

Summary
Conversion of fibroblasts into functional cardiomyocytes represents a potential means of restoring cardiac function after myocardial infarction, but so far this process remains inefficient and little is known about its molecular mechanisms. Here we show that DAPT, a classical Notch inhibitor, enhances the conversion of mouse fibroblasts into induced cardiac-like myocytes by the transcription factors GATA4, HAND2, MEF2C, and TBX5. DAPT cooperates with AKT kinase to further augment this process, resulting in up to 70% conversion efficiency. Moreover, DAPT promotes the acquisition of specific cardiomyocyte features, substantially increasing calcium flux, sarcomere structure, and the number of spontaneously beating cells. Transcriptome analysis shows that DAPT induces genetic programs related to muscle development, differentiation, and excitation-contraction coupling. Mechanistically, DAPT increases binding of the transcription factor MEF2C to the promoter regions of cardiac structural genes. These findings provide mechanistic insights into the reprogramming process and may have important implications for cardiac regeneration therapies.
Keywords: direct cellular reprogramming, cell-fate conversion, transdifferentiation, cardiomyocytes, Notch signaling, DAPT, heart regeneration, regenerative medicine
Graphical Abstract
Highlights
-
•
Notch activation is a barrier for GHMT-induced cardiac cell reprogramming
-
•
Notch blockade by DAPT improves GHMT-induced cardiac reprogramming
-
•
DAPT increases sarcomere organization, calcium flux, and beating in GHMT reprogramming
-
•
DAPT enhances transcriptional activity of MEF2C in GHMT reprogramming
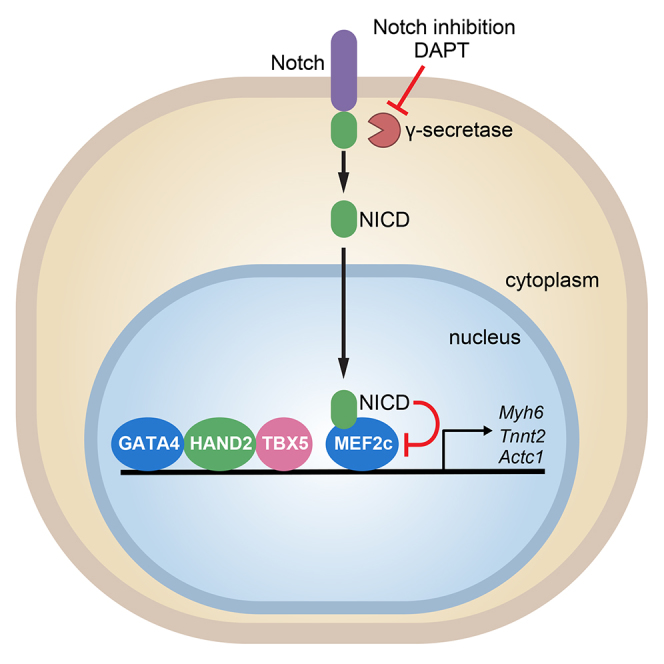
In this article, Olson and colleagues show that Notch signaling activation is a critical barrier for cardiac cell reprogramming. Notch signaling blockade by DAPT, a γ-secretase inhibitor, enhances fibroblast conversion to cardiomyocytes by increasing calcium flux, sarcomere structure, and beating. Mechanistically, Notch inhibition enhances the transcriptional activity of the cardiogenic transcription factor MEF2C, thereby enhancing expression of cardiac genes.
Introduction
Ischemic heart disease resulting in myocardial infarction (MI) remains one of the most common causes of mortality worldwide. Although some reports have indicated that adult human cardiomyocytes possess the capacity to proliferate at a very low rate (Beltrami et al., 2001, Bergmann et al., 2009), this limited capacity is insufficient to compensate for the massive loss of cardiomyocytes that occurs upon MI. Instead, injury activates a fibrotic response that generates a non-contractile scar that impairs normal heart function. Thus, the ability to generate new cardiomyocytes represents an important therapeutic goal, and many recent studies have proposed different strategies to achieve this goal. Cellular transplantation of cardiac progenitor cells, stem cells, or in vitro stem cell-derived cardiomyocytes holds clinical potential, but cell therapy is, so far, relatively inefficient due to failure of stem cells to fully adopt a functional contractile phenotype, insufficient mechanical and electrophysiological integration into the myocardium, and inadequate long-term retention of transplanted cells in the heart (Sanganalmath and Bolli, 2013). Therefore, there is a major need to develop new therapeutic strategies that eliminate the obstacles facing current therapies for post-MI cardiac repair.
During the last decade, cellular reprogramming has emerged as a promising strategy for regenerative medicine. In 2010, the cardiac transcription factors GATA4, MEF2C, and TBX5 were shown to be capable of converting mouse fibroblasts into cardiomyocyte-like cells in vitro (induced cardiac-like myocytes [iCLMs]) (Ieda et al., 2010). Soon thereafter, cardiac reprogramming was achieved in human cells (Nam et al., 2013, Wada et al., 2013) and several variations on the original cocktail of transcription factors were found to enhance reprogramming efficiency (Addis et al., 2013, Christoforou et al., 2013, Hirai et al., 2013, Jayawardena et al., 2012, Protze et al., 2012, Song et al., 2012, Zhou et al., 2015; reviewed in Batty et al., 2016). Conversion of fibroblasts into cardiomyocytes in vivo has also been shown to improve cardiac function following MI in mice (Qian et al., 2012, Song et al., 2012). Very recently, it has been shown that cardiac reprogramming can be achieved by a purely chemical approach, avoiding the introduction of foreign genetic material (Cao et al., 2016). Despite the great potential of these discoveries, cardiac lineage conversion remains relatively inefficient and the underlying molecular mechanisms have not been fully elucidated.
