

Abstract
Background/purpose
Recently, the mutational background of diffuse large B cell lymphoma (DLBCL) has been revealed, identifying specific genetic events that drive lymphomagenesis. However, the prognostic value of these mutations remains to be determined. Prognostic biomarkers in DLBCL are urgently needed, since the current clinical parameter-based factors (e.g., International Prognostic Index (IPI)) are insufficient, particularly in identifying patients with poor prognosis who might benefit from alternative treatments.
Methods
We investigated the prognostic value of somatic mutations in DLBCL in a clinical trial (NCT00544219) patient cohort homogenously treated with six cycles of rituximab, cyclophosphamide, hydroxydaunorubicin, vincristine, and prednisone (R-CHOP), followed by two cycles of R (R-CHOP-14). The primary endpoint was event-free survival (EFS) at 2 years. Secondary endpoints included progression-free survival (PFS) and overall survival (OS). Targeted high-throughput sequencing (HTS) of tumor genomic DNA was performed on all exons or hotspots of 68 genes frequently mutated in B cell lymphomas. Mutational data was correlated with the endpoints to identify prognostic associations.
Results
Targeted HTS detected somatic mutations in 71/76 (93%) of investigated cases. The most frequently mutated genes were KMT2D, SOCS1, GNA13, and B2M. Survival analysis revealed that CREBBP- and EP300-mutated cases had significantly worse OS, PFS, and EFS. In addition, ATM mutations predicted worse outcomes for all three clinical endpoints in germinal center B cell-like DLBCL. In contrast, SOCS1 mutations were associated with better PFS. On multivariable analysis taken into account IPI and failure to achieve complete remission, CREBBP and EP300 mutations remained significant to predict worse OS, PFS, and EFS.
Conclusion
Targeted mutation analysis of a uniformly treated prospective clinical trial DLBCL cohort identifies tumor-based genetic prognostic markers that could be useful in the clinical management of such patients.
Trial registration
ClinicalTrials.gov NCT00544219
Electronic supplementary material
The online version of this article (doi:10.1186/s13045-017-0438-7) contains supplementary material, which is available to authorized users.
Keywords: DLBCL, Prognostic markers, Targeted high-throughput sequencing, NGS, Lymphoma, CREBBP, SOCS1, EP300
Introduction
Diffuse large B cell lymphoma (DLBCL) is a heterogeneous and aggressive lymphoid neoplasm, the treatment of which has significantly improved in the last decade with addition of the anti-CD20 monoclonal antibody rituximab (R) to the chemotherapy regimen consisting of cyclophosphamide, hydroxydaunorubicin, vincristine, and prednisone (R-CHOP) [1]. Since then, R-CHOP has become a standard treatment for the vast majority of primary DLBCL cases with cure rates at about 60%. The remaining 40% of DLBCL either relapses after a period of remission or are refractory to the applied first-line therapy. Patients in this group are treated with aggressive salvage regimens supported by autologous stem cell transplantation (ASCT) [2], but success rates are modest, particularly in primary refractory DLBCL [3, 4]. Therefore, it is important to identify high-risk patients before administration of first-line therapy so that the potentially more aggressive tumors can be treated with alternative regimens [5].
Risk stratification of DLBCL has relied for more than 20 years on the International Prognostic Index (IPI), which is based on evaluation of multiple clinical parameters [6]. Following the addition of rituximab, IPI was revised (R-IPI) [7] and, most recently, further enhanced (NCCN-IPI), to better identify high-risk patients [8]. According to NCCN-IPI, the high-risk group has 5-year overall survival (OS) probability of 33% compared to 54% predicted by IPI, although this remains to be confirmed by the datasets of prospective trials.
In addition to patient-based factors, tumor-based prognostic markers have been proposed. It is known that ~10% of DLBCL cases have MYC rearrangements that are strongly associated with worse outcomes, especially if linked to MYC protein overexpression [9]. It has also been shown that the activated B cell (ABC) cell-of-origin (COO) DLBCL subtype has a worse outcome compared to the germinal center B cell (GCB) subtype [10]. Because original classification based on gene expression profiling on a transcriptome level proved to be technically too challenging for implementation in clinical practice, surrogate immunohistochemistry-based algorithms were developed [11]. However, their utility was limited by suboptimal concordance to the gene expression-based gold standard. Recently, a Lymph2Cx assay, which can measure expression of 20 genes and can be also applied on formalin-fixed paraffin-embedded (FFPE) material, was proposed for reproducible classification of DLBCL into COO subgroups [12]. It remains to be seen if this new technology will be widely accepted in lymphoma centers worldwide. In addition to COO classification, immunohistochemical (IHC) studies have identified multiple protein markers, such as CD5, Ki-67, FOXP1, HLA-I, p21, and CD40, that prospectively showed prognostic value for R-CHOP-treated DLBCL [13–17].
