

INTRODUCTION
Genomic and other molecular factors frequently impact the efficacy, safety, and pharmacokinetic (PK) profiles of therapeutic products. Recent advances in our understanding of how these factors contribute to interindividual variability in drug response has led to the use of genomic strategies to improve clinical trial design in the drug development setting, and also to guide patient care in the clinical setting.1, 2
BACKGROUND
A biomarker is a defined characteristic that is measured as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions.3 The United States Food and Drug Administration (FDA) recognizes the potential of biomarker‐based strategies to optimize patient selection and dosing through drug labeling, application of genomic and other biomarkers in clinical trial design, and other pre‐ and post‐market maneuvers.4, 5 One key area of the drug approval process where genomic biomarkers can be particularly important is benefit–risk assessment, a process the FDA employs in regulatory decision‐making to ensure the safety, efficacy, and quality of human drugs.
To legally market a prescription drug or biological product (referred to collectively as “drug” herein) in the United States, marketing authorization must be granted from the FDA. Under the Kefauver‐Harris Amendments to the Food, Drug, and Cosmetic Act (FD&C Act), the FDA cannot approve an application to market a drug unless there is substantial evidence that the drug will have the effect it purports to have and there are adequate tests demonstrating the proposed drug is safe under the conditions prescribed, recommended, or suggested in its proposed labeling.6 Evidence of efficacy is primarily derived from adequate and well‐controlled clinical trials that demonstrate improvement based on a clinical end point that directly measures how a person feels, functions, or survives (exceptions may be made for drugs in the FDA's Accelerated Approval Program and well‐established surrogate end points).7 In addition to demonstrating substantial evidence of efficacy, trials must also demonstrate that the drug can be safely used in the population for which it is indicated. Some drug‐related adverse events are minor and may be self‐limiting or managed by dose reduction or temporary withholding of therapy, while others are severely debilitating or even fatal. Therefore, it is necessary for regulators to evaluate a drug's benefits in light of its potential risks to patients. This process is commonly referred to as benefit–risk assessment.
Genomic factors may alter the benefit–risk profile of a drug such that it may be considered more or less favorable both at the population level, or the individual patient level. Such differences in benefit–risk may arise from heterogeneity in the treatment effect across subgroups of patients (i.e., the genemic factor provides information about the likelihood of response to a treatment) or prognostic differences in patient subgroups that alter risk‐tolerance to a treatment. In recent years, numerous drugs and biologics have been developed in and approved for targeted subsets of patients because efficacy and safety varied according to the presence or absence of a specific molecular biomarker. This review will discuss the potential impact of genomic factors on benefit–risk assessment from regulatory and clinical perspectives.
BENEFIT–RISK
Demonstrating a favorable benefit–risk profile is important for both drug approval and uptake in the clinical setting. The FDA's benefit–risk assessment is directed by science, medicine, policy, and judgment in accordance with the framework outlined in the FD&C Act, the public Health Service Act, and the regulations issued by the agency to implement these acts.8 The benefit–risk assessment conducted to inform regulatory decision‐making takes into account the evidence of safety and efficacy submitted to support drug approval, as well as the nature and severity of the disease the drug is intended to treat, the safety and efficacy profiles of other available therapies, and the utility of risk management tools.8
Multiple strategies to mitigate drug risk, and thus improve the safety profile of the drug, are utilized during product development and clinical use. In the drug development setting, many types of studies inform the benefit–risk profile of a drug, including nonclinical toxicology studies, clinical pharmacology studies, adequate and well‐controlled clinical trials designed to directly evaluate safety and efficacy, and others. These studies are often the basis for appropriate patient population selection (e.g., type and severity of disease), dose selection, management of drug–drug or drug–food interactions, dose adjustments in special populations (e.g., hepatic and renal impairment), or other maneuvers to enhance benefit–risk.9, 10 Moreover, a Risk Evaluation and Mitigation Strategy (REMS) may be required to help reduce the occurrence of adverse events. REMS may include educational materials for patients and/or physicians, special training or certification for healthcare providers, patient monitoring programs, and other elements.11 In the clinical setting, benefits and risks to the individual patient are weighed in each unique case, and numerous intrinsic and extrinsic factors may be considered when selecting the therapeutic strategy and dose, and patient response may be carefully monitored or titrated to a pharmacodynamic effect to maximize effectiveness or minimize toxicity.9 While all of these strategies have become commonplace in drug development and clinical settings, and are frequently included in drug labeling and clinical practice guidelines, residual risk will always remain. Therefore, additional strategies are needed to optimize the benefit–risk profile.
