

Abstract
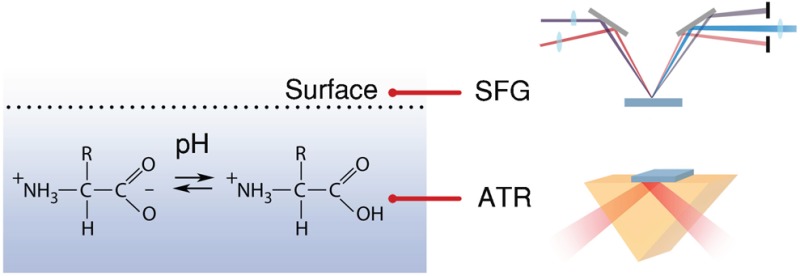
We use surface-specific intensity vibrational sum-frequency generation and attenuated total reflection spectroscopy to probe the ionization state of the amino-acids l-alanine and l-proline at the air/water surface and in the bulk. The ionization state is determined by probing the vibrational signatures of the carboxylic acid group, representing the nondissociated acid form, and the carboxylate anion group, representing the dissociated form, over a wide range of pH values. We find that the carboxylic acid group deprotonates at a significantly higher pH at the surface than in the bulk.
Introduction
The air/water interface is characterized by a discontinuity and asymmetry in intermolecular interactions, which results in molecular properties that differ from the bulk. For instance, it has been shown that molecules residing at the surface can show a different molecular conformation.1 The degree of acid dissociation and thus the ionization state of molecules can also be different at the interface compared to the bulk. The degree of acid dissociation of molecules at the air/water interface constitutes an important parameter for the structural properties of molecules near the surface and for the understanding of the reactivity and potential catalytic role of the interface.2,3
The acid dissociation constants of molecules in aqueous solution are routinely determined using techniques like potentiometric titration, voltammetry, and electrophoresis.4 The dissociation constants determined by these techniques represent bulk values, as the signal originating from bulk molecules typically overwhelms the signal arising from those at the surface. Hence, the degree of acid dissociation at the water surface needs to be probed by highly surface-specific techniques.5−13
In previous spectroscopic studies of acid/base pairs it has been found that the surface favors the neutral form of the acid/base pair in comparison to the bulk.6 For instance, using highly surface-specific second harmonic generation (SHG) and VSFG, it has been shown for phenol and carboxylic acids that the neutral acid species is favored over the anionic conjugate base.7,12,13 Similarly, it was found for molecules containing an acid ammonium group that the neutral amine base species was favored over the cationic quaternary ammonium acid.5,9
In a recent infrared reflection–absorption spectroscopy (IRRAS) study of the amino-acid l-phenylalanine that contains both a carboxylic acid group and an amine group, an increase of the acid dissociation of both the carboxylic acid and the ammonium group compared to the bulk was reported.14 Clearly, this behavior cannot be explained from the concept that the surface would favor the neutral species, as for l-phenylalanine deprotonation of the ammonium group implies that the amino-acid acquires an overall negative charge. Hence, the ammonium group is expected to be less deprotonated at the surface than in the bulk. The observation that both functional groups deprotonate more easily near the surface than in the bulk was explained from the presence of an enhanced concentration of OH– in the probed surface region,14 which for the technique of IRRAS has a depth of ∼1 μm. This probing depth constitutes thousands of molecular layers, thus making it possible that the amount of deprotonation in the top molecular layers differs from the average deprotonation observed in the probed surface region with a depth of ∼1 μm.
Here, we report on a VSFG study of the surface concentrations of the acid and neutral forms of the amino-acids l-alanine and l-proline as a function of the bulk pH. The signal measured with VSFG spectroscopy depends on the asymmetry in the orientation of the probed molecular species. In this experiment, we probe the vibrations of the acid and the base form of the carboxylic acid group and the probing depth is determined by the depth over which the orientation of these species will be asymmetric. For the probed amino acids there will be an asymmetry in the top molecular layers of the water solvent due to the preferred orientation of the hydrophilic and the hydrophobic groups of the amino acid in the top molecular layers of the water solvent, in particular the hydrophobic groups will preferably stick out of the surface. Somewhat deeper in the solvent there is no preferred orientation anymore, as all groups of the amino-acid are interacting with water. Hence, the VSFG signal is only due to amino acids and their conjugate bases located in the top molecular solvent layers, which implies a probing depth <1 nm. We compare these surface concentrations with the corresponding bulk concentrations that we measure with attenuated total reflection (ATR) infrared spectroscopy.