During the last decade, substantial progress has been made toward understanding the genetic basis of DLBCL [18–20]. Shared and COO subtype-specific DNA lesions have been identified, converging into several most frequently dysregulated cellular pathways [21]. Based on these findings, a handful of molecular prognostic markers have been identified, among which, TP53, FOXP1, and MYD88 mutations and CDKN2A deletions were associated with inferior outcomes in R-CHOP-treated DLBCL [22–24]. The prognostic role of such molecular markers is likely to increase in the near future as high-throughput sequencing (HTS) enters routine practice in many institutions. However, more studies, particularly prospective analyses, are required to validate the existing molecular prognostic markers and to discover new ones that would add power to the existing prognostication algorithms.
In this study, we employed targeted HTS to identify somatic mutations in tumors of a well-documented prospective clinical cohort consisting of uniformly treated primary DLBCL patients. By correlating gene mutation status to the robust survival data, we aimed to discover new, and validate known, prognostic markers in DLBCL.
Methods
Study cohort
The clinical trial SAKK 38/07 (NCT00544219), active between 2007 and 2010, included 138 eligible patients with primary untreated DLBCL to prospectively determine the prognostic value of interim PET/CT scans by standardized treatment and evaluation criteria. The main clinical results have been published previously [25].
Tissue specimens of patients who consented for additional translational research were used for a subsequent study investigating the prognostic value of phenotypic and genotypic profiles by IHC and fluorescence in situ hybridization (FISH), the results of which have been recently published in this journal [13].
For the current study, FFPE tissues of 84 primary untreated de novo DLBCL patients with adequate amounts of remaining material were selected. Tumor content was determined by morphological evaluation and was at least 50% in all samples.
Updated clinical data (last follow-up on 31 January 2017) were used for evaluation of the prognostic role of genetic mutations. All patients in the study cohort were uniformly treated with six cycles of R-CHOP, followed by two cycles of R (R-CHOP-14). The primary endpoint was event-free survival (EFS) at 2 years (for definition, see the “Statistical analysis” section), and the secondary endpoints were progression-free survival (PFS) and OS at 2 and 5 years as well as objective responses according to international criteria [26].
DNA extraction and quantification
Genomic DNA was extracted with the GeneRead DNA FFPE kit (Qiagen, Nussloch, Germany) following manufacturers’ recommendations with minor modifications. Briefly, one to three 10–25 μm thick tissue sections were deparaffinized by several xylene washes, rehydrated and digested with proteinase K overnight at 56 °C in a shaking heat block. Following digestion, the samples were incubated for 1 h at 90 °C to reverse fixation-induced DNA crosslinks and inactivate proteinase K. Thereafter, uracil-N-glycosylase (UNG) enzyme was added to remove artificially (formalin) induced uracils and reduce the number of false-positive C > T transitions. After incubation at 37 °C for 1 h, the samples were loaded into a DNA purification column and were washed and eluted in 40 μl of nuclease-free water. DNA yields were quantified with the Qubit High sensitivity DNA assay (Life Technologies, Eugene, OR, USA).
