

Abstract
The neuroprotective effect of polydatin (PD) against hemorrhagic shock-induced mitochondrial injury has been described previously, and mitochondrial dysfunction and apoptosis were reportedly involved in ischemic stroke. In the present study the neuroprotective effect of PD in preventing apoptosis was evaluated following induction of focal cerebral ischemia by middle cerebral artery occlusion (MCAO) in rats. PD (30 mg/kg) was administered by caudal vein injection 10 min prior to ischemia/reperfusion (I/R) injury. 24 h following I/R injury, ameliorated modified neurological severity scores (mNSS) and reduced infarct volume were observed in the PD treated group. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining and Annexin V/propidium iodide assays demonstrated the anti-apoptotic effect of PD in the ischemic cortex. In addition, PD improved I/R injury-induced mitochondrial dysfunction, reflected by morphological observations and measurements of mitochondrial membrane potential and intracellular ATP measurement. Western blot analysis revealed an increase in B-cell lymphoma 2 apoptosis regulator (Bcl-2) expression, and a decrease in Bcl-2-associated protein X apoptosis regulator expression in the PD group in comparison with the vehicle treated group. PD treatment also prevented the release of cytochrome c from mitochondria into the cytoplasm, and blunted the activities of caspase-9 and caspase-3. Furthermore, PD treatment decreased the levels of reactive oxygen species in neurons isolated from the ischemic cortex. The findings of this study, therefore, suggest that PD has a dual effect, ameliorating both oxidative stress and mitochondria-dependent apoptosis, making it a promising new therapy for the treatment of ischemic stroke.
Keywords: polydatin, ischemic stroke, oxidative stress, mitochondria, apoptosis
Introduction
Cerebral ischemia, a condition in which blood flow to the brain is insufficient, is one of the most serious neurological disorders and can lead to stroke when cell death occurs (1–3). Currently available treatments for ischemic stroke, including anticoagulants and thrombolytics, have not displayed substantial improvements in patients with ischemic stroke (4). Therefore, investigations into the pathological mechanisms of ischemic stroke, and identification of effective neuroprotective agents that can be utilized after ischemic stroke occurs are critical to improving outcomes.
Cerebral ischemia is the result of transient or permanent reduction of cerebral blood flow initiated by thrombotic or thromboembolic arterial occlusions. Thrombolytic therapy designed to restore cerebral perfusion in a timely fashion is considered the primary therapeutic strategy for ischemic brain injury (4). However, reperfusion following thrombolytic therapy often leads to a series of cellular, biochemical and metabolic consequences of cerebral ischemia, including generation of intracellular reactive oxygen species (ROS), calcium overload, mitochondrial injury and cell death, which may ultimately lead to irreversible ischemia-reperfusion (I/R) injury (5,6). The mechanisms of cerebral I/R injury are complex, and include cellular responses such as oxidative stress, inflammation and apoptosis (4–6). Previous studies have demonstrated the involvement of oxidative stress and intrinsic apoptotic signaling in the pathophysiology of cerebral I/R injury (7,8). Elevated levels of ROS directly induce damage to proteins, lipids and DNA. ROS also act as a molecular trigger for apoptotic signaling, increasing apoptotic cell death following ischemia (9). Intrinsic apoptosis is regulated by mitochondrial activation and the release of cytochrome c and second mitochondrial derived activator of caspases (Smac), resulting in caspase-3 activation (10,11). The intrinsic or mitochondrial-dependent mechanism of caspase activation depends on the in B-cell lymphoma 2 apoptosis regulator (Bcl-2) family of proteins that regulate mitochondrial outer membrane permeabilization (9). The Bcl family consits anti-apoptotic and pro-apoptotic factors (11,12). The pro-apoptotic members of this family, including Bcl-2-associated protein X (Bax) and Bcl-2 antagonist/killer 1, trigger the release of mitochondrial apoptotic factors into the cytoplasm, leading to caspase activation, whereas the anti-apoptotic family members, such as Bcl-2, act to prevent apoptosis (12).
Polydatin (PD) is a monocrystalline drug isolated from the herb Fallopia japonica, previously known as Polygonum cuspidatum and commonly known as Japanese Knotweed. It is used in traditional Chinese remedies to treat sepsis, burns and I/R injury (12–14). Previous studies have demonstrated the protective effects of PD against acute shock-induced mitochondrial injury in neurons, arteriolar smooth muscle cells and enterocytes, as evidenced by the presence of ROS scavenging and prevention of mitochondrial damage (15–17).
The aim of the present study was to evaluate the potential therapeutic effect of PD following ischemic stroke, and elucidate the mechanisms of PD neuroprotection with regard to oxidative stress and intrinsic apoptotic signaling.
Materials and methods
Middle cerebral artery occlusion (MCAO) model and drug treatment
All animal experiments were approved by the animal experimental committee of Fujian Medical University (Fuzhou, China). A total of 36 adult male Sprague-Dawley rats (250–270 g, 8–10 weeks) were purchased from the Experimental Animal Center at Fujian Medical University, and were allowed to acclimate for 1 week prior to the beginning of experimentation. The animals had ad libitum access to rat chow and water. Animals were divided randomly into three groups: sham-operated (sham); MCAO group treated with 0.5 ml of normal saline (vehicle); and MCAO group treated with 30 mg/kg of polydatin (PD; Neptunus Interlong Bio-technique Co., Ltd., Shenzhen, Guangdong, China). Vehicle and PD treatments were administered via caudal vein injection 10 min before the start of the MCAO.