Experimental Section
The used VSFG setup has been described in detail in previous work.15 In short, it consists of a regenerative Ti:Saphire amplifier, which produces 35 fs pulses at 1 kHz repetition rate with a 3.5 mJ pulse energy. One third of its output is used to produce tunable mid-IR pulses by pumping a home-built optical parametric amplifier and a difference-frequency generation stage. The IR pulses have a bandwidth of ∼200 cm–1 (full-width-at-half-maximum) and are centered at ∼1620 cm–1 to probe the antisymmetric stretch vibration of the carboxylate groups (νAS,COO–), and at ∼1750 cm–1 to probe the carbonyl stretch vibration of carboxylic acid groups (νCOOD). The remaining part of the Ti:Saphire amplifier output is spectrally narrowed with an etalon to ∼15 cm–1 and used together with the broadband IR pulse to produce SFG light from the sample. The incident angles of IR and VIS are 40° and 36°, respectively. All spectra were recorded under SSP (SFG, VIS, IR) polarization combination. The generated SFG light is spectrally dispersed using a monochromator and recorded with an Electron-Multiplied Charge Coupled Device (EMCCD, Andor Technologies).
ATR spectroscopy is a widely used linear infrared spectroscopic technique that is suited for measuring vibrational spectra of samples that absorb too strong for transmission measurements. The layer thickness that is probed in the ATR geometry is determined by the decay length of the evanescent field, which is a function of wavelength, incident angle and the refractive indexes of the ATR crystal and the sample. At ∼1600 cm–1 the probed depth is on the order of ∼1 μm.16 We recorded ATR spectra with 0.4 cm–1 resolution using a Bruker Vertex80v equipped with an ATR module (Platinum ATR Diamond).
The VSFG and the ATR measurements were performed in solutions of D2O instead of H2O to avoid overlap of the νAS,COO– (∼1602 cm–1) and νCOOD (∼1720 cm–1) bands with the water bending mode located at ∼1670 cm–1. We use a pH meter (Mettler Toledo FE20/EL20) that is calibrated for measuring the pH in H2O solutions instead of D2O solutions. The measured pH* of a D2O solution is transformed to the pD value using the following equation:17
| 1 |
The pD value can be transferred to the pH value of a solution in H2O of equal acidity using the following:17
| 2 |
The studied samples are 1.8 M solutions of l-Alanine (≥98%, Sigma-Aldrich) and 1.4 M solutions of l-proline (≥99%, Sigma-Aldrich) in D2O. D2O (≥99.96%) was purchased from Cambridge Isotope Laboratories. We adjusted the pD of the samples by adding NaOD or DCl (Sigma-Aldrich).
Results and Discussion
In Figure 1 we present VSFG (a) and ATR (b) spectra of l-proline at different pH values. We assign the peaks at ∼1400, ∼1602, and ∼1720 cm–1 to the symmetric and antisymmetric stretch vibrations of the carboxylate anion (νSS,COO– and νAS,COO–) and the carbonyl stretch vibration of the carboxylic acid group (νCOOD), respectively. All experiments were performed in deuterated water to avoid spectral overlap with the absorption of the water bending mode. The pH values were determined from the measured pD values of the D2O solutions as desribed in the Experimental Section. pH = 7 and pD = 7.5 both refer to neutral solutions at 293 K ([H3O+] = [OH–] and [D3O+] = [OD–]). It is clearly seen in Figure 1 that the intensity of the νAS,COO– vibrational band increases with increasing pH values, while the intensity of the νCOOD vibrational band decreases, reflecting the shift in the acid–base equilibrium.
Figure 1.