The Notch pathway is an evolutionarily conserved signaling pathway that regulates cell-fate specification, differentiation, and developmental patterning (Guruharsha et al., 2012). Notch is a single-pass transmembrane receptor that is activated upon cell-cell contact, as a result of its interaction with ligands (Jagged or Delta). In mammals, four Notch receptors (Notch 1–4) and five transmembrane ligands (Jagged 1, Jagged 2, Delta-like 1, Delta-like 3, and Delta-like 4) have been identified. Upon activation, the Notch receptor undergoes two sequential cleavages, first by a metalloprotease (ADAM) on its extracellular domain and, subsequently, by a γ-secretase enzyme complex that triggers the release of the Notch intracellular domain (NICD) and its translocation to the nucleus (Andersson et al., 2011). In the canonical Notch pathway, NICD binds to the transcription factor RBPJ-k in the nucleus, displaces co-repressors, and allows the recruitment of co-activators to target gene promoters, leading to transcriptional activation. Typical target genes include those encoding basic-helix-loop-helix repressors of the Hes and Hey families. However, there are several reports supporting the existence of a “non-canonical” Notch pathway, which functions independently of RBPJ-k. Interestingly, Notch activity impairs myoblast differentiation independently of RBPJ-k (reviewed in Andersen et al., 2012), in part by blocking the binding of the transcription factor MEF2C to chromatin (Wilson-Rawls et al., 1999). In the heart, the Notch signaling pathway plays a critical role during development by regulating cell fate and cardiomyocyte proliferation. Accordingly, mutations that affect this signaling pathway are linked with congenital heart disease (reviewed in de la Pompa and Epstein, 2012). However, there is conflicting evidence regarding the role of Notch in cardiac differentiation, showing that Notch activation can both promote and inhibit cardiomyocyte differentiation (Jang et al., 2008, Chen et al., 2008, Koyanagi et al., 2007, Li et al., 2006). The potential influence of Notch signaling on the reprogramming of fibroblasts to iCLMs has not yet been explored.
Here, we show that inhibition of Notch signaling by the γ-secretase inhibitor DAPT ((S)-tert-butyl 2-((S)-2-(2-(3,5-difluorophenyl)acetamido)propanamido)-2-phenylacetate) enhances the reprogramming of mouse fibroblasts to iCLMs, at least in part by increasing the binding of MEF2C to cardiac gene promoters. These findings provide insights into the molecular basis of cardiac lineage conversion and provide a chemical approach to improve the efficiency of this technique.
Results
DAPT, a γ-Secretase Inhibitor, Enhances Cardiac Reprogramming by GHMT
We showed previously that GATA4, HAND2, MEF2C, and TBX5 (hereafter GHMT) can reprogram fibroblasts to iCLMs (Song et al., 2012). In an effort to improve the efficiency of this process, we screened a collection of seven chemical compounds that were previously shown to promote reprogramming of fibroblasts to induced pluripotent stem cells (Table S1) (Brady et al., 2013, Dutta et al., 2011, Esteban et al., 2010, Ichida et al., 2014, Li et al., 2012, Maherali and Hochedlinger, 2009, Shao et al., 2016). Mouse embryonic fibroblasts (MEFs) were isolated from transgenic mice that express GFP under the control of the cardiac α-myosin heavy chain promoter (αMHC-GFP), and reprogramming was induced by transducing the cells with retroviruses encoding for GHMT. After retroviral transduction, the medium was changed to cardiomyocyte induction medium containing DMSO or various chemicals (Table S1). After 7 days of induction, the generation of iCLMs was scored by the presence of GFP-positive cells (Figure 1A). The γ-secretase inhibitor DAPT, a well-established inhibitor of Notch signaling, was the most efficient compound tested, approximately doubling the number of GFP-positive cells after 7 days of reprogramming (Figure 1B).
Figure 1.

DAPT Increases Calcium Flux, Sarcomere Structure, and Spontaneous Beating in iCLMs
(A) Experimental strategy for drug screening to enhance cardiac reprogramming.
(B) Quantification of the number of αMHC-GFP-positive MEFs after 7 days of reprogramming by GHMT and treatment with the indicated chemicals. Values are shown as relative to the mock control; n = 3 biological replicates.
(C) αMHC-GFP MEFs were infected with GHMT, and treated with DMSO or DAPT for 2 weeks. Analysis by qPCR of the mRNA expression of the indicated genes, relative to expression in DMSO-treated cells; n = 3 biological replicates.
(D) Quantification of the number of cells positive for cTnT and α-actinin by immunostaining; n = 3 biological replicates.
(E) Representative image of the immunostaining quantified in (D). Scale bar, 200 μm.
(F) Representative confocal images of immunostaining against α-actinin showing sarcomere structure in iCLMs (treated with DMSO or DAPT) and mouse adult cardiomyocytes. The percentage of cells presenting a sarcomere structure is shown in the upper-right corner of every panel. Scale bar, 20 μm.
(G) Quantification of the percentage of α-actinin-positive cells with sarcomeric structure. The average of three different experiments is shown, with a total n = 471 in vehicle-treated cells and n = 570 in DAPT-treated cells.
(H) Percentage of beating cells, relative to the number of input cells at indicated times; n = 3 biological replicates.
(I) Quantification of Ca2+ flux-positive GCaMP3 cells after 2 weeks of reprogramming; n = 3 biological replicates.
(J) Quantification of Ca2+ flux-positive MEFs treated with 4-Fluo AM dye after 2 weeks of reprogramming; n = 3 biological replicates.
Data are presented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01. See also Figures S1–S3 and Table S1.
To further characterize the role of Notch inhibition in cardiac reprogramming, we performed optimization experiments by varying the concentration of DAPT (2.5, 5, and 10 μM) and the time period of treatment. Optimized conditions were determined to be 2.5 μM DAPT with continuous exposure for a 2-week period (Figure S1). DAPT treatment significantly increased the number of iCLMs, measured by the expression of cardiomyocyte-specific genes (Figure 1C) and by immunostaining for the cardiomyocyte markers cardiac troponin T (cTnT) and α-actinin (Figures 1D and 1E). We performed proliferation analysis by flow cytometry after 7 days of reprogramming and determined that DAPT treatment does not affect the proliferation of iCLMs (cTnT-positive cells) or fibroblasts (cTnT-negative cells); therefore, the increase in the number of iCLMs is not due to a DAPT effect on proliferation (Figure S2A). To confirm our observations, we tested dibenzazepine (DBZ), a structurally unrelated γ-secretase inhibitor, and found that DBZ was also able to enhance reprogramming, although to a lesser extent than DAPT, at least at the concentration tested (10 μM) (Figures S2B and S2C). In addition, the effect of DAPT was tested on reprogramming of adult tail-tip fibroblasts (TTFs), and similar reprogramming enhancement was observed (Figure S2D).
DAPT Increases Calcium Flux, Sarcomere Structure, and Spontaneous Beating of iCLMs
To determine whether DAPT enhances cardiomyocyte features, we assessed sarcomere structure. In the presence of GHMT, DAPT induced a 5-fold increase in the number of cells with an organized sarcomere structure (Figures 1F and 1G). Moreover, although GHMT-reprogrammed cells rarely show spontaneous beating at 25 days, treatment with DAPT induced a 6-fold increase in the number of spontaneously beating cells (Figure 1H). To analyze calcium flux, we used two different methods: a calcium reporter system (αMHC-Cre/Rosa26-Flox-Stop-Flox-GCaMP3 MEFs) for live cell imaging (Figure S3), and the fluorescent calcium indicator dye 4-Fluo AM. Both methods showed that DAPT treatment significantly increased the number of cells with calcium flux (Figures 1I and 1J).