IMPROVING BENEFIT–RISK PROFILE THROUGH GENOMICS
Recently, we have seen a rapid advancement in genomic technologies that has helped usher in a new era of “personalized” or “precision” medicine.12 New “omics” technologies now allow us to analyze patient differences at the genomic, transcriptomic, proteomic, metabolomic, and epigenomic levels.12 In addition, more powerful medical imaging techniques and computational biology approaches allow us unprecedented insights into patient characteristics and disease pathology.13 The medical field is only beginning to leverage the power of these newly available data to assess patient risk, diagnose disease, accurately predict prognosis, and develop effective therapies. However, genomic factors are relatively advanced in their application, and have been successfully incorporated into clinical trial design, drug labeling, and clinical practice to improve the benefit–risk profile associated with numerous therapies. A fundamental understanding of the impact of genomics on drug efficacy, safety, and PK can allow prospective identification of patients likely to respond, patients likely to suffer drug‐related adverse events, and patients who require altered dosing, potentially shifting the benefit–risk profile of the drug (Figure 1). In some cases genomics can have such a profound impact on benefit–risk that recognition and incorporation of genomic factors into drug development may shift the balance of drug approvability or result in warnings or contraindications for use in certain subsets of the population. The following examples illustrate the potential impact of genomic factors on drug efficacy, safety, and dosing, and thus the overall benefit–risk profile of the drug.
Figure 1.
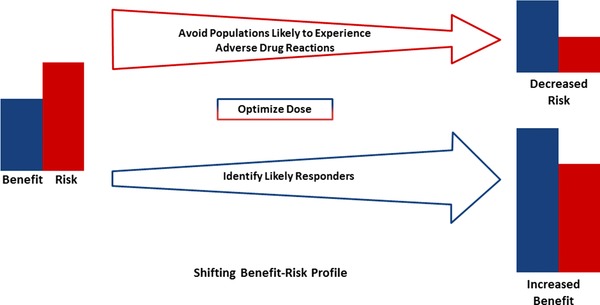
Genomic factors may be utilized to decrease risk and/or increase benefit. Decreasing arrow size represents decreasing risk and increasing arrow size represents increasing benefit. Either reducing risk or increasing benefit can result in a more favorable benefit–risk profile, and a greater likelihood of approvability.
LEVERAGING GENOMIC FACTORS
Using genomic factors to improve efficacy: gefitinib
Gefitinib, a selective inhibitor of epidermal growth factor receptor (EGFR) tyrosine kinase, was approved under the FDA's Accelerated Approval Program in 2003 for the treatment of patients with locally advanced or metastatic non‐small cell lung cancer (NSCLC) after failure of both platinum‐based and docetaxel chemotherapies.14 Under the Accelerated Approval Program, the data providing evidence of gefitinib efficacy in this population came primarily from a phase II trial of gefitinib in the third‐line setting with supportive data from a second phase II trial of gefitinib in both second‐ and third‐line settings.14 In these studies the objective response rate in Caucasian patients was 10–12%, while the response rate in Japanese patients was higher, at over 27%.15, 16 Females and nonsmokers were also noted to have higher response rates.14 Following Accelerated Approval, a confirmatory trial (Iressa Survival Evaluation in Lung Cancer (ISEL)) was conducted in second‐ and third‐line patients, and gefitinib failed to demonstrate improvement in overall survival compared to placebo (5.6 months in patients treated with gefitinib vs. 5.1 months in patients treated with placebo; hazard ratio 0.89 (95% confidence interval (CI) 0.77–1.02), P = 0.087).17 The findings from the ISEL trial, as well as several other negative postmarketing studies, led to gefitinib use being restricted in 2005 to patients already receiving and benefiting from gefitinib therapy, and eventually its voluntary withdrawal from the market in 2012. Interestingly, exploratory subgroup analyses again demonstrated higher objective response rates in patients of Asian origin, as well as women, never‐smokers, and patients with adenocarcinoma.17 However, the mechanism underlying these subgroup differences was not well understood at the time.
