation in pluripotent stem cells. Cell Metab. 2013;18:325–332. doi: 10.1016/j.cmet.2013.06.005. [DOI] [PubMed] [Google Scholar]
- Zhang T., Cooper S., Brockdorff N. The interplay of histone modifications - writers that read. EMBO Rep. 2015;16:1467–1481. doi: 10.15252/embr.201540945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.