MCAO was produced using the intraluminal filament technique as previously described (5). Rats were anesthetized with an intraperitoneal injection of ketamine/xylazine (0.1 ml/100 g). The right common carotid artery was exposed via a midline neck incision, and was dissected from surrounding nerves and fascia up to its bifurcation at the base of the skull. The external carotid artery and internal carotid artery were then isolated in turn. A nylon monofilament was inserted through the proximal external carotid artery into the internal carotid artery and advanced until a slight resistance was felt, indicating occlusion of the origin of the MCA. The nylon monofilament remained in place for 2 h. The nylon monofilament was then retracted so as to allow reperfusion of the ischemic region. In the sham group, the procedure was followed as for MCAO rats up to the occlusion of the MCA; however, the filament was removed within 60 sec. All the rats were sacrificed by decollation 24 h subsequent to MCAO or sham treatment for neuron isolation and assessment.
Measurement of infarct volume
Three rats in each group were randomly selected for 2,3,5-triphenyltetrazolium chloride (TTC) staining. Freshly isolated brains were stored at 20°C for 15 min. Brains were then cut into 2 mm thick coronal sections. Sections were placed in 2% TTC and warmed in a heated water bath at 37°C for 30 min. Sections were then fixed with 4% paraformaldehyde overnight at 4°C. Stained sections were photographed, and the area of ischemic brain injury was calculated by a blinded observer using ImageJ software version 1.48 (National Institutes of Health, Bethesda, MD, USA). Infarct volume is expressed as infarct area percentage (%).
Evaluation of neurological deficit
A modified neurological severity score (mNSS) test was performed by a blinded investigator 24 h subsequent to the induction of MCAO as described previously (4). The mNSS is a composite of motor, sensory, balance and reflex tests, and is graded on a scale of 0–18, with a normal score being 0 and a maximal deficit score being 18. For injury severity scores, 1 point is awarded for the inability to perform the test, or for lack of a tested reflex; higher scores reflect a more severe injury.
Terminal deoxynucleotidyl transferase dUTP (deoxyuridine 5-triphosphate-digoxigenin) nick end labeling (TUNEL) staining
Cortex tissues on histopathological slides were stained using a TUNEL kit (BioVision, Inc., Milpitas, CA, USA) according to the manufacturer's protocols, counterstained with Hoechst 33258, and then examined under an ECLIPSEFN1 fluorescent microscope (Nikon Corporation, Japan). Apoptotic cells appeared fluorescent green, and the number of apoptotic cells per 10 visual fields was determined at ×200 magnification.
Morphological observation
Morphological changes to neuronal mitochondria were observed by transmission electron microscopy (TEM). Cortical tissues were fixed with 2.5% glutaraldehyde for 24 h at room temperature and stained with cacodylate-buffered osmium tetroxide for 4 h in 4°C. Sections (60 nm thickness) were prepared and examined under an electron microscope at ×10,000 magnification (Philips CM10; Philips, Eindhoven, The Netherlands).
Measurement of cytosolic cytochrome c
The levels of cytochrome c in the cytoplasm were evaluated with a cytochrome c ELISA kit (cat. no. K257-100, BioVision, Inc.). Cortical tissues were dissected 24 h after MCAO, then weighed, and lysed at 4°C for 10 min in a cold lysis buffer (10 mM Tris-HCl, 0.3 M sucrose, 10 µM aprotinin, 10 µM pepstatin, 10 µM leupeptin, and 1 mM PMSF, pH 7.5). Tissue homogenates were centrifuged at 10,000 × g for 60 min at 4°C, the supernatant containing the cytosolic fraction collected, and protein concentration determined by bicinchoninic acid (BCA) assay. Samples were then treated with a conjugate reagent, were transferred to a cytochrome c antibody-coated microwell plate, and incubated at room temperature for 60 min. Wells were washed, treated with substrate and incubated for 30 min at 4°C. Following termination of the reaction, the optical density was read at 450 nm using a microplate reader (SpectraMax M5; Molecular Devices LLC, Sunnyvale, CA, USA). A serial dilution of cytochrome c calibrator was assayed in parallel with the samples, and the concentration of cytochrome c in the samples was determined by calibration of the standard curve.
Measurement of caspase-3 and −9 activation. Activity of caspase-3 and caspase-9 was evaluated using a commercial caspase activity assay kit (cat. nos. K105-100 and K118-100, BioVision Inc.). Cortical tissues were dissected 24 h subsequent to MCAO, and were homogenized in lysis buffer supplemented with dithiothreitol as described in the manufacturer's instructions. Following 15 min incubation on ice, the supernatant was transferred into another centrifuge tube, and centrifuged at 1,000 × g at 4°C for 5 min. Protein levels in the supernatant were determined by BCA assay. Aliquots of each sample (20–50 µg protein) were incubated with 90 µl of reaction buffer and 10 µl of the substrate N-Acetyl-Asp-Glu-Val-Asp phospho-nitroanilide at 37°C for 2 h. The absorbance of the released phospho-nitroanilide was measured at at 405 nm for caspase-3 and 505 nm for caspase-9 with a spectrophotometer (SpectraMax M5; Molecular Devices, LLC).
Neuron isolation procedures
Adult rat cortical neurons were isolated in the shortest time possible, with minimal neuronal hypoxia or ischemia, according to the previously described method (16). Cortical tissues from the ischemic hemisphere were dissected 24 h subsequent to MCAO. The cortex was cut into fragments and the cells were dissociated by incubation with 2 mg/ml papain in DMEM for 30 min at 37°C. To achieve optimal purification, the immune adherence method was used to isolate neurons. The digested cortical suspension was poured into anti-neural cell adhesion molecule-coated petri dishes (cat. no. AB5032, Millipore, Billerica, MA, USA) and placed in a shaker (4°C, 50 × g). Following 1 h incubation, the adhered cells were collected. Trypan Blue staining was used to exclude non-intact cells.