While gefitinib and other EGFR tyrosine kinase inhibitors were being evaluated in clinical trials, studies were also under way to identify the molecular mechanisms underlying the variable response to these agents. A study that sequenced DNA extracted from NSCLC tumor specimens identified somatic EGFR mutations in eight of nine patients who had achieved a clinically significant response to gefitinib, while no EGFR mutations were observed in seven patients with NSCLC who had not responded to gefitinib.18 Of the eight tumor specimens positive for EGFR mutations, four had in‐frame deletions in exon 19 of EGFR, and the remaining samples had substitutions in exons 21 (L858R (2) and L861Q (1)) or exon 18 (G719C (1)) of EGFR. In vitro studies found that EGFR mutated cell lines had enhanced sensitivity to gefitinib, which supported the clinical results.18 Shortly thereafter, additional clinical studies also identified EGFR mutations in tumors from the majority of NSCLC patients who achieved a clinical response to gefitinib, but not in gefitinib‐refractory tumors, and additional in vitro experiments provided further mechanistic evidence that certain somatic EGFR mutations conferred sensitivity to gefitinib.19, 20 In addition, EGFR mutations were identified more frequently in tumors from patients in Japan compared to the United States, from female compared to male patients, in adenocarcinomas compared to other histologies, and from never‐smokers compared to former or current smokers.19, 20 These observed differences in EGFR mutation frequencies across race, sex, and smoking status provided a biologically plausible explanation for the previously observed differences in response to EGFR tyrosine kinase inhibitors across these demographic subsets.
Findings from additional studies confirmed the presence of somatic EGFR tyrosine kinase inhibitor sensitizing mutations in about 10–20% of NSCLC samples from patients in North America and Western Europe and 40–60% of patients of East Asian descent.21 Common EGFR tyrosine kinase inhibitor sensitizing mutations include a collection of EGFR exon 19 deletions (∼45% of cases), and the EGFR L858R substitution mutation (40–45% of cases) in exon 21.22 Less frequent EGFR mutations that are potentially sensitizing to EGFR tyrosine kinase inhibitors include G719X (G719A, G719C, and G719S, ∼3% of cases), L861Q, and exon 19 insertions; however, the extent to which EGFR tyrosine kinase inhibitors are effective in patients whose tumors harbor these other EGFR mutations is less clear and these mutations are sometimes referred to as “intermediate sensitizing.”23 In addition, several EGFR mutations have been identified that confer resistance to first‐ and second‐generation EGFR tyrosine kinase inhibitors, most notably the EGFR T790M mutation and a collection of exon 20 insertion mutations.23
Based on this growing body of knowledge, a clinical trial (Iressa Pan‐Asia Study (IPASS)) was conducted comparing gefitinib to carboplatin/paclitaxel as first‐line treatment in a clinically enriched population of nonsmokers or former light smokers in East Asia who had adenocarcinoma of the lung.24 The trial met its primary end point in the intent‐to‐treat population (hazard ratio for progression or death, 0.74; 95% CI, 0.65 to 0.85; P < 0.001) and showed an objective response rate of 43% for gefitinib. However, an exploratory subgroup analysis of patients with EGFR mutation‐positive (defined as the presence of any of 29 EGFR mutations that were tested) vs. EGFR mutation‐negative tumors showed a robust impact of EGFR mutation status on gefitinib efficacy, with a significant interaction between treatment and EGFR mutation status with respect to progression‐free survival. For patients whose tumors were EGFR mutation‐positive, progression‐free survival was significantly longer in patients who received gefitinib with a hazard ratio of 0.48 (95% CI, 0.36 to 0.64; P < 0.001) for progression or death and an objective response rate of 71%. In contrast, for patients with EGFR mutation‐negative tumors, progression‐free survival was significantly longer in patients who received carboplatin/paclitaxel, with a hazard ratio for progression‐free survival with gefitinib of 2.85 (95% CI, 2.05 to 3.98; P < 0.001) and an objective response rate of 1.1%. Subsequently, an open‐label, single‐arm (Iressa Follow‐Up Measure (IFUM)) trial of gefitinib as first‐line treatment in Caucasian patients with EGFR mutation‐positive NSCLC confirmed an objective response rate of ∼70% in patients with EGFR tyrosine kinase inhibitor sensitizing mutations.25
In 2014, a new drug application (NDA) was submitted to the FDA to market gefitinib for treatment of patients with metastatic NSCLC whose tumors have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations as detected by an FDA‐approved test. Gefitinib was subsequently approved in July 2015 based on the data derived from the IFUM trial, with supportive data from the retrospective genetic subgroup analysis of IPASS.26 The indication statement of the drug labeling specifies that gefitinib is indicated for first‐line treatment of patients with metastatic NSCLC whose tumors have EGFR exon 19 deletions or exon 21 (L858R) substitution mutations. Only two patients each with tumors harboring L861Q or G719X substitutions were enrolled in the IFUM trial; the objective responses and duration of response observed in these patients are described in the Clinical Trials section of the drug labeling.26
The regulatory history of gefitinib, from approval to market withdrawal to reintroduction in a molecularly defined subset of patients as a “targeted therapy,” exemplifies the impact that molecular factors can have on benefit–risk assessment. In an unselected population, gefitinib demonstrated efficacy in a small proportion of patients. However, when the molecular driver of drug response was identified and gefitinib was evaluated in the subset of NSCLC patients with EGFR tyrosine kinase inhibitor‐sensitizing mutations, the response rate increased substantially. This higher response rate resulted in an improved benefit–risk profile in the molecularly defined patient population, and created a pathway for gefitinib to be approved, providing an additional treatment option for this subset of patients (the EGFR tyrosine kinase inhibitors erlotinib and afatinib were approved prior to the market reentry of gefitinib).