To assess transcriptional events prior to the acquisition of spontaneous beating, we performed RNA-sequencing (RNA-seq) analysis of MEFs infected with GHMT and treated with vehicle (DMSO) or DAPT for 15 days (Table S2). Using a fold change cutoff of 1.5 and a false discovery rate of <0.01, 115 differentially expressed genes were identified, 27 of which were upregulated and 88 downregulated in GHMT cells treated with DAPT (Figure 2A; Tables S3 and S4). Among the upregulated genes we found numerous cardiomyocyte-related genes, such as Myh6, Actc1, Actn2, Pln, Myh7, and Myl1 (Table S3). DAPT did not enhance the expression of the GHMT reprogramming factors. Gene ontology analysis showed that most of the significantly increased biological processes were related to muscle and heart physiology, cardiac and muscle differentiation, and excitation-contraction coupling (Figure 2B). Moreover, DAPT treatment of GHMT-expressing cells efficiently upregulated cardiomyocyte-related genes and downregulated fibroblast genes (Figure 2C). Similarly, using the gene set enrichment analysis (GSEA) computational method, we obtained a highly significant upregulation of gene sets related to cardiomyocytes, such as “muscle contraction” and “respiratory electron transport” gene sets, and a clear downregulation of the “cycling genes” set (Figures 2D and S4).
Figure 2.

DAPT Treatment Induces Genetic Programs Related to Muscle Development, Differentiation, and Excitation-Contraction Coupling
RNA-seq was performed on MEFs infected with GHMT and treated for 15 days with DMSO (vehicle) or DAPT.
(A) Heatmap generated with differentially expressed genes.
(B) Graphical representation of gene ontology analysis using PANTHER Overrepresentation Test.
(C) Hierarchical clustering and heatmap using cardiomyocyte- and fibroblast-specific genes.
(D) Enrichment plots of the indicated gene sets and their nominal p values. See also Figure S4 and Tables S2–S4.
DAPT Cooperates with AKT1 to Enhance Cardiac Reprogramming
Previously, we showed that protein kinase AKT1 enhances cardiac reprogramming through mTOR and FOXO3A (Zhou et al., 2015). We tested whether DAPT cooperates with AKT1 during cardiac reprogramming by infecting αMHC-GFP MEFs with GHMT plus AKT1 (AGHMT), and treating the cells with vehicle or DAPT. Notably, AKT1 and DAPT together increased reprogramming efficiency up to 70%, as revealed by immunostaining against cTnT and α-actinin (Figures 3A and 3B). In addition, DAPT treatment of AGHMT cells increased the number of cells with calcium flux (Figure 3C). Moreover, 18 days after AGHMT infection, 40% of the cells treated with DAPT displayed spontaneous beating in culture (Figure 3D). Given this striking effect, we analyzed by western blot the expression of the ryanodine receptor and the ER Ca2+-ATPase SERCA2, both critical regulators of intracellular calcium handling and contractility. Expression of both proteins was increased by DAPT treatment, reaching higher levels than with AGHMT alone (Figure 3E). We conclude that DAPT cooperates with AKT1 to boost cardiac reprogramming.
Figure 3.

Notch Inhibition Cooperates with AKT1 to Enhance Cardiac Reprogramming
αMHC-GFP MEFs (or GCaMP MEFs in C) were infected with GHMT or AGHMT, and treated with DMSO (vehicle) or DAPT.
(A) Representative immunostaining images of GFP, α-actinin, and cTnT at day 15 of reprogramming. Scale bar, 200 μm.
(B) Quantification of cells positive for α-actinin and cTnT as determined by immunostaining; n = 3 biological replicates.
(C) Quantification of Ca2+ flux-positive cells in GCaMP MEFs at day 15; n = 3 biological replicates.
(D) Percentage of beating cells, relative to the number of input cells; n = 3 biological replicates.
(E) Immunoblot against the Ca2+ handling proteins ryanodine receptor (RyR) and SERCA2 at day 15 of reprogramming. Densitometric quantification is shown as the average of every replicate, relative to GAPDH.
Data are presented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Inhibition of the Canonical Notch Pathway Does Not Enhance Cardiac Reprogramming
By RNA-seq analysis, we surprisingly noted that none of the most common Notch target genes were downregulated by DAPT. As shown in Table S4, this was seen for the Hes/Hey gene family and the Notch target genes c-Myc and p21. Consistently, these data were validated by qPCR (Figure 4A). Moreover, using GSEA several gene sets related to the canonical Notch pathway were analyzed, none of which were downregulated in DAPT-treated samples (Figure 4B). However, among the downregulated genes there were two genes that have been previously described as Notch targets: Skp2 (Del Debbio et al., 2016, Dohda et al., 2007, Sarmento et al., 2005) and IL33 (Sundlisaeter et al., 2012). SKP2 is a component of the ubiquitin protein ligase complex SCF, and specifically recognizes the phosphorylated cyclin-dependent kinase p27 to promote its degradation. Given that p27 is known to be involved in cell-cycle arrest and cell differentiation, we tested whether DAPT could enhance reprogramming by increasing the protein levels of p27. However, western blot analysis indicated that p27 protein levels were decreased, rather than increased, upon DAPT treatment (Figure S5A), and cardiac reprogramming was enhanced, rather than impaired, in p27 knockout MEFs compared with wild-type MEFs (Figure S5B).
Figure 4.

Inhibition of the Canonical Notch Pathway Does Not Enhance Reprogramming
(A) qPCR analysis of Notch target genes in MEFs reprogrammed by GHMT, at day 15; n = 3 biological replicates.
(B) Enrichment plots of Notch pathways gene sets, and its nominal p value.
(C–E) αMHC-GFP MEFs were infected with GHMT and an shRNA scramble (scr) or an shRNA against Rbpj-k. At day 15, Rbpj-k mRNA expression was analyzed by qPCR (C), and different cardiac markers were analyzed by qPCR (D) or by immunostaining (E); n = 3 biological replicates. Scale bar, 100 μm.
Data are presented as means ± SD. ∗∗∗p < 0.001. See also Figure S5.