Targeted HTS sequencing variant calling and filtering
A target enrichment panel was designed to cover mutational hotspots or all exons of genes most frequently mutated in B cell lymphoid neoplasms according to the COSMIC database (release v70) and manual review of the literature [27]. Sequencing libraries were constructed exactly as described previously [27]. One microliter of the prepared library was used for the Bioanalyzer High Sensitivity DNA assay (Agilent, USA) to confirm expected DNA fragment length distribution. Quantification was performed with the Ion Library Quantitation kit (Thermo Fisher Scientific, Carlsbad, CA, USA) following the original protocol. Libraries were diluted to 40 pM, loaded to the Ion540 sequencing chips by automated IonChef instrument, and sequenced with the Ion Torrent S5 XL machine (Thermo Fisher Scientific, Carlsbad, CA, USA). The depth of coverage, coverage uniformity, and number of variants called per sample are summarized in Additional file 1: Table S1. Mutation identification was performed by the Variant caller plug-in v5.0 of the Torrent Suite (Thermo Fisher Scientific) using default low stringency parameters for somatic mutation calling. Mutations were annotated using the Ion Reporter variant annotation workflow v5.0 and dbNSFP v3.0 database [28]. MetaLR rank score was used to predict the functional impact of non-synonymous point mutations to the encoded protein [29]. After annotation, the variants were subjected to additional, more stringent, and quality- and relevance-based filtering by criteria as detailed in Table 1. All variants with variant allelic frequency >5% were used in downstream analysis.
Table 1.
Criteria used for mutation filtering (variant inclusion)
| Criterion name | Threshold value |
|---|---|
| General quality | |
| Phred-based quality | >50 |
| Strand bias | ≤0.75 |
| Number of reads supporting called variant | ≥10 |
| Functional relevance | |
| Variant allelic frequency | ≥5% |
| Localization | Exonic and splice site |
| Variant effect | Non-synonymous |
| SNP exclusion | |
| Variant allelic frequency | <95% |
| Database annotation and alternative allelic frequency (1000 genomes project, European descendent samples) | Not listed in dbSNP v138 or listed, but MAF ≤0.01% |
| Variants in detected in the control cohort of 23 non-tumoral samples from lymphoma patients | Not overlapping |
Finally, aligned BAM files were manually inspected at sites of all remaining variants to exclude false-positive mutations or other artifacts introduced during library preparation [30].
Statistical analysis
All statistical analyses were performed using the Statistical Package of Social Sciences (IBM SPSS version 22.0, Chicago, IL, USA) for Windows. EFS was calculated from registration to progressive disease or relapse, death of any cause, and initiation of any non-protocol anti-cancer treatment because of lymphoma symptoms or need of concomitant radiotherapy. PFS was calculated from registration to progressive disease or relapse, and death of any cause. OS was calculated from registration to death. Patients not experiencing an event were censored at last follow-up. The survival probabilities were determined using the Kaplan–Meier method, and groups were compared using the log-rank test. Factors of prognostic significance in univariable models underwent multivariable analysis using the Cox proportional hazards model. For other endpoints, differences between groups were tested either with t test, Wilcoxon rank-sum test, or Fisher’s exact test, as appropriate. In all tests, p values are two-sided, considered significant if <0.05, and not corrected for multiple testing. For survival analysis within the COO subgroups, p values were corrected for multiple testing and were considered significant if <0.017.
Results
Patients and clinico–pathologic characteristics
In total, 84 patient samples were included. Four samples yielded insufficient DNA for HTS library preparation, and another four samples failed quality control after sequencing (low coverage uniformity, low percentage of reads mapped to target), leaving 76 cases for further study. Patient characteristics are presented in Table 2. Survival data was available for all 76 patients. The 2-year OS, PFS, and EFS of the entire cohort were 91% (95% CI 84 to 98%), 78% (95% CI 68 to 88%), and 63% (95% CI 53 to 75%), respectively. There was no significant difference in any of the survival endpoints between the two different immunohistochemically determined COO subtypes. IHC and FISH data on all samples were collected as part of the previous translational study focusing on the prognostic value of genotypic and phenotypic characteristics [13]. “Double-hit score,” representing IHC-based evaluation of tumor cell expression of BCL2 and MYC (Table 2), showed that 10/76 (13%) cases were positive for both MYC and BCL2. None of the investigated cases had chromosomal translocations affecting both MYC and BCL2 genes. Thirty of 76 (39%) and 1/76(1%) of cases were positive for FOXP1 and CD5, respectively, both associated with worse DLBCL outcomes in our previous study [13]. All investigated cases were Epstein–Barr virus (EBV)-negative as determined by in situ hybridization for EBV early RNA (EBER).
Table 2.