Immunofluorescent test
Isolated neurons were washed with PBS, permeabilized with 4% formaldehyde and 0.5% Triton X-100 for 30 min in 4°C. Cells were washed in PBS twice, blocked in 5% bovine serum albumin (Merck Millipore, Darmstadt, Germany) for 1 h in 4°C, and incubated with neurofilament antibody (1:500, cat. no. ab8135, Abcam, Cambridge, UK) at 4°C overnight. Following a wash with PBS, the cells were stained with a FITC-conjugated secondary antibody (1:2,000, cat. no. ab6717, Abcam) for 1 h at room temperature. Then, cells were captured using intravital upright microscope (ECLIPSEFN1; Nikon Corporation, Tokyo, Japan). Results indicated that the purity of neuron preparations was >90%, as required for onward experiments.
Measurement of cell apoptosis
Cell apoptosis was detected using a FITC-Annexin V apoptosis detection kit (BD Biosciences, San Jose, CA, USA). Neurons were isolated, washed twice with PBS, and suspended in 1X binding buffer at a concentration of approximately 1×105 cells/ml. FITC-Annexin V (5 µl) and 10 µl of 50 µg/ml propidium iodide (PI); (Sigma-Aldrich; Merck Millipore) were added to the cell suspension. Following incubation at room temperature for 20 min in the dark, cell fluorescence was determined using a flow cytometer (Becton Dickinson FACScan; BD Biosciences).
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential (∆Ψm) was assessed by flow cytometry, using the potential-sensitive fluorescent dye, JC-1. This dual-emission probe changes color from red-orange to green as the mitochondrial membrane depolarizes. Neurons were incubated with 5 µmol/l JC-1 for 15 min at 37°C. The cells were then washed with PBS and analyzed by flow cytometry (Becton Dickinson FACScan, BD Biosciences). Following gating using forward scatter and side scatter with 488 nm excitation wavelength, the fluorescence intensity were detected via FL1 (green, 530 nm) and FL2 (red, 590 nm). A minimum of 10,000 cells were analyzed for each sample. The cells with green fluorescence were evaluated to reflect the percentage of cells with low ΔΨm.
Measurement of cellular ATP
Intracellular ATP was determined using a CellTiter-Glo luciferase-based assay (Promega Corporation, Madison, WI), according to the manufacturer's instructions. CellTiter-Glo reagent (100 µl) was added to 10,000 isolated neurons in each well of an opaque 96-well plate. The plates were incubated at room temperature for 10 min, and luminescence (excitation wavelength at 488 nm, emission wavelength at 530 nm) was determined with a microplate reader (SpectraMax M5; Molecular Devices, LLC).
Measurement of ROS levels
Intracellular ROS production was determined using a 2′-7′-dichlorofluorescein diacetate (DCFH-DA) ROS detection kit (Sigma-Aldrich; Merck Millipore). Neurons were isolated and treated with DCFH-DA (10 µM) for 20 min at 37°C. Following incubation, the cells were washed and analyzed using a microplate reader (Spectra Max, M5; Molecular Devices, LLC). The relative intensity of DCF fluorescence compared with sham group cells was determined at a wavelength of 535 nm.
Measurement of Bax and Bcl-2
Cortical tissues were isolated 24 h after MCAO and homogenized in ice-cold tissue lysis buffer [3 M NaCl, 1 M Tris-HCl (pH 7.4), 0.5 M EDTA, 100 mM PMSF, 10% Triton X-100] for 30 min. The samples were then centrifuged at 10,000 × g for 20 min at 4°C. Supernatant total protein concentration was determined by bicinchoninic acid assay. Samples were then denatured at 100°C for 5 min, and separated on 10% SDS-PAGE gels and transferred to polyvinylidene difluoride (PVDF) membranes for immunoblotting. Membranes were blocked with blocking solution (5% non-fat milk diluted with PBS) at room temperature for 2 h, followed by incubation with Bax (cat. no. ab32503), Bcl-2 (cat. no. ab59348), or GAPDH (cat. no. ab8245) primary antibodies (1:1,000 dilution, Abcam) overnight at 4°C. Membranes were then washed in PBS-Tween (PBST including 0.05% Tween-20), incubated with secondary antibody (cat. no. ab6721, Abcam; 1:5,000 dilution with PBS) for 2 h at room temperature, then washed 3 times in PBST. The PVDF membranes were incubated with ultrasensitive enhanced chemiluminescence (cat. no. abs920B-500, Absin Bioscience, Inc., Shanghai, China) for 1 min at 4°C. Next, the images of PVDF membranes were obtained by a gel imaging system (Image Station 4000R, Kodak, Rochester, NY, USA).
Statistical analysis
Data are presented as the mean + standard deviation. Differences between groups were determined using one-way analysis of variance with the least significant difference multiple-comparison test, or Student's t-test when appropriate. P<0.05 was considered to indicate a statistically significant difference.
Results
Neuroprotection by PD following experimental stroke
To assess the protective effects of PD following stroke, neurological scores and brain infarct volumes were evaluated 24 h subsequent to MCAO using mNSS (Fig. 1A) and TTC staining (Fig. 1B) respectively. As demonstrated in Fig. 1, compared with the sham group, the vehicle group displayed significant neurological deficits (P<0.001; Fig. 1A) and brain infarcts (P<0.001; Fig. 1B) as evidenced by the white patches in brain tissues. Administration of PD significantly decreased the infarct volume (P=0.002 Fig. 1B) and ameliorated the behavioral deficit (P Fig. 1A) compared with the vehicle group.
Figure 1.