IL33, a member of the interleukin-1 family, resides in the nucleus bound to chromatin where it modifies transcription by undefined mechanisms. However, IL33 is released from cells upon tissue injury, inducing an inflammatory response. We validated by qPCR and ELISA the reduction of IL33 in GHMT-DAPT samples (Figures S5C and S5D), but did not see a decrease in reprogramming efficiency when recombinant IL33 protein was added to the reprogramming medium (Figure S5E), or an increase in reprogramming upon IL33 downregulation by small hairpin RNAs (shRNAs) (Figures S5E and S5F); therefore, IL33 does not seem to play an important role in cardiac reprogramming.
To test whether RBPJ-k, a major downstream regulator of the canonical Notch pathway, mediated the effects of DAPT, we inhibited Rbpj-k expression by shRNAs, achieving a 70% reduction in mRNA expression (Figure 4C). Decrease of Rbpj-k expression did not improve cardiac reprogramming mediated by GHMT, as measured by qPCR and immunostaining of different cardiac markers (Figures 4D and 4E). We conclude that inhibition of the canonical Notch pathway is not responsible for the enhancement of reprogramming by DAPT.
DAPT Enhances MEF2C Binding to Cardiac Gene Promoters
Notch activation is known to impair myoblast differentiation (Shen et al., 2006, Wilson-Rawls et al., 1999). We previously showed that this effect is due, at least in part, to the interaction of NICD with the MEF2C DNA binding domain, which reduces MEF2C binding to chromatin, thereby decreasing expression of MEF2C target genes (Wilson-Rawls et al., 1999). To examine whether Notch has a similar role in cardiac reprogramming, we overexpressed NICD in MEFs during reprogramming and found that it impaired cardiac reprogramming by GHMT, as seen by qPCR and immunostaining (Figures 5A and 5B). To analyze whether DAPT treatment and the subsequent decrease in NICD affects the binding of MEF2C to its target genes, we analyzed the promoter regions of the cardiac genes that were differentially regulated by DAPT using our RNA-seq data (Tables S3 and S4). We performed chromatin immunoprecipitation (ChIP) using an MEF2C antibody in MEFs infected with GHMT and treated or untreated with DAPT. Using qPCR, we analyzed MEF2C binding to specific promoter regions in the presence or absence of DAPT. Our results showed that addition of DAPT substantially increased the binding of MEF2C to the promoter of the Myh6, Tnnt2, and Actc1 genes (Figure 5C). Importantly, this increase in binding was not due to an induction of MEF2C protein by DAPT, as demonstrated by western blot analysis (Figure 5D). These observations suggest that Notch inhibition results in full activation of the cardiac differentiation factor MEF2C, thus providing a mechanistic explanation for the effects of DAPT on cardiac reprogramming.
Figure 5.

NICD Inhibition Increases the Transcriptional Activity of MEF2C at Cardiac Gene Promoters
(A and B) αMHC-GFP MEFs were infected with GHMT, or GHMT + NICD, and treated or untreated with DAPT. At day 15, different cardiac markers were analyzed (A) by immunostaining or (B) by qPCR; n = 3 biological replicates. Scale bar, 100 μm.
(C and D) MEFs were infected with empty vector or GHMT, and treated with vehicle (DMSO) or with DAPT for 15 days. (C) Chromatin immunoprecipitation of MEF2C followed by qPCR of the indicated genomic regions. An exonic region of Actc1 was used as a control; n=3, three independent immuoprecipitations. (D) Immunoblot using MEF2C antibody. Densitometric quantification, relative to GAPDH, is shown.
(E) Model illustrating proposed molecular mechanism by which Notch inhibition enhances cardiac reprogramming.
Data are presented as means ± SD. ∗p < 0.05, ∗∗∗p < 0.001.
Discussion
Heart failure remains the primary cause of mortality and morbidity worldwide, and cardiac reprogramming represents a new approach to cardiac repair. Although this technique has been significantly improved over the past 5 years, some barriers still remain for its clinical translation, including the poor efficiency of conversion and the failure to create consistent populations of mature contractile cells (Sadahiro et al., 2015). In the present study, we demonstrate that the γ-secretase inhibitor DAPT promotes cardiac reprogramming from mouse fibroblasts to induced cardiomyocytes, not only by increasing the number of induced cardiomyocytes but also by producing a remarkable increase in the number of spontaneously beating cells. Accordingly, DAPT treatment induces the formation of an advanced sarcomere structure and increased calcium flux.
Our results suggest that the Notch signaling pathway is active during the cardiac reprogramming process, which is consistent with Notch having a critical role during heart development by regulating cardiomyocyte cell fate and proliferation (reviewed in de la Pompa and Epstein, 2012). Our results are in line with previous studies reporting that Notch inactivation promotes cardiac differentiation from pluripotent stem cells (Jang et al., 2008, Nemir et al., 2006). Conversely, other reports suggest that Notch activation is important for cardiac lineage differentiation in other progenitor cell types (Chen et al., 2008, Koyanagi et al., 2007, Li et al., 2006), and it has been suggested that Notch favors or impairs differentiation in a cell-type- and time-specific manner (Liu et al., 2014). In fact, Notch inactivation could have different effects depending on the reprogramming strategy; some studies have already reported different chemical strategies to improve cardiac reprogramming (Cao et al., 2016, Ifkovits et al., 2014, Wang et al., 2014), but the role of the Notch pathway during cardiac reprogramming has not been reported. Regarding its role in vivo, some reports indicate that activation of Notch signaling has a cardioprotective role after heart injury (Ferrari and Rizzo, 2014, Gude and Sussman, 2012), but to our knowledge Notch inactivation has not been coupled, so far, with cardiac reprogramming as a cardiac repair strategy.
When combined with AKT1, during GHMT-mediated reprogramming we revealed that DAPT induces up to 70% conversion efficiency, with ∼45% of reprogrammed cells displaying spontaneous beating. Previously, we reported that AKT1 enhances reprogramming through mTORC1 and FOXO3A (Zhou et al., 2015). We examined whether DAPT could enhance reprogramming by the activation of the phosphatidylinositol 3-kinase (PI3K)-AKT1 pathway, but did not observe any increase in Akt activation upon DAPT treatment (data not shown). Notch inhibition and AKT1 seem to employ independent but cooperative mechanisms to boost the production of iCLMs by GHMT. Given that the inefficiency of the cardiac reprogramming process is an important limitation, these results represent a significant advance over other existing lineage conversion techniques.