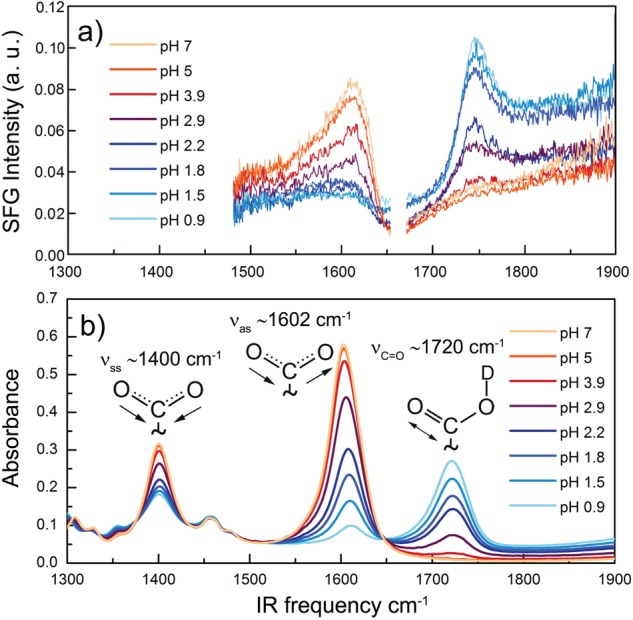
Spectra of l-proline in the frequency region of the carbonyl and carboxylate anion vibrations a) VSFG and b) ATR spectra at different pH values. The white area in the VSFG spectra represents a region where no VSFG response was measured.
To quantify the observed changes of the intensities of νAS,COO– and νCOOD vibrational bands, we fit the VSFG spectra at each measured pH value. The VSFG spectra contain of resonant and nonresonant contributions. To extract the amplitudes of the resonant contributions of the νAS,COO– and νCOOD bands at different pH values, we fit the VSFG spectra with a nonresonant background and Lorentzian line shapes for the resonances:
| 3 |
where χNR(2) and χR are the second-order nonresonant and resonant nonlinear susceptibility elements, ANR and φNR are the nonresonant amplitude and phase. Ai, ωi, and Γi are the amplitude, center frequency, and width of the i-th resonance, respectively.
In fitting the VSFG data, the center frequencies and widths were fixed, and the amplitudes of the resonant and the nonresonant contributions were left free. The measured VSFG spectrum at frequencies >1800 cm–1 at pH 7 was subtracted from all spectra before fitting, to remove the weak signal of the low-frequency wing of the broad resonant contribution originating from the vibrational band of the OD stretch vibrations of D2O centered at ∼2400 cm–1. In Figure 2 we show the fitted VSFG spectra of l-proline (red lines) and in Table 1 we present the parameters obtained from the fit.
Figure 2.

VSFG spectra of l-proline in the frequency regions of the νAS,COO– (a) and the νCOOD (b) vibrations. The red lines represent the results of the fit described in the text.
Table 1. Fitting Parameters for the VSFG Spectra of l-Proline in νCOOD and νAS,COO– Vibrational Regions.
| νCOOD ω0 = 1746 cm–1 Γ = 20 cm–1 | νAS,COO– ω0 = 1628 cm–1 Γ = 22 cm–1 | ||||||
|---|---|---|---|---|---|---|---|
| pH | ANR | φNR | AR | ANR | φNR | AR | |
| 0.9 | 0.12 ± 0.01 | –2 ± 1 | 3.99 ± 0.06 | 0.16 ± 0.01 | 179 ± 12 | 0.13 ± 0.03 | |
| 1.5 | 0.12 ± 0.01 | –4 ± 1 | 3.73 ± 0.04 | 0.16 ± 0.01 | 174 ± 4 | 0.44 ± 0.03 | |
| 1.8 | 0.10 ± 0.03 | –8 ± 1 | 3.43 ± 0.06 | 0.16 ± 0.01 | 175 ± 2 | 0.74 ± 0.02 | |
| 2.2 | 0.08 ± 0.01 | –21 ± 2 | 2.76 ± 0.06 | 0.16 ± 0.01 | 175 ± 2 | 1.1 ± 0.03 | |
| 2.9 | 0.08 ± 0.01 | –24 ± 2 | 1.87 ± 0.05 | 0.17 ± 0.01 | 177 ± 2 | 1.9 ± 0.03 | |
| 3.9 | 0 | 0 | 0.4 ± 0.1 | 0.16 ± 0.01 | 183 ± 1 | 2.73 ± 0.03 | |
| 5 | 0.16 ± 0.01 | 180 ± 1 | 3.93 ± 0.03 | ||||
| 7 | 0.16 ± 0.01 | 179 ± 1 | 4.27 |
The ATR spectra were fitted with 3 Lorentzian functions in the frequency region from 1500 to 1800 cm–1. Central frequencies and widths of each Lorentzian peak were kept constant throughout the fits of all spectra. One of the peaks (centered at 1400 cm–1) lays outside the fitted region, but was needed to achieve an accurate fit of low-frequency region of the ATR spectra. Figure 3 shows fitting results of ATR spectra of l-proline and Table 2 presents the obtained parameters of each Lorentzian band.