Patient characteristics
| Age, median (range) | 59 (18–81) | |
|---|---|---|
| Gender, N (%) | F | 34 (45) |
| M | 42 (55) | |
| Stage, N (%) | I | 7 (9) |
| II | 24 (31) | |
| III | 22 (30) | |
| IV | 23 (30) | |
| IPI, N (%) | 0–1 | 35 (46) |
| 2 | 19 (25) | |
| 3 | 11 (14) | |
| 4–5 | 11 (14) | |
| Treatment | R-CHOP-14 | 76 |
| Treatment response according to international criteria [26], N (%) | CR | 63 (83) |
| PR | 12 (16) | |
| SD | 1 (1) | |
| PD | 0 (0) | |
| Survival, median (IQR) | PFS | 55.05(56.7) |
| EFS | 55.05 (56.7) | |
| OS | 61.9 (9.65) | |
| Cell-of-origin (Tally) [11], N (%) | non-GCB | 44 (58) |
| GCB | 32 (42) | |
| Double-hit score, N (%) | 0 | 35 (46) |
| 1 | 31 (41) | |
| 2 | 10 (13) | |
| Translocations N (%) | MYC | 5 (9) |
| BCL2 | 6 (11) |
Mutational data
In total, 317 somatic alterations were detected. Missense mutations were the most frequent, 232/317 (73%), followed by 40 (13%) nonsense mutations and 37 (12%) frameshift insertions/deletions (Fig. 1a–b and Additional file 1: Table S2). Sixty-eight percent (46/68) of the investigated genes were mutated at least once, and at least one somatic mutation was identified in 71/76 (93%) of investigated tumor samples. The most frequently mutated gene was KMT2D (34% of cases), followed by cytokine signaling 1 (SOCS1) (28%), GNA13 (17%), and B2M (15%) (Fig. 1c). EZH2 (p = 0.0176) along with GNA13 (p = 0.0181) and SGK1 (p = 0.028) were more frequently mutated in GCB–DLBCL. On average, GCB–DLBCL cases had more mutated genes than ABC-DLBCL cases (mean 3.9 vs 2.8, p = 0.018). However, there was no significant difference between COO subtypes considering the cumulative number of mutations per case (some genes had multiple mutations in the same tumor).
Fig. 1.
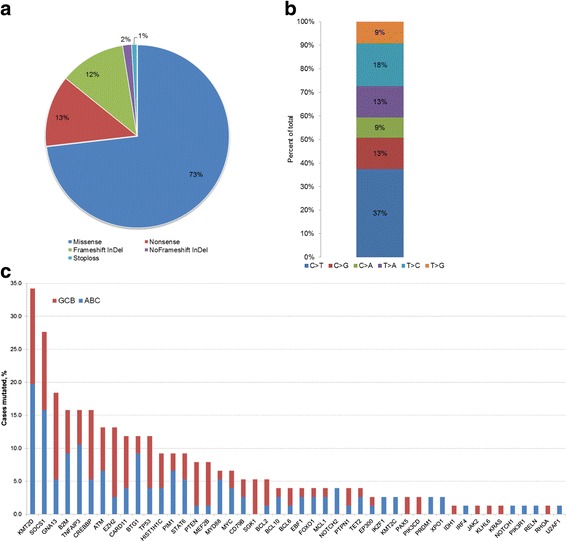
Overview of detected mutations. a Classification of the detected mutations according to their type. b Frequency of different nucleotide substitutions among all types of point mutations (n = 270). c Frequency of gene mutations in respect to cell-of-origin classification
We used the ensemble MetaLR score that integrates nine different functional prediction algorithms and population allele frequencies to predict the functional impact of the detected missense mutations. Ninety-eight percent of all missense mutations were successfully annotated, and 86/227 (38%) were reliably predicted to be deleterious. Functional effects did not distribute evenly among investigated genes. In some (GNA13, TP53, CREBBP, TNFAIP), all or nearly all substitutions were either nonsense or predicted damaging missense mutations, whereas in others (PIM1, BCL2), the majority of mutations were “neutral,” and thus likely played a passenger role in lymphomagenesis (Fig. 2).
Fig. 2.