When molecular drivers of disease are identified prior to the large phase III clinical trials designed to demonstrate efficacy and safety of the drug, enrichment strategies can be employed resulting in more efficient clinical trials with a greater likelihood of success, with fewer patients exposed to ineffective therapies. Indeed, the initial phase III clinical trials of the EGFR tyrosine kinase inhibitor afatinib were conducted in a selected population of NSCLC patients whose tumors harbored EGFR mutations (including some mutations ultimately found to confer resistance), and demonstrated efficacy while enrolling a smaller number of patients than the initial phase III studies of gefitinib and erlotinib that were conducted in unselected patient populations.17, 27, 28
Using genomic factors to decrease risk: abacavir
In addition to identifying patients most likely to benefit from a drug, genomic information can also be utilized to mitigate severe safety issues associated with drugs, thus improving a drug's benefit–risk profile by decreasing risk. One prominent example is the antiretroviral drug abacavir, a nucleoside reverse transcriptase inhibitor used to treat human immunodeficiency virus (HIV) infection (in combination with other antiretroviral agents). Abacavir was initially approved for treatment of HIV in 1998; however, hypersensitivity reactions were observed in ∼5% of patients who received abacavir, with some being severe and potentially fatal.29 Abacavir‐associated hypersensitivity reactions are typically characterized by fever, nausea, vomiting, skin rash, and more rarely respiratory symptoms and hypotension; symptoms generally resolve following discontinuation of the drug. When initially described, the mechanism underlying development of the hypersensitivity reactions was unknown, and thus the reactions were referred to as “idiosyncratic.”29
In 2002, two studies identified multiple genetic variants in the human leukocyte antigen (HLA)‐B region were associated with clinically diagnosed hypersensitivity reactions; the HLA‐B*5701 allele demonstrated the strongest association, but was present in only 55–78% of the clinically diagnosed cases in these studies.30, 31 Following development of a diagnostic test to immunologically confirm abacavir hypersensitivity, the Prospective Randomized Evaluation of DNA Screening in a Clinical Trial (PREDICT)‐1 study demonstrated that in abacavir‐naïve patients, prospective screening for HLA‐B*5701 and exclusion of HLA‐B*5701 allele carriers from receiving abacavir significantly reduced immunologically confirmed hypersensitivity reactions from 2.7% in the control arm to 0% in the prospectively genotyped arm and clinically diagnosed hypersensitivity reactions from 7.8% in the control arm to 3.4% in the prospectively genotyped arm.32 Moreover, HLA‐B*5701 screening had a negative predictive value of 100% for immunologically confirmed hypersensitivity reactions, indicating that HIV patients without the HLA‐B*5701 variant have a low risk of developing hypersensitivity to abacavir.
Based on the exceptional clinical evidence provided by PREDICT‐1, the FDA updated abacavir labeling in 2008 to include information describing the risk of hypersensitivity reactions in patients who carry the HLA‐B*5701 allele (as a Boxed Warning). The labeling was again updated in 2015 to include the presence of the HLA‐B*5701 allele as a contraindication to abacavir therapy.