Figure 3.
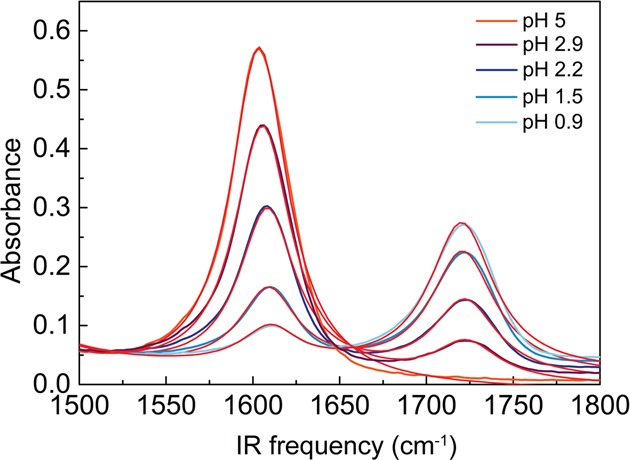
Experimentally measured ATR spectra of l-proline and Lorentzian model fits.
Table 2. Fitting Parameters for the l-Proline ATR Spectra.
| ω0 = 1400 cm–1 Γ = 140 cm–1 | ω0 = 1608 cm–1 Γ = 40 cm–1 | ω0 = 1720 cm–1 Γ = 45 cm–1 | |
|---|---|---|---|
| pH | peak area | peak area | peak area |
| 7 | 8.3 ± 0.7 | 36.8 ± 0.2 | |
| 5 | 8.1 ± 0.6 | 36 ± 0.2 | |
| 3.9 | 8.8 ± 0.7 | 33.8 ± 0.2 | 1.5 ± 0.3 |
| 2.9 | 8.9 ± 0.5 | 27 ± 0.2 | 4.6 ± 0.2 |
| 2.2 | 8.7 ± 0.4 | 17.6 ± 0.1 | 9.1 ± 0.2 |
| 1.8 | 7.9 ± 0.4 | 13 ± 0.1 | 11.4 ± 0.1 |
| 1.5 | 7.4 ± 0.3 | 8.4 ± 0.1 | 14.5 ± 0.1 |
| 0.9 | 7.0 ± 0.4 | 0.06 ± 0.08 | 17.7 ± 0.3 |
In Figure 4 we plot the pH dependence of the normalized areas (normalized to the maximum area) of the νAS,COO– and νCOOD vibrations obtained from fitting the VSFG (a) and ATR (b) spectra. From the curves shown in Figure 4 we find that for l-proline [COOD] is equal to [COO–] at a pH value of ∼2.2 ± 0.2 in the bulk and at a pH value of ∼3 ± 0.2 at the surface.
Figure 4.
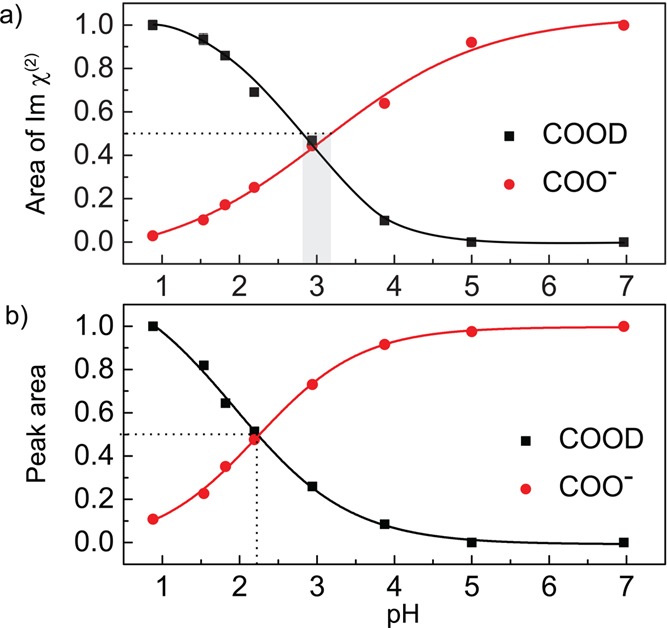
Areas (normalized) of the bands associated with the νAS,COO– and νCOOD vibrations obtained from fitting a) VSFG and b) ATR spectra.