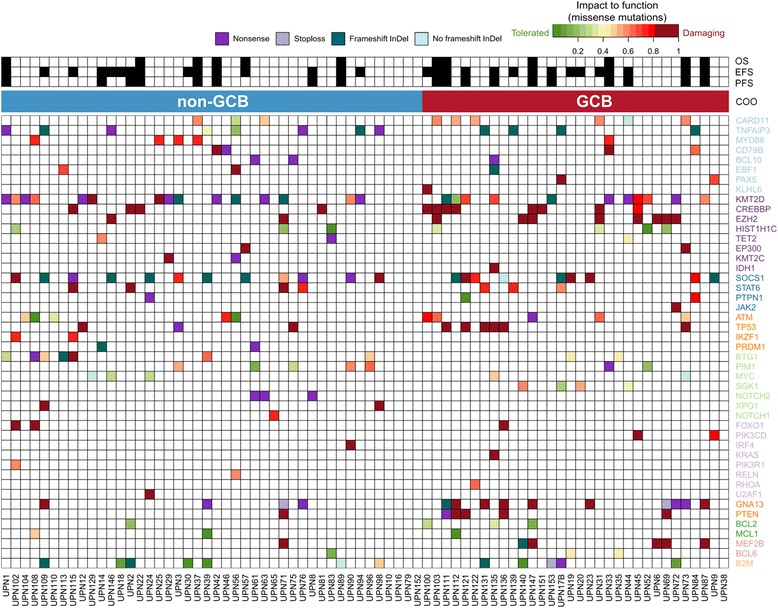
Overview of detected mutations and clinical endpoints. In the upper part of the figure, the occurrence of an event for each clinical endpoint is indicated by a filled-in square. In the lower part, mutations are color-coded by their type. Additionally, missense mutations are color-coded according to their predicted impact on protein function (MetaLR rank score, see the “Methods” section) Genes (in rows) are grouped according to their involvement in the cellular pathway and ordered according to their mutation frequencies within each group. Cases are ordered according to the cell-of-origin subtype. If multiple mutations occurred within one gene in the same case, the most damaging mutation is shown. COO cell-of-origin, EFS event-free survival, GCB germinal center B cell type, OS overall survival, PFS progression-free survival
Survival analysis
We performed Kaplan–Meier survival analysis of all genes that were mutated in at least 4 cases within our cohort and, also, on combinations of mutated genes that act on the same pathway. Only significant findings are reported.
SOCS1 mutated cases had better PFS compared to wild-type cases (p = 0.022), but the difference was not significant for OS and EFS (p = 0.11 and p = 0.182, respectively) (Fig. 3a). In total, 47 mutations of SOCS1 were detected, affecting 21 cases (12 non-GCB and 9 GCB). Mutations were distributed evenly along the coding sequence with two hotspots at Ala17 and Phe79 (four and three mutations, respectively) (Additional file 2: Figure S1). In all but 2 cases, at least one truncating (frameshift insertion/deletion or nonsense) or predicted damaging missense mutation was present, suggesting a deleterious effect on protein synthesis and/or function. Nineteen of 47 (40%) SOCS1 mutations were potentially induced by aberrant somatic hypermutation (aSHM), as they occurred within the RGYW/WRCY DNA sequence motif known to be targeted by activation-induced deaminase.
Fig. 3.
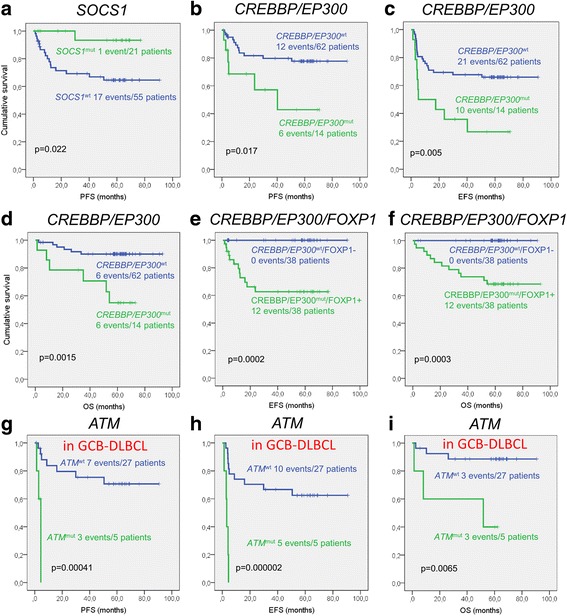
Survival analysis according to mutational status. a Progression-free survival (PFS) in SOCS1-mutated and non-mutated cases. b–d Comparison of overall survival (OS), PFS, and event-free survival (EFS) in CREBBP- or EP300-mutated and wild-type cases. e–f Separation between cases with good and bad prognosis was further increased by combining CREBBP or EP300 mutational status and FOXP1 protein overexpression. g–i Prognostic significance of ATM mutations in GCB–DLBCL
Since CREBBP and EP300 have close functional interactions in acetylating histone and nonhistone proteins, we decided to evaluate their combined prognostic value. Cases with mutations in CREBBP or EP300 had significantly worse OS, EFS, and PFS (p = 0.0015, p = 0.005, and p = 0.017, respectively) (Fig. 3b–d). In total, 14 cases were affected, with one heterozygous mutation in each case. All detected variants were missense substitutions localized in the acetyltransferase HAT domain with predicted deleterious effects on protein function (Additional file 2: Figure S1). However, the existence of more mutations cannot be excluded, since the applied gene panel only partly investigated the coding sequence of both CREBBP and EP300. CREBBP mutations alone had a negative impact on OS, but this did not reach statistical significance (p = 0.061). Analysis of potential linkage of these mutations to aSHM showed no such evidence.