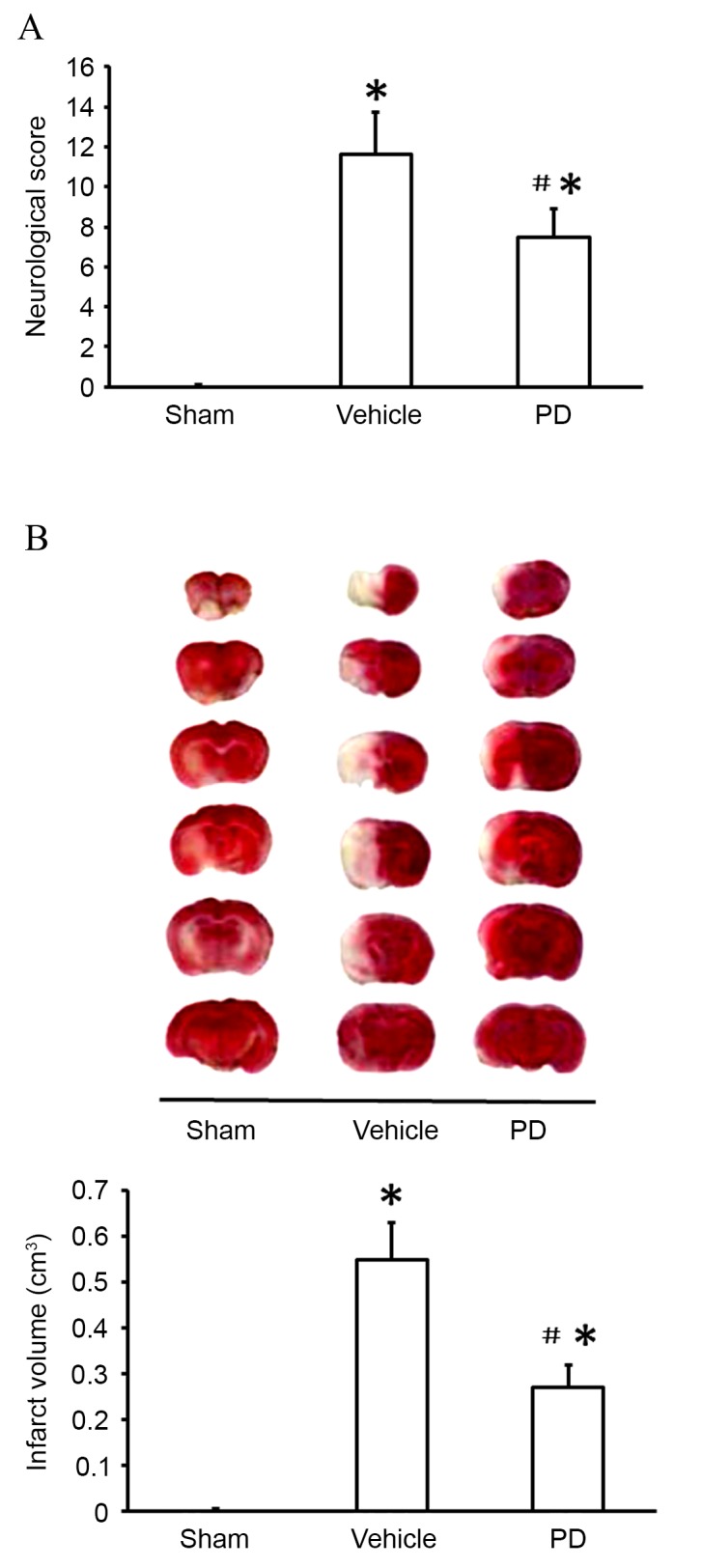
PD is neuroprotective following middle cerebral artery occlusion. (A) Evaluation of neurological deficit by modified neurological severity score. (B) Representative brain sections with 2,3,5-triphenyltetrazolium chloride staining and quantification of infarct volume. Data are presented as the mean + standard deviation (n=6 in each group). *P<0.05, vs. sham group; #P<0.05, vs. vehicle group. PD, polydatin.
Neuroprotection of PD on I/R injury-induced neuron apoptosis
Cell viability and apoptosis was detected by TUNEL staining (Fig. 2A and B) and the FITC-Annexin V/PI double staining assay (Fig. 2C) respectively. The number of TUNEL-positive cells was significantly increased in the ischemic region of the cortex in vehicle treated animals compared with sham treated animals (P<0.001; Fig. 2B); this was reduced in the cerebral cortex of the PD-treated compared with vehicle group (P=0.002 Fig. 2B). In the Annexin V-FITC assay, the rate of cell apoptosis was significantly increased following MCAO compared with the sham group (P<0.001; Fig. 2C); treatment with PD decreased cell apoptosis compared with the vehicle group (P=0.011; Fig. 2C).
Figure 2.

PD treatment reduces apoptosis in the cortex following middle cerebral artery occlusion. (A) TUNEL staining in the cortex (white arrows indicate the TUNEL-positive cells). Original magnification, ×200 magnification. (B) Quantification of TUNEL-positive cells averaged over 10 microscopic fields per animal. (C) Isolated neuronal apoptosis as evaluated by the Annexin V/PI double stain assay. Data are presented as the mean + standard deviation (n=6 in each group). *P<0.05 vs. sham group; #P<0.05 vs. vehicle group. TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Neuroprotection of PD on I/R injury-induced mitochondrial dysfunction
In the present study, TEM was used to examine mitochondrial morphology. Cells in the sham group displayed normal mitochondria with intact membranes and cristae (Fig. 3). Mitochondria from the vehicle group appeared swollen and irregularly shaped, with disrupted and poorly defined cristae. These mitochondrial alterations following MCAO were partially prevented by PD treatment (Fig. 3). Mitochondrial membrane potential (∆Ψm) was also measured using JC-1, a potential-sensitive fluorescent dye that forms aggregates in normally polarized mitochondria and monomers in damaged and depolarized mitochondria. The percentage of neurons with low ∆Ψm was significantly increased in the vehicle group compared with the sham group (P<0.001; Fig. 4A). The PD group contained significantly fewer cells with low ∆Ψm than the vehicle group (P=0.005; Fig. 4A). Furthermore, intracellular ATP levels were significantly lower in the vehicle group compared with the sham group (P<0.001; Fig. 4B), indicating mitochondrial dysfunction. Following PD treatment, intracellular ATP levels were significantly increased compared with vehicle (P=0.014; Fig. 4B). These data indicate that PD attenuates MCAO-induced mitochondrial dysfunction.