It should be noted that the VSFG intensity depends not only on the number density of the molecules at the interface but also on the molecular orientation. A change in the VSFG intensities of the COO– and COOD vibrations with a variation of the pH may thus also result from a change in the molecular orientation. We checked the potential occurrence of this effect by probing the VSFG spectrum of the CH vibrational bands of l-proline (Figure 5). The intensity of the CH vibrational bands of l-proline do not show any prominent changes when varying the pH. This observation indicates that l-proline does not change its orientation at the water surface when the pH is varied.
Figure 5.
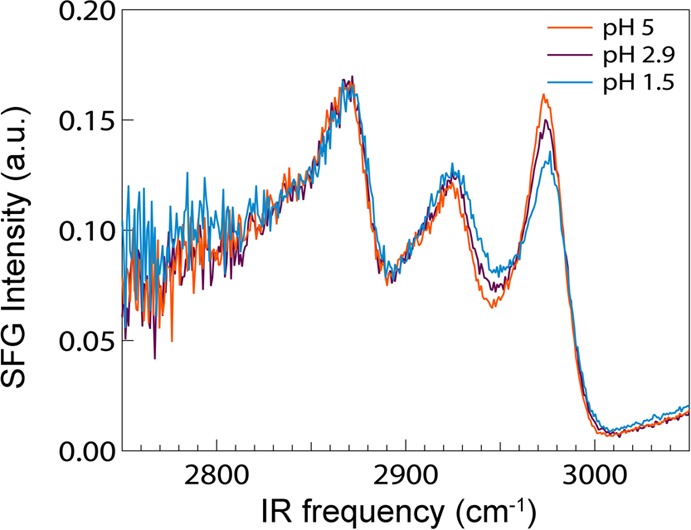
VSFG spectra of l-proline (a) in the frequency region of the C—H stretch vibrations at different pH values.
We also studied the pH dependence of the VSFG intensities of the COO– and COOD bands of l-alanine. As for proline, we find the pH at which the number densities of the COO– and COOD groups are the same to be higher at the surface than in the bulk (∼2.8 ± 0.1 vs 2.5 ± 0.1). The data and fitting results for l-alanine can be found in the SI.
We thus find clear evidence that the carboxylic acid groups of amino-acids require a higher pH to deprotonate at the surface than in the bulk. This result does not agree with the earlier reported results,7,12,13 in which it was found that at the water surface the neutral form of an acid/base pair is favored over the charged form. When the carboxylic acid group is deprotonated, the amino acid acquires an overall neutral charge, as it contains both an anionic carboxylate group and a cationic quaternary ammonium group. We observe that for a given pH, the overall neutral species has a lower concentration at the surface than in the bulk, which implies that in the top molecular layers the cationic (acidic) species of l-proline and l-alanine is favored over the neutral (zwitterionic) species.
The enhanced surface density of the carboxylic acid group over the carboxylate anion group can be explained as follows. The deprotonation of the carboxylic acid group yields a negatively charged carboxylate group. This process is highly favored by the formation of strong hydrogen bonds between the negatively charged carboxylate group and the surrounding water molecules. As a result, amino-acids occur as zwitterions in solution whereas in the gas phase nonzwitterionic structures are present. This difference is rationalized by the fact that water strongly stabilizes the zwitterionic form of an amino-acid by forming strong hydrogen bonds.18 For example, the zwitterion of l-glycine in the gas phase was calculated to be unstable, whereas it is more stable than neutral glycine when solvated by water.19 It has been found that five water molecules are needed to stabilize the zwitterionic form of l-proline.20 At the water surface the solvation interactions cannot extend as far as in the bulk and thus the carboxylate group will be less stabilized, which results in a less favorable solvation of the negative charge of the carboxylate anion. As a consequence, higher bulk pH values are required to induce the dissociation of carboxylic acid groups of molecules at the surface. This explanation also implies that the deprotonation of the carboxylic acid group is not determined by the overall charge of the molecule, but rather by the energetics of the local solvation interactions of the acid/base groups with the surrounding water molecules.