In our previous study, we reported that overexpression of FOXP1 evaluated by IHC was prognostic of worse OS [13]. We combined mutation- and IHC-based prognostic markers to determine whether this improved identification of DLBCL cases with poor outcomes. Fifty-one percent (39/76) of cases overexpressed FOXP1 and/or had mutations of CREBBP/EP300. Five tumors shared both of these features. Survival analysis showed significantly worse OS, PFS, and EFS in cases with either FOXP1 overexpression or CREBBP or EP300 mutations (p = 0.0003, p = 0.006, and p = 0.0002, respectively) (Fig. 3e–f).
ATM mutations emerged as prognostic factor for worse EFS, PFS, and OS within the GCB–DLBCL subgroup (p = 0.000002, p = 0.00041, and p = 0.0065, respectively; Fig. 3g–i). In total, 11 mutations affecting 10 DLBCL cases were found (5 in GCB and 5 in non-GCB instances). Only in one case ATM mutations were reliably predicted to be deleterious, while the functional significance of other mutations remained unclear. Also, there was no evidence of targeting of this gene by aberrant somatic hypermutation as only 2/11 mutations occurred within the RGYW motif.
In multivariable analysis that included IPI and failure to achieve complete remission (FACR), CREBBP and EP300 mutational status (but not SOCS1 status) emerged as the most significant factor predicting worse OS (p = 0.013, HR 4.12, CI 1.38–14.15) (Table 3). While these mutations were also significant for predicting worse EFS and PFS (p = 0.021 and p = 0.041, respectively), FACR was a superior predictor of the latter two endpoints (p = 0.0006 and p = 0.001).
Table 3.
Stepwise Cox regression analysis of survival
| OS | EFS | PFS | ||||
|---|---|---|---|---|---|---|
| Variable | p value | HR (95% CI) | p value | HR (95% CI) | p value | HR (95% CI) |
| CREBBP mut or EP300 mut | 0.013 | 4.41 (1.38–14.16) | 0.021 | 2.53 (1.15–5.54) | 0.041 | 2.94 (1.05–8.27) |
| Failure to achieve complete remission | 0.032 | 0.27 (0.08–0.89) | 0.0006 | 0.22 (0.097–0.53) | 0.001 | 0.17 (0.056–0.49) |
| International Prognostic Index (IPI) | 0.86 | 0.96 (0.61–1.51) | 0.38 | 0.88 (0.66–1.17) | 0.646 | 1.09 (0.75–1.60) |
Discussion
There is a critical need for additional tumor-based prognostic markers in DLBCL to identify high-risk DLBCL patients prior to first treatment that might benefit from alternative risk-adjusted therapies. Tumor mutations represent promising candidates for outcome prognostication due to the unequivocal nature of results obtained by respective mutational detection techniques and the increased availability and robustness of HTS. We have demonstrated that targeted mutational analysis of a relatively small but uniformly treated and prospectively followed up patient cohort can reveal significant prognostic associations in DLBCL. This was only possible (a lesson learned) because of the proper trial design contemplating central collection of tissue for translational analysis as an integral study part. Additional lessons learned from this prospective trial such as the importance of a central diagnostic pathology review as well as handling of biological entities and subentities in the spectrum of so-called high grade B cell lymphomas have been discussed in a paper of ours recently published in this journal [13].