Figure 3.

PD protects against middle cerebral artery occlusion induced morphological changes in mitochondria. Mitochondrial morphology was examined by transmission electron microscopy. Swollen mitochondria are indicated by arrows. PD; polydatin.
Figure 4.
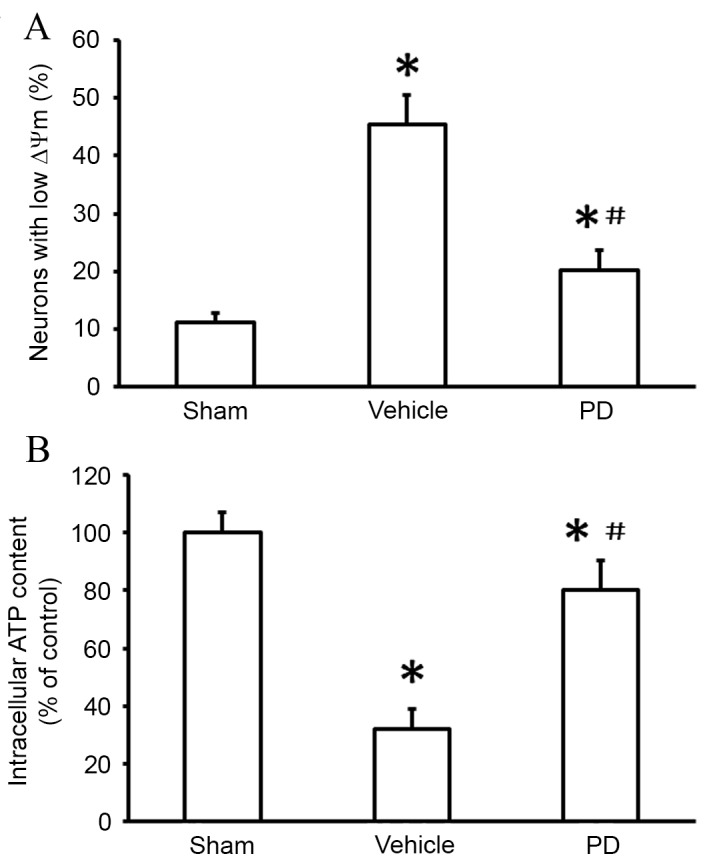
PD protects against mitochondrial dysfunction in isolated neurons following middle cerebral artery occlusion. (A) ∆Ψm was measured using the fluorescent probe JC-1 and analyzed by flow cytometry. (B) Neuronal ATP levels were determined by a luciferase-based assay. Data are presented as the mean + standard deviation (n=6 in each group). *P<0.05, vs. sham group; #P<0.05, vs. vehicle group. PD, polydatin.
PD prevents I/R injury-induced mitochondria-dependent apoptotic signaling
Mitochondrial regulation of apoptosis is partially mediated by the release of cytochrome c and apoptosis-inducing factors, which are regulated by the Bcl-2 family of proteins (9). In the present study, expression of the Bcl-2 family regulators, Bax and Bcl-2, and levels of cytochrome c release, were investigated. Compared with the sham group, vehicle treatment following MCAO resulted in significant upregulation of Bax protein expression levels (P<0.001; Fig. 5A and B), and significant downregulation of Bcl-2 protein expression levels (P<0.001; Fig. 5A and C). PD treatment resulted in significantly lower levels of Bax protein expression compared with vehicle (P=0.012; Fig. 5A and B) and significantly higher levels of Bcl-2 protein expression than vehicle (P=0.019; Fig. 5A and C).
Figure 5.

PD treatment reduces Bax upregulation, Bcl-2 downregulation and cytochrome c release in cortex following middle cerebral artery occlusion. (A) Bax and Bcl-2 protein expression levels in the cortex were detected by western blot. (B) Quantification of Bax protein expression levels relative to GAPDH. (C) Quantification of Bcl-2 protein expression levels relative to GAPDH. (D) Cytosolic cytochrome c levels were determined using a cytochrome c ELISA kit. Data are presented as the mean + standard deviation (n=6 in each group). *P<0.05 vs. sham group; #P<0.05 vs. vehicle group. PD, polydatin; Bax, Bcl-associated protein X apoptosis regulator; Bcl-2, B-cell lymphoma 2 apoptosis regulator.
Cytochrome c release activates downstream signaling cascades that trigger the intrinsic apoptotic pathway (9). In the present study, caspase-3 and caspase-9 activity were examined, as they are critical to the apoptosis cascade (18). MCAO increased activation of caspase-3 compared with sham (P<0.001; Fig. 6A); this upregulation was inhibited by treatment with PD (P=0.021 vs. vehicle; Fig. 6A). A significant increase in caspase-9 activity was observed following MCAO in the vehicle group compared with the sham group (P<0.001; Fig. 6B), which was abrogated by PD treatment (P=0.011 vs. vehicle; Fig. 6B).
Figure 6.