In agreement with our functional assays, transcriptome analysis by RNA-seq showed that DAPT treatment induces genetic programs related to muscle development, differentiation, and excitation-contraction coupling, and at the same time downregulates genetic programs related to cell proliferation. Of note, no gene set related to the Notch canonical pathway was significantly downregulated, and downregulation of Rbpj-k, a main downstream Notch regulator, did not produce a significant effect on cardiac reprogramming, so we conclude that the Notch canonical pathway is not responsible for DAPT enhancement of reprogramming. However, we have shown that NICD overexpression impairs GHMT-mediated reprogramming. By ChIP experiments, we demonstrated that MEF2C binds to the promoter of the cardiac genes Myh6, Tnnt2, and Actc1, and that this binding is enhanced by DAPT treatment. These results are in line with our previous observations that NICD blocks the binding of MEF2C to chromatin and suppresses its transcriptional activity in myotubes (Wilson-Rawls et al., 1999). Interestingly, it has been demonstrated that the co-transcriptional activator MAML binds to MEF2C to promote myotube differentiation and NICD blocks MEF2C-MAML binding, impairing differentiation (Shen et al., 2006). In light of this and our current findings, we propose a mechanistic model in which DAPT treatment results in Notch inhibition and, consequently, full activation of MEF2C transcriptional activity on cardiac gene promoters (Figure 5E), although we recognize that it is possible that other mechanisms may contribute to this process.
Cardiac reprogramming constitutes a promising strategy for heart repair. Since it was described in 2010, several signaling pathways have been demonstrated to affect this process, such as transforming growth factor β (Ifkovits et al., 2014), Wnt (Lalit et al., 2016), and PI3K-AKT (Zhou et al., 2015) pathways. Changing the cell fate (in this case from fibroblasts to cardiomyocytes) is a complex and tightly regulated process whereby many different pathways, acting at different levels, must be orchestrated to first lose the initial cell identity and successfully adopt a different one. The finding that Notch inactivation by DAPT promotes cardiac reprogramming provides a “druggable” inroad into the reprogramming process. It will be of clinical interest to test whether Notch inactivation favors cardiac reprogramming in vivo in the context of a myocardial infarct, mediated either by transcription factors or purely by chemicals. Of note, Notch inhibitors are currently in several preclinical and clinical trials for cancer treatment (Yuan et al., 2015). Although cellular reprogramming as a regenerative strategy still faces many challenges, our findings represent a significant advance in terms of efficiency and quality of cardiac reprogramming, as well as a better understanding of the molecular mechanisms that govern this process.
Experimental Procedures
All experiments involving animals were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center.
Cell Culture
Wild-type, αMHC-GFP transgenic (Song et al., 2012), or p27 knockout (Fero et al., 1996) MEFs and wild-type TTFs were isolated as described by Nam et al. (2014), and cultured in DMEM containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. The following chemical compounds were added to the induction medium: 2.5 μM DAPT (Selleckchem); 10 μM DBZ (Selleckchem); 2 μM 616452 (Calbiochem); 5 μM GO 6983 (Sigma); 10 ng/μL L-ascorbic acid (Sigma); 1 μM OAC2 (Sphinx Scientific); 100 nM JQ1 (BPS Bioscience); 10 ng/mL human interleukin-6 (Life Technologies); or 10 μg/mL recombinant mouse IL33 protein (R&D Systems).
Mouse Adult Cardiomyocyte Isolation
Adult mouse cardiomyocytes were isolated as described previously (Makarewich et al., 2014). Anesthesia was induced using 3% isoflurane and maintained using 1% isoflurane. Mouse hearts were rapidly excised and the aorta was cannulated on a constant-flow Langendorff apparatus. The heart was digested by perfusion of Tyrode's solution containing 0.2 mg/mL Liberase DH (Roche) and 0.02 mM CaCl2, 10 mM glucose, 5 mM HEPES, 5.4 mM KCl, 1.2 mM MgCl2, 150 mM NaCl, and 2 mM sodium pyruvate (pH 7.4). When the tissue softened, the left ventricle was isolated and gently minced, filtered, and equilibrated in Tyrode's solution with 200 μM CaCl2 and 1% BSA at room temperature.
Cardiac Reprogramming, Beating Cell Analysis, and Calcium Flux Measurements
Generation of retroviral constructs of mouse Gata4, Hand2, Mef2c, Tbx5, and Akt1 has been previously described (Song et al., 2012, Zhou et al., 2015). The retroviral constructs for the expression of shRNAs targeting Rbpj-k and IL33 were purchased from Origene. 293T cells were transiently transfected using Fugene6 with 4 μg of pCLEco (which express the viral genes gag, pol and envEco) and 4 μg of the vector of interest. Forty-eight hours later, the supernatant containing the viral particles was collected and filtered, and polybrene was added to a final concentration of 8 μg/mL. This supernatant was added to the fibroblasts (MEFs or TTFs), seeded the previous day at a concentration of 7,500 cells/cm2. This procedure was repeated 12 hr and 24 hr later. Forty-eight hours later, viral medium was replaced with induction medium, composed of DMEM/199 (4:1), 10% FBS, 5% horse serum, 1% penicillin/streptomycin, 1% non-essential amino acids, 1% essential amino acids, 1% B-27, 1% insulin-selenium-transferrin, 1% vitamin mixture, and 1% sodium pyruvate (Invitrogen). The medium was changed every 2 days until the cells were harvested. Beating cell analyses were performed as described by Zhou et al. (2015). For calcium flux measurements, cells were loaded with 5 μM Fluo-4 AM (Molecular Probes), or we used αMHC-Cre/Rosa26A-Flox-Stop-Flox-GCaMP3 MEFs and calcium flux was quantified as described in Zhou et al. (2015).
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.2% Triton X-100 in PBS for 15 min at room temperature. Cells were blocked with 10% goat serum in PBS for 1 hr and then incubated with primary antibodies against cTnT (mouse monoclonal, Thermo Scientific, MS-295-P; 1:400), α-actinin (mouse monoclonal, Sigma, A7811; 1:400), and GFP (A11122, Life Technologies) for 2 hr at room temperature followed by incubation with appropriate Alexa fluorogenic secondary antibodies (Invitrogen) at room temperature for 1 hr.
Proliferation Assays
Wild-type MEFs were infected with GHMT and treated with 2.5 μM DAPT or DMSO for 7 days. We analyzed the proliferation using the Click-iT Edu Flow Cytometry Assay Kit (Invitrogen), treating with 20 μM 5-ethynyl-2′-deoxyuridine for 24 hr and following the manufacturer's protocol.