We found that deleterious mutations in two acetyltransferase genes, CREBBP and EP300, which belong to the KAT3 family of histone/protein lysine acetyltransferases, predict worse OS, PFS, and EFS in DLBCL independent of IPI and FACR. Point mutations or deletions of CREBBP/EP300 reportedly affect 39% of all DLBCL cases [31]. In line with other studies, we confirm that CREBBP mutations are more frequent than EP300 and tend to occur more often in GCB–DLBCL [18]. In the context of cancerogenesis, CREBBP and EP300 act as tumor suppressors. It has been shown that loss of one CREBBP allele leads to reduced acetylation and inactivation of p53, impaired expression of glucocorticoid-receptor-responsive genes, and upregulation of BCL6 [31, 32]. Finally, recent data suggest that heterozygous deleterious CREBBP mutations lead to decreased global histone H3 lysine 14 (H3K14), K18, and K27 acetylation and reduced MHC class II expression (Hashwah et al., currently under peer review). Association with poorer outcomes were suggested in studies that found CREBBP mutations in 20% of relapsed/refractory GCB–DLBCL [33], and in a large proportion of relapsed acute lymphoblastic leukemia patients [31, 34]. Despite this important clue, however, the prognostic value of CREBBP/EP300 mutations in DLBCL has not been previously reported.
SOCS1 mutations predicted excellent PFS in our cohort, but differences in OS and EFS were not significant. Suppressor of cytokine signaling 1 (SOCS1 ) is a known inhibitor of JAK/STAT-dependent signal transduction, which binds to phosphorylated JAK and marks it for proteosomal degradation [35, 36]. Mutations of SOCS1 have been previously shown to be associated with favorable survival in DLBCL [37] and are also frequently detected in Hodgkin lymphoma and primary mediastinal B cell lymphoma (PMBCL), both with a relatively favorable prognosis [38]. We also previously showed that SOCS1 mutations occurred exceptionally in non-relapsing primary DLBCL whereas being completely absent in relapsing DLBCL cases, supporting the association with favorable prognosis [27]. Schif et al. reported that DLBCL bearing truncating SOCS1 mutations have excellent OS, whereas those with only missense mutations have markedly worse prognosis [37]. Our data, however, does not confirm such distinction: 6 of 19 SOCS1-mutated cases had only missense mutations, but their PFS was equally favorable as those with truncating mutations. The SOCS1 mutational frequency in our cohort (28%) was higher than reported average (~13%) [39]. All detected mutations had relatively high variant allelic frequencies (median 28%, range 6–78%); therefore, our higher mutation rate cannot be explained by comparably high sequencing depth and sensitive detection of subclonal mutations that could have been missed by exome-scale sequencing studies. A potential explanation might be bias of our cohort toward better than average survival, e.g., due to study protocol exclusion of patients with performance status >2 on the ECOG scale, with symptomatic central nervous system disease, or with HIV, and/or hepatitis infection, thus being potentially enriched for cases with better prognosis that consequently more often bear SOCS1 mutations.
The prognostic value of multiple other gene mutations such as TP53, MYD88, FOXP1, and FOXP2 in DLBCL has been suggested previously [22, 23, 40, 41]. In our cohort, TP53-mutated cases had worse OS, but this difference did not reach statistical significance. Also, the low number (n = 5) of MYD88 L265P-mutated cases did not allow detection of any reliable prognostic associations. Analogously, despite ATM mutations were consistently associated with worse outcomes in GCB–DLBCL in our study, this observation was based on a small number of events and cases bearing mutations and therefore remains to be validated on other larger collectives. While we were unable to identify mutations of FOXP1, we previously reported that overexpression of the FOXP1 protein is associated with worse OS in the investigated DLBCL cohort [13]. Here, we show that combination of HTS- and IHC-based prognostic markers (CREBBP/EP300 mutations and FOXP1 overexpression) enables even better stratification between patients with good and worse prognosis.
It is unlikely that any single biomarker will significantly improve risk stratification in DLBCL due to the profound heterogeneity of this disease. This point is illustrated by 3 cases in our cohort with mutations of both SOCS1 and CREBBP (Fig. 2, cases UPN57, UPN112, and UPN115) that clinically behaved like CREBBP-mutated cases and had worse prognosis. It is more likely that a combination of several different types of markers established on uniformly treated prospective cohorts and selected by robust statistical methods would provide models that could be prospectively validated on larger DLBCL collections. A good example of such an updated composite prognostic model is the m7-FLIPI, a recently developed score for follicular lymphoma, which incorporates mutations and clinical factors and provides superior prognostication compared to traditionally used clinical factor-based FLIPI [42].