The discovery of the association between HLA‐B*5701 genotype and hypersensitivity reactions, and subsequent labeling updates to inform healthcare providers of the risk for hypersensitivity in this patient subgroup, has helped mitigate a major safety concern when genotyping is conducted prior to administration.33 The ability of HLA‐B*5701 screening to identify patients at risk for abacavir hypersensitivity, and avoid abacavir use in these patients, has been recognized by healthcare providers leading to widespread implementation of screening programs prior to abacavir use. The most recent version of the Guidelines for the Use of Antiretroviral Agents in HIV‐1 Infected Adults and Adolescents (developed by the Department of Health and Human Services) classifies abacavir as a part of a “Recommended regimen” when initiating antiretroviral therapy in treatment‐naïve patients, and recommends screening for HLA‐B*5701 prior to starting patients on abacavir and avoiding abacavir in HLA‐B*5701‐positive patients.34
Using genomic factors to identify the appropriate dose: eliglustat
There are numerous examples where genetic polymorphisms in drug metabolizing enzymes impact a drug's PK properties. In some cases, drug labeling recommends dose adjustment based on genotype, usually in an attempt to match exposure of certain subgroups that demonstrate altered PK properties (e.g., “poor metabolizers”) to the majority of the population who receives the drug. In these cases, prospective dosage adjustment may help avoid drug toxicity or ensure therapeutically effective concentrations are achieved (e.g., in the case of ultrarapid metabolizers), thus improving benefit–risk profile by reducing risk or increasing likelihood of benefit, respectively.
One recent example where polymorphic drug metabolism resulted in different benefit–risk profiles across population subsets is the glucosylceramide synthase inhibitor eliglustat. In a clinical study to evaluate the potential of eliglustat to cause QT prolongation, eliglustat administration resulted in concentration‐dependent prolongation of the PR, QTc, and/or QRS cardiac intervals that could potentially result in cardiac arrhythmias.35 Eliglustat is primarily metabolized by the polymorphic drug metabolizing enzyme CYP2D6, and studies demonstrated eliglustat PK is highly dependent on CYP2D6 genotype.36 Although eliglustat exposures in CYP2D6 poor metabolizers (PMs) were ∼7‐fold higher than exposures in CYP2D6 extensive metabolizers (EMs), at the reduced dosage of 100 mg once daily (compared to 100 mg twice daily for EMs and intermediate metabolizers (IMs)), the maximum concentrations achieved in CYP2D6 PMs at the 100 mg once daily dose was considered to result in a low likelihood of QT‐related safety concerns.36 However, even at the higher dose of 200 mg twice daily, exposures in CYP2D6 ultrarapid metabolizers (UMs) were 57% and 82% lower than for EMs and IMs, respectively, at 100 mg twice daily.36 Eliglustat was ultimately approved for “treatment of adult patients with Gaucher disease type 1 who are CYP2D6 extensive metabolizers (EMs), intermediate metabolizers (IMs), or poor metabolizers (PMs) as detective by an FDA‐cleared test,” with the reduced dosage recommended for CYP2D6 PMs.35 Since it is unclear whether or not CYP2D6 UMs would achieve adequate concentrations of eliglustat to achieve therapeutic effect, the drug is not indicated for this subset of the population, and this is described in the Limitations of Use section of labeling. CYP2D6 genotype information is also provided in the Dosage and Administration section to inform dosing across CYP2D6 genotype subgroups and in the Drug Interactions section to inform dosing across CYP2D6 genotype subgroups when eliglustat is administered with other drugs that affect its metabolism. Eliglustat clearly demonstrates how genetic factors that impact a drug's PK properties can result in different benefit–risk profiles across genetic subgroups, and that prospective dose adjustment can improve the benefit–risk profile in some genetic subgroups.