Quantitative mRNA Measurement, Western Blot Analysis, and ELISA
Total RNA was extracted using TRIzol (Invitrogen) following the provider's recommendations and retrotranscribed into cDNA using iScript Supermix (Bio-Rad) following the manufacturer's instructions. Real-time qPCR was performed using Taq-man probes (Applied Biosystems) for Tnnt2, Myh6, and Actc1, and Kapa Sybr Fast (Kapa Biosystems) for IL33 using the primers Fw 5′-TCC AAC TCC AAG ATT TCC CCG-3′ and Rv 5′-CAT GCA GTA GAC ATG GCA GAA-3′. For input normalization, we used Gapdh Fw 5′-AGG TCG GTG TGA ACG GAT TTG-3′ and Rv 5′-TGT AGA CCA TGT AGT TGA GGT CA-3′. Western blot analyses were performed as described by Nelson et al. (2016), using SERCA2 ATPase antibody (1:1,000, MA3-919, Thermo Scientific), ryanodine receptor antibody (1:1,000, MA3-916, Thermo Scientific), and MEF2C antibody (1:500, ab79436, Abcam). ELISA was performed using Mouse IL33 DuoSet from R&D.
RNA-Seq and Transcriptome Analysis
Total RNA was isolated using TRIzol following the provider's instructions. Illumina RNA-seq was performed by the University of Texas Southwestern Microarray Core Facility. Quality assessment of the RNA-seq data was done using the NGS QC Toolkit (Patel and Jain, 2012) with default setting. Quality filtered reads generated by the tool were then aligned to the mouse reference genome GRCm38 (mm10) using the TopHat2 (v. 2.0.0) aligner (Kim et al., 2013) using default settings. Differential gene expression analysis was done using the R package DESeq (v. 1.6.3) (Love et al., 2014). Read counts obtained from featureCounts (Liao et al., 2014) were normalized by taking the median of each gene count across samples and dividing each sample gene count by the relative ratio of library sizes between the calculated median and sample size. The averaged normalized expression values of the samples were used to calculate fold change and p values. Cutoff values of fold change greater than 1.5 and p values less than 0.05 were then used to select for differentially expressed genes between sample group comparisons. Significant pathway enrichment analysis was performed using PANTHER Overrepresentation Test (release 20150430, http://geneontology.org). Notch pathways were extracted from MSigDB (Subramanian et al., 2005) and enrichment plots were performed using GSEA (Mootha et al., 2003).
Chromatin Immunoprecipitation
ChIP assays were performed as previously described (Rubins et al., 2005), with some modifications. In brief, cells were crosslinked with 1% formaldehyde-PBS for 15 min with constant shaking, and formaldehyde was neutralized by the addition of glycine to a final concentration of 0.125 M for another 5 min. Cells were washed with cold PBS and collected in 300 μL of Chip Whole Lysis Buffer (10 mM Tris-HCl [pH 8.0], 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40, 1% SDS, and 0.5% sodium deoxycholate). Chromatin fragmentation was performed by sonication using a Diagnode BioRupter (three cycles of 7.5 min with 30 s on/off). Protein concentration was quantified and 450 μg were immunoprecipitated by using 3 μg of anti-MEF2C (ab79436, Abcam). The antibody/chromatin complexes were rotated end-to-end overnight at 4°C. Antibody/chromatin complexes were pulled down using Dynabeads protein G-conjugated magnetic beads (Life Technologies). Chromatin was washed, eluted, and reverse-crosslinked. Chromatin fragments were then analyzed by qPCR using SYBR Green fluorescence. Primers are available upon request.
Statistical Analysis
Statistical significance was determined using the Student's t test (unpaired two-tailed). All data are presented as mean ± SD (error bars). For every reprogramming experiment, three different plates per group were reprogrammed in parallel (n = 3 biological replicates). Representative reprogramming experiments (out of at least three experiments) are shown, unless otherwise specified. Significance in figures is depicted by ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Author Contributions
M.A. designed and performed experiments, data analysis, discussion, and writing; H.H., H.Z., and M.G.M contributed to experimental work and discussion; B.C. performed RNA-seq analysis; R.B.-D. contributed discussion and writing; E.N.O. supervised the study and contributed to writing.
Acknowledgments
We thank Manuel Serrano for reagents and Jose Cabrera for help with images. This work was supported by grants from the NIH (grants HL-077439, HL-111665, HL-093039, DK-099653, HL-130253, and U01-HL-100401), Foundation Leducq Networks of Excellence (grant 14CVD04), Cancer Prevention and Research Institute of Texas, the Robert A. Welch Foundation (grant 1-0025 to E.N.O.), and the Spanish Ministry of Economy (MINECO, Ramon y Cajal Grant to M.A.). H.H. was supported by an Uehara Memorial Foundation Postdoctoral Fellowship. H.Z. was supported by a pre-doctoral fellowship (14PRE20030030) from the American Heart Association. M.G.M. was supported by a PEW Latin American Fellowship. E.N.O. is a cofounder and member of the Scientific Advisory Board of Tenaya Therapeutics and holds equity in the company.
Published: March 2, 2017
Footnotes
Supplemental Information includes five figures, four tables, and two movies and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2017.01.025.
Contributor Information
Maria Abad, Email: mabad@vhio.net.
Eric N. Olson, Email: eric.olson@utsouthwestern.edu.
Accession Numbers
The accession number for the RNA-seq data reported in this paper is GEO: GSE93585.