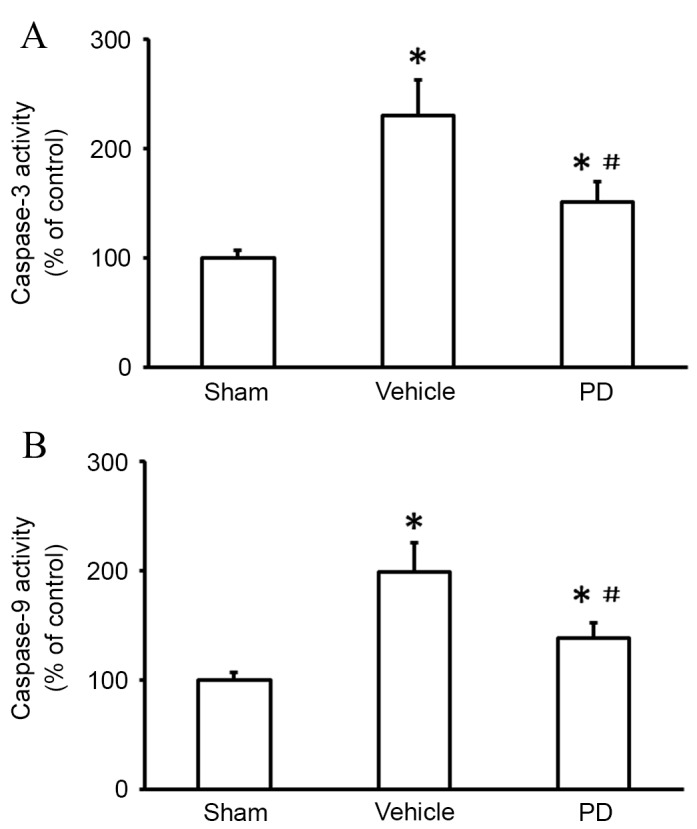
PD protects against caspase-3 and caspase-9 activation in the cortex following middle cerebral artery occlusion. (A) Caspase-3 and (B) caspase-9 activity were measured using fluorometric assay kits. Data are presented as the mean + standard deviation (n = 6 in each group). *P<0.05 vs. sham group; #P<0.05 vs. vehicle group. PD, polydatin.
Neuroprotection of PD on I/R injury-induced ROS production
To determine whether the neuroprotective effect of PD was associated with its antioxidant properties, ROS production was measured using a DCFH-DA assay 24 h after MCAO. ROS production was significantly higher in the vehicle group than the sham group (P<0.001, Fig. 7); this increase was significantly lowered by PD treatment compared with vehicle (P=0.008; Fig. 7).
Figure 7.
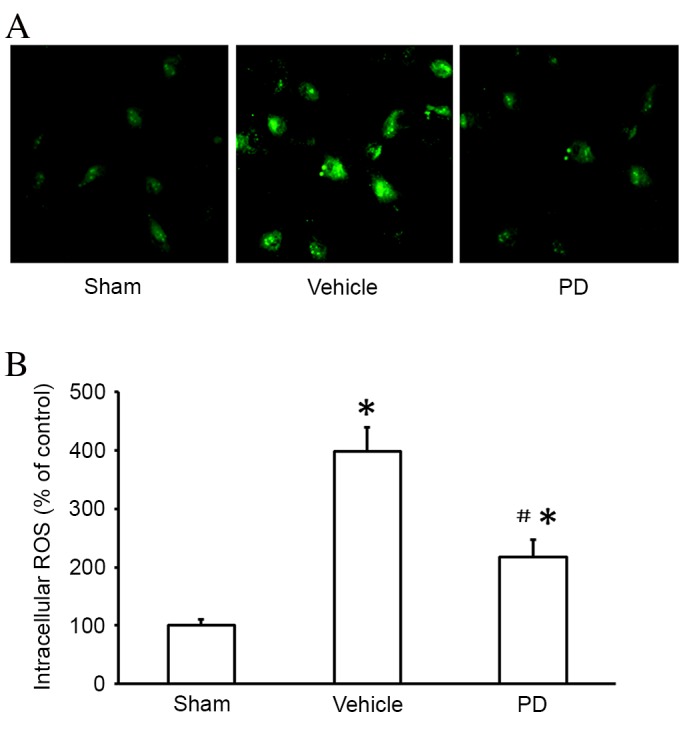
PD reduces mitochondrial ROS production in isolated neurons following middle cerebral artery occlusion. (A) Intracellular ROS were detected by DCFH-DA and observed by fluorescent microscopy. Original magnification, ×400. (B) Quantification of MitoSOX Red fluorescence by flow cytometry. Data are presented as the mean + SD (n=6 in each group). *P<0.05 vs. sham group; #P<0.05 vs. vehicle group. PD, polydatin; ROS, reactive oxygen species.
Discussion
Previous studies by this group have demonstrated the protective effects of PD on sepsis, burns and hemorrhagic shock (12,13,16). However, the effects of PD on the ischemic brain have, to the best of our knowledge, not been examined previously. In the current study, PD treatment was demonstrated to confer a neuroprotective effect on cortical neurons in a MCAO rat model. This finding highlighted modulation of mitochondria-dependent apoptosis as a mechanism by which PD reduces neuronal damage and oxidative stress following I/R injury.
Oxidative stress is a key factor in cerebral I/R damage, with ROS contributing to apoptotic cell death in ischemia via several pathways (19,20). Previous studies suggested that PD acts as a free radical scavenger (14,16). In the current study, increased ROS generation in the ischemic brain was demonstrated to be partially reversed by PD, which could be attributed to an antioxidant action. However, the molecular mechanism of PD against oxidants remains to be elucidated and should be investigated in future studies.