LEVERAGING GENOMIC FACTORS IN DRUG DEVELOPMENT: INCREASING EFFICIENCY BY IMPROVING BENEFIT–RISK
Genomic information can be leveraged throughout all phases of the drug development process to improve efficiency and to optimize the efficacy and safety profiles of the drug.4 In early‐phase clinical studies genomic factors may be assessed to help determine the most appropriate dose to carry forward into later‐phase efficacy studies, and to determine whether or not stratified dosing is necessary for genomic subsets (e.g., patients with functional genetic polymorphisms in drug metabolizing enzymes). By identifying the most appropriate dose for all patients, the drug is more likely to achieve adequate concentrations for drug efficacy, while avoiding concentration‐dependent toxicities.37 In addition, genomic factors may be analyzed in proof‐of‐concept studies to begin to assess whether drug efficacy is limited to a molecular subset of patients, and these factors may be utilized to enrich later‐phase efficacy studies for likely responders to the drug. Genomic factors could also be utilized to mitigate risks in clinical trials by excluding patients who may be more likely to experience drug‐related adverse events; however, the genomic factors contributing to rare adverse drug reactions are usually unknown prior to phase III clinical trials. As such, retrospective genomic analyses in clinical trials may be more useful to identify fundamental mechanisms of observed safety issues that arise in the pre‐ or postmarket setting.38
In recent years, genomic factors have been incorporated into innovative trial designs, including “basket” and “umbrella” trials, most notably in the oncology field.39 Basket trials enroll patients with numerous different malignancies based on a shared molecular classification and allow initial assessment of the drug in each tumor type, thus improving patient population selection for later‐phase efficacy studies. The V‐BASKET trial, for example, is enrolling patients with solid tumors or multiple myeloma (except melanoma and papillary thyroid cancer) who have BRAF V600 mutation‐positive cancers and treating them with the BRAF inhibitor vemurafenib.40 In contrast to basket trials, umbrella trials enroll a single tumor histology type, but stratify the histology‐defined population into multiple molecularly defined subpopulations that are matched to drugs likely to have activity against tumors that harbor that molecular alteration, thereby potentially improving the likelihood of response. Examples of umbrella trials include the BATTLE and I‐SPY trials for NSCLC and breast cancer, respectively.40 By incorporating basket and umbrella trial designs, drug developers may ultimately improve the benefit–risk profile of a drug by identifying patient populations with enhanced response rates in early‐phase trials, and focusing later‐phase efficacy trials on the patient populations who are most likely to respond. Basket and umbrella trial designs, in principle, should also increase clinical trial efficiency by decreasing screening failures, speeding enrollment, and allowing multiple patient subpopulations to be studied under a single protocol.
LOOKING TOWARD THE FUTURE
While there are many successful examples of genomic information being used in clinical trials and clinical practice to make drugs safer and more effective, implementation of genomic technologies is likely to expand significantly in the foreseeable future. As genomic testing technologies continue to become more accessible to healthcare providers in the course of their clinical practice, use of genomic information to prospectively select appropriate medications and dosages for all patients may become a common part of clinical practice. Moreover, direct‐to‐consumer genetic testing, preemptive testing of multiple clinically relevant genomic variants, and inclusion of the genomic information in patients’ medical records could facilitate increased use of genomics in both drug development and clinical settings as healthcare providers become more familiar with using genomic information and genetic testing results are readily available at the time of prescribing.41 The increased utilization of genomic information in drug development and clinical decision‐making may serve to improve the benefit–risk profile of drugs.
In drug development, increased utilization of genomic information to develop drugs for the patient populations that demonstrate maximum efficacy with fewer adverse events will likely result in a continued increase in precision medicine approvals. Indeed, a recent report indicated that 42% of all compounds in the drug development pipeline, and 73% of all oncology compounds, utilize biomarker data during drug development.42 The same report indicated that investment in this area has nearly doubled over the past 5 years, and that the majority of biopharmaceutical researchers predict that development will continue to increase over the next 5 years. Based on these recent trends, it is likely that we will continue to see an increase in the successful utilization of genomic factors to optimize benefit–risk, and thus the approval of safer and more effective therapies.
SUMMARY AND CONCLUSIONS
Despite decades of progress in improving the safety and effectiveness of therapies, all medications present at least some risk to patients, and no medications are effective in all patients. Therefore, strategies to decrease adverse events and increase the likelihood of efficacy can be useful to improve patient care. Genomic technologies have given us a powerful tool to improve the precision with which we diagnose and treat patients, and we are beginning to see the utility of genomic information to transform drug development and clinical practice. Genomics may be leveraged in the preapproval setting by regulators and pharmaceutical companies to improve benefit–risk at the population level by helping identify populations (or subpopulations) most likely to respond and those most likely to suffer drug‐related adverse events. In the clinical practice setting, genomic information, in combination with other patient factors, may be leveraged by healthcare providers to determine the most appropriate drug and dose for each patient, thus improving the individual benefit–risk profile. Accordingly, use of genomics to inform both drug development strategies and patient care decisions can help achieve a common goal of providing patients with safer, more effective therapies.
Conflict of Interest/Disclosures
This article reflects the views of the authors and should not be construed to represent the FDA's views or policies. The authors declared no conflict of interest.