Supplemental Information
{kind=link}
{kind=link}
References
- Addis R.C., Ifkovits J.L., Pinto F., Kellam L.D., Esteso P., Rentschler S., Christoforou N., Epstein J.A., Gearhart J.D. Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success. J. Mol. Cell Cardiol. 2013;60:97–106. doi: 10.1016/j.yjmcc.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen P., Uosaki H., Shenje L.T., Kwon C. Non-canonical Notch signaling: emerging role and mechanism. Trends Cell Biol. 2012;22:257–265. doi: 10.1016/j.tcb.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson E.R., Sandberg R., Lendahl U. Notch signaling: simplicity in design, versatility in function. Development. 2011;138:3593–3612. doi: 10.1242/dev.063610. [DOI] [PubMed] [Google Scholar]
- Batty J.A., Lima J.A., Jr., Kunadian V. Direct cellular reprogramming for cardiac repair and regeneration. Eur. J. Heart Fail. 2016;18:145–156. doi: 10.1002/ejhf.446. [DOI] [PubMed] [Google Scholar]
- Beltrami A.P., Urbanek K., Kajstura J., Yan S.M., Finato N., Bussani R., Nadal-Ginard B., Silvestri F., Leri A., Beltrami C.A. Evidence that human cardiac myocytes divide after myocardial infarction. N. Engl. J. Med. 2001;344:1750–1757. doi: 10.1056/NEJM200106073442303. [DOI] [PubMed] [Google Scholar]
- Bergmann O., Bhardwaj R.D., Bernard S., Zdunek S., Barnabe-Heider F., Walsh S., Zupicich J., Alkass K., Buchholz B.A., Druid H. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady J.J., Li M., Suthram S., Jiang H., Wong W.H., Blau H.M. Early role for IL-6 signalling during generation of induced pluripotent stem cells revealed by heterokaryon RNA-Seq. Nat. Cell Biol. 2013;15:1244–1252. doi: 10.1038/ncb2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao N., Huang Y., Zheng J., Spencer C.I., Zhang Y., Fu J.D., Nie B., Xie M., Zhang M., Wang H. Conversion of human fibroblasts into functional cardiomyocytes by small molecules. Science. 2016;352:1216–1220. doi: 10.1126/science.aaf1502. [DOI] [PubMed] [Google Scholar]
- Chen V.C., Stull R., Joo D., Cheng X., Keller G. Notch signaling respecifies the hemangioblast to a cardiac fate. Nat. Biotechnol. 2008;26:1169–1178. doi: 10.1038/nbt.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforou N., Chellappan M., Adler A.F., Kirkton R.D., Wu T., Addis R.C., Bursac N., Leong K.W. Transcription factors MYOCD, SRF, Mesp1 and SMARCD3 enhance the cardio-inducing effect of GATA4, TBX5, and MEF2C during direct cellular reprogramming. PLoS One. 2013;8:e63577. doi: 10.1371/journal.pone.0063577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Pompa J.L., Epstein J.A. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev. Cell. 2012;22:244–254. doi: 10.1016/j.devcel.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Debbio C.B., Mir Q., Parameswaran S., Mathews S., Xia X., Zheng L., Neville A.J., Ahmad I. Notch signaling activates stem cell properties of muller glia through transcriptional regulation and Skp2-mediated degradation of p27Kip1. PLoS One. 2016;11:e0152025. doi: 10.1371/journal.pone.0152025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohda T., Maljukova A., Liu L., Heyman M., Grander D., Brodin D., Sangfelt O., Lendahl U. Notch signaling induces SKP2 expression and promotes reduction of p27Kip1 in T-cell acute lymphoblastic leukemia cell lines. Exp. Cell Res. 2007;313:3141–3152. doi: 10.1016/j.yexcr.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Dutta D., Ray S., Home P., Larson M., Wolfe M.W., Paul S. Self-renewal versus lineage commitment of embryonic stem cells: protein kinase C signaling shifts the balance. Stem Cells. 2011;29:618–628. doi: 10.1002/stem.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban M.A., Wang T., Qin B., Yang J., Qin D., Cai J., Li W., Weng Z., Chen J., Ni S. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell. 2010;6:71–79. doi: 10.1016/j.stem.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Fero M.L., Rivkin M., Tasch M., Porter P., Carow C.E., Firpo E., Polyak K., Tsai L.H., Broudy V., Perlmutter R.M. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996;85:733–744. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- Ferrari R., Rizzo P. The Notch pathway: a novel target for myocardial remodelling therapy? Eur. Heart J. 2014;35:2140–2145. doi: 10.1093/eurheartj/ehu244. [DOI] [PubMed] [Google Scholar]
- Gude N., Sussman M. Notch signaling and cardiac repair. J. Mol. Cell Cardiol. 2012;52:1226–1232. doi: 10.1016/j.yjmcc.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruharsha K.G., Kankel M.W., Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 2012;13:654–666. doi: 10.1038/nrg3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H., Katoku-Kikyo N., Keirstead S.A., Kikyo N. Accelerated direct reprogramming of fibroblasts into cardiomyocyte-like cells with the MyoD transactivation domain. Cardiovasc. Res. 2013;100:105–113. doi: 10.1093/cvr/cvt167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichida J.K., Tcw J., Williams L.A., Carter A.C., Shi Y., Moura M.T., Ziller M., Singh S., Amabile G., Bock C. Notch inhibition allows oncogene-independent generation of iPS cells. Nat. Chem. Biol. 2014;10:632–639. doi: 10.1038/nchembio.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M., Fu J.D., Delgado-Olguin P., Vedantham V., Hayashi Y., Bruneau B.G., Srivastava D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375–386. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifkovits J.L., Addis R.C., Epstein J.A., Gearhart J.D. Inhibition of TGFbeta signaling increases direct conversion of fibroblasts to induced cardiomyocytes. PLoS One. 2014;9:e89678. doi: 10.1371/journal.pone.0089678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang J., Ku S.Y., Kim J.E., Choi K., Kim Y.Y., Kim H.S., Oh S.K., Lee E.J., Cho H.J., Song Y.H. Notch inhibition promotes human embryonic stem cell-derived cardiac mesoderm differentiation. Stem Cells. 2008;26:2782–2790. doi: 10.1634/stemcells.2007-1053. [DOI] [PubMed] [Google Scholar]
- Jayawardena T.M., Egemnazarov B., Finch E.A., Zhang L., Payne J.A., Pandya K., Zhang Z., Rosenberg P., Mirotsou M., Dzau V.J. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res. 2012;110:1465–1473. doi: 10.1161/CIRCRESAHA.112.269035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyanagi M., Bushoven P., Iwasaki M., Urbich C., Zeiher A.M., Dimmeler S. Notch signaling contributes to the expression of cardiac markers in human circulating progenitor cells. Circ. Res. 2007;101:1139–1145. doi: 10.1161/CIRCRESAHA.107.151381. [DOI] [PubMed] [Google Scholar]