It should be noted that the difference in acid and base concentrations between the surface and the bulk does not imply that the equilibrium constant for acid dissociation would be different at the surface. The equilibrium constant is related to the activities of the acid, the base, and the proton, and the activities of all species are the same for the surface and the bulk, as the surface and the bulk are in thermodynamic equilibrium. The low surface concentration of the base thus implies that the activity coefficient of the base is significantly higher at the surface than in the bulk, which we explain from the relatively poor hydration of the carboxylate anion at the surface compared to the bulk. The fact that the activities of the acid, base, and proton are the same at the surface as in the bulk also implies that the higher carboxylic acid concentration (and lower carboxylate base concentration) at the surface cannot be caused by a potentially higher surface concentration of protons. Even if the water surface would contain a higher proton concentration than the bulk, which has been found in MD simulations,21−23 and which finds support in VSFG and SHG experiments of the water/air interface,24−26 the activity of the proton is not different from the bulk, meaning that there is no excess proton-induced shift of the acid–base equilibrium at the surface toward the acid species.
Conclusions
We study the surface concentrations of the acid and neutral (zwitterionic) forms of the amino-acids l-alanine and l-proline as a function of pH with surface vibrational sum-frequency generation spectroscopy (VSFG). To this purpose we measure VSFG spectra of specific vibrations of the carboxylic acid group and the carboxylate anion base. The surface acid and base concentrations are compared with the corresponding bulk concentrations that are determined using attenuated total reflection (ATR) infrared spectroscopy. We observe that the concentration of the acid is significantly enhanced over the base at the surface in comparison to the bulk. We find that the number densities of the acid and the neutral form of l-proline are the same at the surface for a pH value of 3 ± 0.2, whereas these number densities are the same in the bulk at a pH of 2.2 ± 0.2. We thus observe that at a given pH the concentration of the acid is significantly enhanced over the base at the surface in comparison to the bulk. For l-alanine these values are 2.8 ± 0.1 for the surface and 2.5 ± 0.1 for the bulk. The decreased concentration of carboxylate anions at the water surface compared to the bulk can be explained from a difference in solvation. In the bulk the carboxylate ion is better solvated than at the surface, thus favoring the deprotonated species over the carboxylic acid form. The enhanced concentration of the carboxylic acid over the carboxylate anion at the water surface may have consequences for the conformation and the functioning of biomolecules at aqueous surfaces.
Acknowledgments
This work is part of the research program of the “Stichting voor Fundamenteel Onderzoek der Materie (FOM)”, which is financially supported by the “Nederlandse organisatie voor Wetenschappelijk Onderzoek (NWO)”. K.M. gratefully acknowledges the European Commission for funding through a Marie Curie fellowship. We thank Dr. Niklas Ottosson for fruitful discussions on the thermodynamic aspects of the experiments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b12079.