References
- 1. Tremaine, L. et al The role of adme pharmacogenomics in early clinical trials: Perspective of the industry pharmacogenomics working group (i‐pwg). Pharmacogenomics. 18, 2055–2067 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Squassina, A. et al Realities and expectations of pharmacogenomics and personalized medicine: Impact of translating genetic knowledge into clinical practice. Pharmacogenomics. 8, 1149–1167 (2010). [DOI] [PubMed] [Google Scholar]
- 3. FDA‐NIH Biomarker Working Group . BEST (Biomarkers, EndpointS, and other Tools) Resource (Internet). Silver Spring (MD): Food and Drug Administration (US); 2016‐. Glossary. 2016. Jan 28 (Updated 2016 Apr 28). Available from: https://www.ncbi.nlm.nih.gov/books/NBK338448/ Co‐published by National Institutes of Health (US), Bethesda (MD). [PubMed]
- 4. Zineh, I. & Pacanowski, M.A. Pharmacogenomics in the assessment of therapeutic risks versus benefits: Inside the United States Food and Drug Administration. Pharmacotherapy. 8, 729–735 (2011). [DOI] [PubMed] [Google Scholar]
- 5. Pacanowski, M.A. , Leptak, C. & Zineh, I. Next‐generation medicines: Past regulatory experience and considerations for the future. Clin. Pharmacol. Ther. 3, 247–249 (2014). [DOI] [PubMed] [Google Scholar]
- 6. U.S. Food and Drug Administration . Promoting Safe and Effective Drugs for 100 Years. http://www.fda.gov/AboutFDA/WhatWeDo/History/ProductRegulation/PromotingSafeandEffectiveDrugsfor100Years/. Accessed July, 2016.
- 7. Temple, R. Are surrogate markers adequate to assess cardiovascular disease drugs? JAMA. 8, 790–795 (1999). [DOI] [PubMed] [Google Scholar]
- 8. U.S. Food and Drug Administration . Structured Approach to Benefit–Risk Assessment in Drug Regulatory Decision‐Making. www.fda.gov/downloads/ForIndustry/UserFees/PrescriptionDrugUserFee/UCM329758.pdf. Accessed July, 2016.
- 9. Blaus, A. et al Personalized cardiovascular medicine today: A Food and Drug Administration/Center for Drug Evaluation and Research perspective. Circulation. 15, 1425–1432 (2015). [DOI] [PubMed] [Google Scholar]
- 10. Zineh, I. & Woodcock, J. Clinical pharmacology and the catalysis of regulatory science: Opportunities for the advancement of drug development and evaluation. Clin. Pharmacol. Ther. 6, 515–525 (2013). [DOI] [PubMed] [Google Scholar]
- 11. Slatko, G.H. Risk evaluation and mitigation strategy (rems): FDA perspective on what physicians need to know. Am. Fam. Physician. 9, 771–772 (2015). [PubMed] [Google Scholar]
- 12. Topol, E.J. Individualized medicine from prewomb to tomb. Cell. 1, 241–253 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. U.S. Food and Drug Administration . Paving the way for personalized medicine. Accessed from http://www.fda.gov/downloads/scienceresearch/specialtopics/personalizedmedicine/ucm372421.pdf. (2013).
- 14. Cohen, M.H. et al United States Food and Drug Administration drug approval summary: Gefitinib (zd1839; IRESSA) tablets. Clin. Cancer Res. 4, 1212–1218 (2004). [DOI] [PubMed] [Google Scholar]
- 15. Kris, M.G. et al Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non‐small cell lung cancer: A randomized trial. JAMA. 16, 2149–2158 (2003). [DOI] [PubMed] [Google Scholar]
- 16. Fukuoka, M. et al Multi‐institutional randomized phase ii trial of gefitinib for previously treated patients with advanced non‐small‐cell lung cancer (the Ideal 1 Trial) (corrected). J. Clin. Oncol. 12, 2237–2246 (2003). [DOI] [PubMed] [Google Scholar]
- 17. Thatcher, N. et al Gefitinib plus best supportive care in previously treated patients with refractory advanced non‐small‐cell lung cancer: Results from a randomised, placebo‐controlled, multicentre study (iressa survival evaluation in lung cancer). Lancet. 9496, 1527–1537 (2005). [DOI] [PubMed] [Google Scholar]
- 18. Lynch, T.J. et al Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N. Engl. J. Med. 21, 2129–2139 (2004). [DOI] [PubMed] [Google Scholar]
- 19. Pao, W. et al EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. U. S. A. 36, 13306–13311 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paez, J.G. et al EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 5676, 1497–1500 (2004). [DOI] [PubMed] [Google Scholar]