- Lalit P.A., Salick M.R., Nelson D.O., Squirrell J.M., Shafer C.M., Patel N.G., Saeed I., Schmuck E.G., Markandeya Y.S., Wong R. Lineage reprogramming of fibroblasts into proliferative induced cardiac progenitor cells by defined factors. Cell Stem Cell. 2016;18:354–367. doi: 10.1016/j.stem.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Yu B., Zhang Y., Pan Z., Xu W., Li H. Jagged1 protein enhances the differentiation of mesenchymal stem cells into cardiomyocytes. Biochem. Biophys. Res. Commun. 2006;341:320–325. doi: 10.1016/j.bbrc.2005.12.182. [DOI] [PubMed] [Google Scholar]
- Li W., Tian E., Chen Z.X., Sun G., Ye P., Yang S., Lu D., Xie J., Ho T.V., Tsark W.M. Identification of Oct4-activating compounds that enhance reprogramming efficiency. Proc. Natl. Acad. Sci. USA. 2012;109:20853–20858. doi: 10.1073/pnas.1219181110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liu Y., Li P., Liu K., He Q., Han S., Sun X., Li T., Shen L. Timely inhibition of Notch signaling by DAPT promotes cardiac differentiation of murine pluripotent stem cells. PLoS One. 2014;9:e109588. doi: 10.1371/journal.pone.0109588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maherali N., Hochedlinger K. Tgfbeta signal inhibition cooperates in the induction of iPSCs and replaces Sox2 and cMyc. Curr. Biol. 2009;19:1718–1723. doi: 10.1016/j.cub.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarewich C.A., Zhang H., Davis J., Correll R.N., Trappanese D.M., Hoffman N.E., Troupes C.D., Berretta R.M., Kubo H., Madesh M. Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ. Res. 2014;115:567–580. doi: 10.1161/CIRCRESAHA.115.303831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Nam Y.J., Song K., Luo X., Daniel E., Lambeth K., West K., Hill J.A., DiMaio J.M., Baker L.A., Bassel-Duby R. Reprogramming of human fibroblasts toward a cardiac fate. Proc. Natl. Acad. Sci. USA. 2013;110:5588–5593. doi: 10.1073/pnas.1301019110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam Y.J., Lubczyk C., Bhakta M., Zang T., Fernandez-Perez A., McAnally J., Bassel-Duby R., Olson E.N., Munshi N.V. Induction of diverse cardiac cell types by reprogramming fibroblasts with cardiac transcription factors. Development. 2014;141:4267–4278. doi: 10.1242/dev.114025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson B.R., Makarewich C.A., Anderson D.M., Winders B.R., Troupes C.D., Wu F., Reese A.L., McAnally J.R., Chen X., Kavalali E.T. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 2016;351:271–275. doi: 10.1126/science.aad4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemir M., Croquelois A., Pedrazzini T., Radtke F. Induction of cardiogenesis in embryonic stem cells via downregulation of Notch1 signaling. Circ. Res. 2006;98:1471–1478. doi: 10.1161/01.RES.0000226497.52052.2a. [DOI] [PubMed] [Google Scholar]
- Patel R.K., Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7:e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protze S., Khattak S., Poulet C., Lindemann D., Tanaka E.M., Ravens U. A new approach to transcription factor screening for reprogramming of fibroblasts to cardiomyocyte-like cells. J. Mol. Cell Cardiol. 2012;53:323–332. doi: 10.1016/j.yjmcc.2012.04.010. [DOI] [PubMed] [Google Scholar]
- Qian L., Huang Y., Spencer C.I., Foley A., Vedantham V., Liu L., Conway S.J., Fu J.D., Srivastava D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485:593–598. doi: 10.1038/nature11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubins N.E., Friedman J.R., Le P.P., Zhang L., Brestelli J., Kaestner K.H. Transcriptional networks in the liver: hepatocyte nuclear factor 6 function is largely independent of Foxa2. Mol. Cell Biol. 2005;25:7069–7077. doi: 10.1128/MCB.25.16.7069-7077.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadahiro T., Yamanaka S., Ieda M. Direct cardiac reprogramming: progress and challenges in basic biology and clinical applications. Circ. Res. 2015;116:1378–1391. doi: 10.1161/CIRCRESAHA.116.305374. [DOI] [PubMed] [Google Scholar]
- Sanganalmath S.K., Bolli R. Cell therapy for heart failure: a comprehensive overview of experimental and clinical studies, current challenges, and future directions. Circ. Res. 2013;113:810–834. doi: 10.1161/CIRCRESAHA.113.300219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmento L.M., Huang H., Limon A., Gordon W., Fernandes J., Tavares M.J., Miele L., Cardoso A.A., Classon M., Carlesso N. Notch1 modulates timing of G1-S progression by inducing SKP2 transcription and p27 Kip1 degradation. J. Exp. Med. 2005;202:157–168. doi: 10.1084/jem.20050559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z., Yao C., Khodadadi-Jamayran A., Xu W., Townes T.M., Crowley M.R., Hu K. Reprogramming by de-bookmarking the somatic transcriptional program through targeting of BET bromodomains. Cell Rep. 2016;16:3138–3145. doi: 10.1016/j.celrep.2016.08.060. [DOI] [PubMed] [Google Scholar]
- Shen H., McElhinny A.S., Cao Y., Gao P., Liu J., Bronson R., Griffin J.D., Wu L. The Notch coactivator, MAML1, functions as a novel coactivator for MEF2C-mediated transcription and is required for normal myogenesis. Genes Dev. 2006;20:675–688. doi: 10.1101/gad.1383706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K., Nam Y.J., Luo X., Qi X., Tan W., Huang G.N., Acharya A., Smith C.L., Tallquist M.D., Neilson E.G. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature. 2012;485:599–604. doi: 10.1038/nature11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundlisaeter E., Edelmann R.J., Hol J., Sponheim J., Kuchler A.M., Weiss M., Udalova I.A., Midwood K.S., Kasprzycka M., Haraldsen G. The alarmin IL-33 is a notch target in quiescent endothelial cells. Am. J. Pathol. 2012;181:1099–1111. doi: 10.1016/j.ajpath.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Wada R., Muraoka N., Inagawa K., Yamakawa H., Miyamoto K., Sadahiro T., Umei T., Kaneda R., Suzuki T., Kamiya K. Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc. Natl. Acad. Sci. USA. 2013;110:12667–12672. doi: 10.1073/pnas.1304053110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Cao N., Spencer C.I., Nie B., Ma T., Xu T., Zhang Y., Wang X., Srivastava D., Ding S. Small molecules enable cardiac reprogramming of mouse fibroblasts with a single factor, Oct4. Cell Rep. 2014;6:951–960. doi: 10.1016/j.celrep.2014.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson-Rawls J., Molkentin J.D., Black B.L., Olson E.N. Activated notch inhibits myogenic activity of the MADS-Box transcription factor myocyte enhancer factor 2C. Mol. Cell Biol. 1999;19:2853–2862. doi: 10.1128/mcb.19.4.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X., Wu H., Xu H., Xiong H., Chu Q., Yu S., Wu G.S., Wu K. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. 2015;369:20–27. doi: 10.1016/j.canlet.2015.07.048. [DOI] [PubMed] [Google Scholar]
- Zhou H., Dickson M.E., Kim M.S., Bassel-Duby R., Olson E.N. Akt1/protein kinase B enhances transcriptional reprogramming of fibroblasts to functional cardiomyocytes. Proc. Natl. Acad. Sci. USA. 2015;112:11864–11869. doi: 10.1073/pnas.1516237112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.