Figure S1: VSFG and ATR spectra of l-alanine. Table 1: Fitting parameters of VSFG spectra of l-alanine. Figure S2: VSFG spectra of l-alanine with fits. Table 2: Fitting parameters of ATR spectra of l-alanine. Figure S3: ATR spectra of l-alanine with fits. Figure S4: Dissociation curves obtained from fitting results of VSFG and ATR spectra of l-alanine (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Yano Y. F. J. Phys.: Condens. Matter 2012, 24, 503101. 10.1088/0953-8984/24/50/503101. [DOI] [PubMed] [Google Scholar]
- Hub J. S.; Wolf M. G.; Caleman C.; van Maaren P. J.; Groenhof G.; van der Spoel D. Chem. Sci. 2014, 5, 1745–1749. 10.1039/c3sc52862f. [DOI] [Google Scholar]
- Jubb A. M.; Allen H. C. J. Phys. Chem. C 2012, 116, 13161–13168. 10.1021/jp302585h. [DOI] [Google Scholar]
- Reijenga J.; van Hoof A.; van Loon A. Anal. Chem. Insights 2013, 8, 53–71. 10.4137/ACI.S12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Ong S.; Wang H.; Eisenthal K. B. Chem. Phys. Lett. 1993, 214, 203–207. 10.1016/0009-2614(93)90082-C. [DOI] [Google Scholar]
- Eisenthal K. B. Chem. Rev. 1996, 96, 1343–1360. 10.1021/cr9502211. [DOI] [PubMed] [Google Scholar]
- Miranda P.; Du Q.; Shen Y. Chem. Phys. Lett. 1998, 286, 1–8. 10.1016/S0009-2614(97)01476-0. [DOI] [Google Scholar]
- Dynarowicz-Latka P.; Cavalli A. Jr; Oliveira O. N. O. N. O. Thin Solid Films 2000, 360, 261–267. 10.1016/S0040-6090(99)00889-5. [DOI] [Google Scholar]
- Wang H.; Zhao X.; Eisenthal K. B. J. Phys. Chem. B 2000, 104, 8855–8861. 10.1021/jp0018091. [DOI] [Google Scholar]
- Ariga K.; Nakanishi T.; Hill J. P.; Shirai M.; Okuno M.; Abe T.; Kikuchi J.-i. J. Am. Chem. Soc. 2005, 127, 12074–12080. 10.1021/ja053226g. [DOI] [PubMed] [Google Scholar]
- Fears K. P.; Creager S. E.; Latour R. A. Langmuir 2008, 24, 837–843. 10.1021/la701760s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Y.; Subir M.; McArthur E. A.; Turro N. J.; Eisenthal K. B. Chem. Phys. Lett. 2009, 477, 241–244. 10.1016/j.cplett.2009.07.011. [DOI] [Google Scholar]
- Tang C. Y.; Huang Z.; Allen H. C. J. Phys. Chem. B 2010, 114, 17068–17076. 10.1021/jp105472e. [DOI] [PubMed] [Google Scholar]
- Griffith E. C.; Vaida V. J. Am. Chem. Soc. 2013, 135, 710–716. 10.1021/ja308089n. [DOI] [PubMed] [Google Scholar]
- Strazdaite S.; Versluis J.; Backus E. H.; Bakker H. J. J. Chem. Phys. 2014, 140, 054711. 10.1063/1.4863558. [DOI] [PubMed] [Google Scholar]
- Kiefer J.; Frank K.; Schuchmann K. F. Appl. Spectrosc. 2011, 65, 1024–1028. 10.1366/11-06323. [DOI] [PubMed] [Google Scholar]
- Krȩżel A.; Bal W. J. Inorg. Biochem. 2004, 98, 161–166. 10.1016/j.jinorgbio.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Price W. D.; Jockusch R. A.; Williams E. R. J. Am. Chem. Soc. 1997, 119, 11988–11989. 10.1021/ja9711627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen J. H.; Gordon M. S. J. Am. Chem. Soc. 1995, 117, 8159–8170. 10.1021/ja00136a013. [DOI] [Google Scholar]
- Yang G.; Zhou L.; Chen Y. SpringerPlus 2016, 5, 19. 10.1186/s40064-015-1661-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse Y.-L. S.; Chen C.; Lindberg G. E.; Kumar R.; Voth G. A. J. Am. Chem. Soc. 2015, 137, 12610–12616. 10.1021/jacs.5b07232. [DOI] [PubMed] [Google Scholar]
- Jagoda-Cwiklik B.; Cwiklik L.; Jungwirth P. J. Phys. Chem. A 2011, 115, 5881–5886. 10.1021/jp110078s. [DOI] [PubMed] [Google Scholar]
- Berkelbach T. C.; Lee H.-S.; Tuckerman M. E. Phys. Rev. Lett. 2009, 103, 238302. 10.1103/PhysRevLett.103.238302. [DOI] [PubMed] [Google Scholar]
- Levering L. M.; Sierra-Hernández M. R.; Allen H. C. J. Phys. Chem. C 2007, 111, 8814–8826. 10.1021/jp065694y. [DOI] [Google Scholar]
- Tian C.; Ji N.; Waychunas G. A.; Shen Y. R. J. Am. Chem. Soc. 2008, 130, 13033–13039. 10.1021/ja8021297. [DOI] [PubMed] [Google Scholar]
- Petersen P. B.; Saykally R. J. J. Phys. Chem. B 2005, 109, 7976–7980. 10.1021/jp044479j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.