- 21. Midha, A. , Dearden, S. & McCormack, R. EGFR mutation incidence in non‐small‐cell lung cancer of adenocarcinoma histology: A systematic review and global map by ethnicity (MUTMAPII). Am. J. Cancer Res. 9, 2892–2911 (2015). [PMC free article] [PubMed] [Google Scholar]
- 22. Sharma, S.V. , Bell, D.W. , Settleman, J. & Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer. 3, 169–181 (2007). [DOI] [PubMed] [Google Scholar]
- 23. Ohashi, K. , Maruvka, Y.E. , Michor, F. & Pao, W. Epidermal growth factor receptor tyrosine kinase inhibitor‐resistant disease. J. Clin. Oncol. 8, 1070–1080 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mok, T.S. et al Gefitinib or carboplatin‐paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 10, 947–957 (2009). [DOI] [PubMed] [Google Scholar]
- 25. Douillard, J.Y. et al First‐line gefitinib in caucasian egfr mutation‐positive NSCLC patients: A phase‐IV, open‐label, single‐arm study. Br. J. Cancer. 1, 55–62 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kazandjian, D. , Blumenthal, G.M. , Yuan, W. , He, K. , Keegan, P. & Pazdur, R. FDA approval of gefitinib for the treatment of patients with metastatic egfr mutation‐positive non‐small cell lung cancer. Clin. Cancer Res. 6, 1307–1312 (2016). [DOI] [PubMed] [Google Scholar]
- 27. Gilotrif (afatinib) (package insert). Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals; 2013.
- 28. Tarceva (erlotinib) (package insert). South San Francisco, CA: Genentech; 2004.
- 29. Hetherington, S. et al Hypersensitivity reactions during therapy with the nucleoside reverse transcriptase inhibitor abacavir. Clin. Ther. 10, 1603–1614 (2001). [DOI] [PubMed] [Google Scholar]
- 30. Hetherington, S. et al Genetic variations in HLA‐B region and hypersensitivity reactions to abacavir. Lancet. 9312, 1121–1122 (2002). [DOI] [PubMed] [Google Scholar]
- 31. Mallal, S. et al Association between presence of hla‐B*5701, HLA‐DR7, and HLA‐DQ3 and hypersensitivity to HIV‐1 reverse‐transcriptase inhibitor abacavir. Lancet. 9308, 727–732 (2002). [DOI] [PubMed] [Google Scholar]
- 32. Mallal, S. et al HLA‐B*5701 screening for hypersensitivity to abacavir. N. Engl. J. Med. 6, 568–579 (2008). [DOI] [PubMed] [Google Scholar]
- 33. Young, B. et al First large, multicenter, open‐label study utilizing HLA‐B*5701 screening for abacavir hypersensitivity in North America. AIDS. 13, 1673–1675 (2008). [DOI] [PubMed] [Google Scholar]
- 34. Panel on Antiretroviral Guidelines for Adults and Adolescents . Guidelines for the use of antiretroviral agents in HIV‐1‐infected adults and adolescents. Department of Health and Human Services. Available at http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed December 2016.
- 35. Cerdelga (eliglustat) (package insert). Waterford, Ireland: Genzyme Ireland; 2014.
- 36. FDA Center for Drug Evaluation and Research Clinical Pharmacology and Biopharmaceutics Reviews . Application number 205494 Orig1s000 (eliglustat tartrate). Accessed from http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205494Orig1s000TOC.cfm. 2014.
- 37. Wilkinson, G.R. Drug metabolism and variability among patients in drug response. N. Engl. J. Med. 21, 2211–2221 (2005). [DOI] [PubMed] [Google Scholar]
- 38. Pirmohamed, M. , Ostrov, D.A. & Park, B.K. New genetic findings lead the way to a better understanding of fundamental mechanisms of drug hypersensitivity. J. Allergy Clin. Immunol. 2, 236–244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Billingham, L. , Malottki, K. & Steven, N. Research methods to change clinical practice for patients with rare cancers. Lancet Oncol. 2, e70–80. (2016). [DOI] [PubMed] [Google Scholar]
- 40. Biankin, A.V. , Piantadosi, S. & Hollingsworth, S.J. Patient‐centric trials for therapeutic development in precision oncology. Nature. 7573, 361–370 (2015). [DOI] [PubMed] [Google Scholar]
- 41. Dunnenberger, H.M. et al Preemptive clinical pharmacogenetics implementation: Current programs in five US medical centers. Annu. Rev. Pharmacol. Toxicol. 89–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tufts Center for the study of drug development. Impact Report. 17, 1–4 (2015). [Google Scholar]