Mitochondria are essential to the production of cellular energy, the generation of ROS, a by-product of normal mitochondrial respiration is increased when the specific respiratory chain is impaired under stress conditions, and the regulation of apoptosis (14,15). Therefore, mitochondria are important to neuronal survival (18). Not only are mitochondria a source of ROS, they are also targets of oxidative stress (16,21). The mitochondrial transmembrane potential (∆Ψm), the difference in potential caused by the ion concentration gradient across the mitochondrial membrane, reflects mitochondrial function (17). A decline in ∆Ψm is correlated with the opening of the permeability transition pore, leading to the release of caspase-activating proteins from the mitochondria (21). Furthermore, ∆Ψm is reduced in response to the arrest of the activity of respiratory complexes I–V, and contributes to inhibition of ATP synthesis (15). Therefore, reduced ∆Ψm, mitochondrial swelling with poorly defined cristae, and reduced intracellular ATP content are all considered signs of mitochondrial injury (16). In the present study, cells in the vehicle group displayed a significant decrease in ∆Ψm and ATP levels compared with the sham-operated group, and the mitochondria appeared swollen and were irregularly shaped, with disrupted and poorly defined cristae. PD treatment prevented these morphological and functional changes, and resulted in ∆Ψm and ATP levels similar to the sham-operated group, suggesting that PD prevents mitochondrial dysfunction following I/R injury.
Mitochondrial damage and transmembrane potential depolarization lead to the release of cytochrome c (22). Once released to the cytosol, cytochrome c activates downstream caspases (9). Caspase-9 is an important initiator of the cytochrome c-dependent caspase cascade, while caspase-3 is involved in apoptotic signaling transduction (4). In the present study, PD treatment was demonstrated to have an effect on proteins involved in mitochondria-dependent apoptosis. I/R injury (caused by MCAO) was demonstrated to induce the release of cytochrome c from mitochondria, leading to activation of caspases 9 and 3. PD treatment inhibited these changes. These findings support the hypothesis that PD treatment prevents the release of apoptotic factors from mitochondria, following reducing the caspases activation, leading to reduced apoptosis following ischemia and reperfusion.
Mitochondria-regulated apoptosis is controlled by members of the Bcl-2 family of proteins (23). The Bcl family consists of both antiapoptotic (Bcl-2, Bcl-xL) and proapoptotic (BAK, BAX) factors (24). The antiapoptotic members of this family prevent apoptosis by sequestering proforms of death-driving cysteine proteases or by preventing the release of mitochondrial apoptogenic factors. Conversely, the proapoptotic members trigger the release of mitochondrial apoptogenic factors into the cytoplasm through the mitochondrial permeability transition pore, thereby leading to caspases activation (12,13). The present study demonstrated that Bcl-2 expression was significantly increased following PD treatment and MCAO, while Bax expression was decreased in the PD group compared with the vehicle group following MCAO. These results indicate that the anti-apoptotic effects of PD treatment may be mediated by changes in Bcl-2 and Bax expression.
In conclusion, the present study has produced evidence of significant neuroprotection in a MCAO rat model following PD treatment. A potential mechanism of this action of PD is through amelioration of oxidative stress and mitochondria-dependent apoptosis. The underlying molecular mechanisms require further clarification, more specifically investigation of the effect of PD on brain injury and its possible clinical application.
Acknowledgements
This work was supported by the Medical Innovation Fund of Fujian Province, China (grant no. 2015-CX-22), the Finance Department of Health Special Fund of Fujian Province, China (grant no. BPB-LXZ2014), the National Natural Science Foundation of China (grant no. 81500066) and the Natural Science Foundation of Fujian Province (grant no. 2016J01451).
Glossary
Abbreviations
- Bcl-2
B-cell lymphoma 2 apoptosis regulator
- Bax
Bcl-2 associated protein X apoptosis regulator
- I/R
ischemia/reperfusion
- MCAO
middle cerebral artery occlusion
- NF
neurofibromatosis
- PD
polydatin
- ROS
reactive oxygen species
- Smac
second mitochondrial derived activator of caspases
- TEM
transmission electron microscopy
- TTC
2,3,5-triphenyltetrazolium chloride
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
References
- 1.Cai Q, Chen Z, Song P, Wu L, Wang L, Deng G, Liu B, Chen Q. Co-transplantation of hippocampal neural stem cells and astrocytes and microvascular endothelial cells improve the memory in ischemic stroke rat. Int J Clin Exp Med. 2015;8:13109–13117. [PMC free article] [PubMed] [Google Scholar]
- 2.Lin R, Lin Y, Tao J, Chen B, Yu K, Chen J, Li X, Chen LD. Electroacupuncture ameliorates learning and memory in rats with cerebral ischemia-reperfusion injury by inhibiting oxidative stress and promoting p-CREB expression in the hippocampus. Mol Med Rep. 2015;12:6807–6814. doi: 10.3892/mmr.2015.4321. [DOI] [PubMed] [Google Scholar]
- 3.Ljubisavljevic MR, Javid A, Oommen J, Parekh K, Nagelkerke N, Shehab S, Adrian TE. The Effects of different repetitive transcranial magnetic stimulation (rTMS) protocols on cortical gene expression in a rat model of cerebral ischemic-reperfusion injury. PLoS One. 2015;10:e139892. doi: 10.1371/journal.pone.0139892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang W, Chen X, Pan J, Ge H, Yin K, Wu Z, Li X, Sha D, Xu Y. Malibatol A protects against brain injury through reversing mitochondrial dysfunction in experimental stroke. Neurochem Int. 2015;80:33–40. doi: 10.1016/j.neuint.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Palencia G, Medrano JÁ, Ortiz-Plata A, Farfán DJ, Sotelo J, Sánchez A, Trejo-Solís C. Anti-apoptotic, anti-oxidant and anti-inflammatory effects of thalidomide on cerebral ischemia/reperfusion injury in rats. J Neurol Sci. 2015;351:78–87. doi: 10.1016/j.jns.2015.02.043. [DOI] [PubMed] [Google Scholar]
- 6.Fang L, Gao H, Zhang W, Zhang W, Wang Y. Resveratrol alleviates nerve injury after cerebral ischemia and reperfusion in mice by inhibiting inflammation and apoptosis. Int J Clin Exp Med. 2015;8:3219–3226. [PMC free article] [PubMed] [Google Scholar]
- 7.Tao T, Liu Y, Zhang J, Xu Y, Li W, Zhao M. Therapeutic hypercapnia improves functional recovery and attenuates injury via antiapoptotic mechanisms in a rat focal cerebral ischemia/reperfusion model. Brain Res. 2013;1533:52–62. doi: 10.1016/j.brainres.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 8.Lu YP, Liu SY, Sun H, Wu XM, Li JJ, Zhu L. Neuroprotective effect of astaxanthin on H(2)O(2)-induced neurotoxicity in vitro and on focal cerebral ischemia in vivo. Brain Res. 2010;1360:40–48. doi: 10.1016/j.brainres.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 9.Li G, Li T, Li Y, Cai S, Zhang Z, Zeng Z, Wang X, Gao Y, Li Y, Chen Z. Ulinastatin inhibits oxidant-induced endothelial hyperpermeability and apoptotic signaling. Int J Clin Exp Pathol. 2014;7:7342–7350. [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J, Bai J, Jiang G, Li X, Wang J, Wu D, Owusu L, Zhang E, Li W. Anti-tumor effect of Pinus massoniana Bark Proanthocyanidins on ovarian cancer through induction of cell apoptosis and inhibition of cell migration. PLoS One. 2015;10:e142157. doi: 10.1371/journal.pone.0142157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li T, Yuan G, Zhang L, Ye L, Li S, Fan Y, Sun J. ApoG2 inhibits the antiapoptotic protein, Mcl1 and induces mitochondria-dependent apoptosis in human colorectal cancer cells. Mol Med Rep. 2015;12:6976–6984. doi: 10.3892/mmr.2015.4299. [DOI] [PubMed] [Google Scholar]
- 12.Li T, Liu Y, Li G, Wang X, Zeng Z, Cai S, Li F, Chen Z. Polydatin attenuates ipopolysaccharide-induced acute lung injury in rats. Int J Clin Exp Pathol. 2014;7:8401–8410. [PMC free article] [PubMed] [Google Scholar]
- 13.Li T, Cai S, Zeng Z, Zhang J, Gao Y, Wang X, Chen Z. Protective effect of polydatin against burn-induced lung injury in rats. Respir Care. 2014;59:1412–1421. doi: 10.4187/respcare.02831. [DOI] [PubMed] [Google Scholar]
- 14.Jiang X, Liu W, Deng J, Lan L, Xue X, Zhang C, Cai G, Luo X, Liu J. Polydatin protects cardiac function against burn injury by inhibiting sarcoplasmic reticulum Ca2+ leak by reducing oxidative modification of ryanodine receptors. Free Radic Biol Med. 2013;60:292–299. doi: 10.1016/j.freeradbiomed.2013.02.030. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Song R, Bian HN, Brunk UT, Zhao M, Zhao KS. Polydatin, a natural polyphenol, protects arterial smooth muscle cells against mitochondrial dysfunction and lysosomal destabilization following hemorrhagic shock. Am J Physiol Regul Integr Comp Physiol. 2012;302:R805–R814. doi: 10.1152/ajpregu.00350.2011. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Song R, Chen Y, Zhao M, Zhao KS. Polydatin-a new mitochondria protector for acute severe hemorrhagic shock treatment. Expert Opin Investig Drugs. 2013;22:169–179. doi: 10.1517/13543784.2013.748033. [DOI] [PubMed] [Google Scholar]
- 17.Zeng Z, Chen Z, Xu S, Song R, Yang H, Zhao KS. Polydatin alleviates small intestine injury during hemorrhagic shock as a SIRT1 activator. Oxid Med Cell Longev. 2015;2015:965961. doi: 10.1155/2015/965961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng Z, Chen Z, Li T, Zhang J, Gao Y, Xu S, Cai S, Zhao KS. Polydatin: A new therapeutic agent against multiorgan dysfunction. J Surg Res. 2015;198:192–199. doi: 10.1016/j.jss.2015.05.041. [DOI] [PubMed] [Google Scholar]
- 19.Sun J, Li YZ, Ding YH, Wang J, Geng J, Yang H, Ren J, Tang JY, Gao J. Neuroprotective effects of gallic acid against hypoxia/reoxygenation-induced mitochondrial dysfunctions in vitro and cerebral ischemia/reperfusion injury in vivo. Brain Res. 2014;1589:126–139. doi: 10.1016/j.brainres.2014.09.039. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Yu W, Li XT, Qi SH, Li B. The effects of propofol on mitochondrial dysfunction following focal cerebral ischemia-reperfusion in rats. Neuropharmacology. 2014;77:358–368. doi: 10.1016/j.neuropharm.2013.08.029. [DOI] [PubMed] [Google Scholar]
- 21.Srivastava SK, Bhardwaj A, Arora S, Tyagi N, Singh S, Andrews J, McClellan S, Wang B, Singh AP. MicroRNA-345 induces apoptosis in pancreatic cancer cells through potentiation of caspase-dependent and -independent pathways. Br J Cancer. 2015;113:660–668. doi: 10.1038/bjc.2015.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Todd K, Ghiso J, Rostagno A. Oxidative stress and mitochondria-mediated cell death mechanisms triggered by the familial Danish dementia ADan amyloid. Neurobiol Dis. 2015;85:130–143. doi: 10.1016/j.nbd.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao ZH, Zheng QY, Li GQ, Hu XB, Feng SL, Xu GL, Zhang KQ. STAT1-mediated down-regulation of Bcl-2 expression is involved in IFN-γ/TNF-α-induced apoptosis in NIT-1 cells. PLoS One. 2015;10:e120921. doi: 10.1371/journal.pone.0120921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kvansakul M, Hinds MG. The Bcl-2 family: Structures, interactions and targets for drug discovery. Apoptosis. 2015;20:136–150. doi: 10.1007/s10495-014-1051-7. [DOI] [PubMed] [Google Scholar]