Conclusion
Deleterious mutations in the HAT domain of the acetyltransferases CREBBP and EP300 are associated with worse OS, PFS, and EFS in DLBCL. ATM mutations are prognostic for worse survival at all clinical endpoints in GCB–DLBCL, but due to low case number remains to be verified on larger collectives. The previously reported beneficial prognostic role of SOCS1 mutations in DLBCL is valid for predicting better PFS.
Acknowledgements
We are grateful to Prof. Anne Müller for her insights on the functional relevance of CREBBP/EP300 mutations and fruitful scientific discussions. Also, special thanks to Valeria Perrina and Sibylle Tschumi for their help in performing high-throughput sequencing.
Funding
The study was supported by the Oncosuisse grant Nr. OCS 02072-04-2007 and Krebsliga beider Basel.
Availability of data and materials
Targeted high-throughput sequencing data was deposited in the Sequence Read Archive (SRA) Ref Nr.: SRP098666.
Authors’ contributions
DJus performed the sequencing experiments and data analysis and wrote the manuscript. DJuc performed the sequencing experiments and analysis. DK and KM performed the statistical analysis and revised the manuscript. SD performed the immunohistochemistry analysis and revised the manuscript. AT designed the study; performed the immunohistochemistry, FISH, and statistical analysis; patrially wrote and revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Approval for the clinical trial was obtained from the ethics committee of each participating institution.
Consent for translational study was obtained from all patients, and it was approved by the Ethics Committee of Northwestern and Central Switzerland (EKNZ 2014-252).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ASCT
Autologous stem cell transplantation
- aSHM
Aberrant somatic hypermutation
- BAM
Binary sequence alignment/map format
- CI
Confidence interval
- COO
Cell-of-origin
- DLBCL
Diffuse large B cell lymphoma
- EBER
Epstein-Barr virus early RNA
- ECOG
Eastern Cooperative Oncology Group
- EFS
Event-free survival
- FACR
Failure to achieve complete remission
- FFPE
Formalin-fixed paraffin-embedded
- FISH
Fluorescence in situ hybridization
- FLIPI
Follicular Lymphoma International Prognostic Index
- GCB
Germinal center B cell
- HTS
High-throughput sequencing
- IHC
Immunohistochemistry
- IPI
International Prognostic Index
- OS
Overall survival
- PET/CT
Positron emission tomography/computed tomography
- PMBCL
Primary mediastinal B cell lymphoma
- PFS
Progression-free survival
- R-CHOP
Rituximab, cyclophosphamide, hydroxydaunorubicin, vincristine, and prednisone
Additional files
Statistics of high-throughput sequencing and variant calling. Table S2. Somatic mutations detected by high-throughput sequencing in 76 cases of primary DLBCL. (XLSX 384 kb)
Mapping of the detected mutations to the protein sequences within respective domains in the ten most frequently mutated and relevant genes is this study. Mutations in CREBBP and EP300 always occurred within the HAT domain responsible for acetylation. In SOCS1, mutations were distributed evenly along the protein sequence. Y-axis represents the number of mutations detected at a particular amino acid position. Green–missense mutation; black–frameshift insertion/deletion; purple–nonsense mutation. (PDF 442 kb)
Contributor Information
Darius Juskevicius, Email: darius.juskevicius@usb.ch.
David Jucker, Email: david.jucker@usb.ch.
Dirk Klingbiel, Email: dirk.klingbiel@sakk.ch.
Christoph Mamot, Email: christoph.mamot@ksa.ch.
Stephan Dirnhofer, Email: stephan.dirnhofer@usb.ch.
Alexandar Tzankov, Email: alexandar.tzankov@usb.ch.
References
- 1.Coiffier B, Thieblemont C, Van Den Neste E, Lepeu G, Plantier I, Castaigne S, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood. 2010;116:2040–5. doi: 10.1182/blood-2010-03-276246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedberg JW. Relapsed/refractory diffuse large B-cell lymphoma. Hematology. 2011;2011:498–505. doi: 10.1182/asheducation-2011.1.498. [DOI] [PubMed] [Google Scholar]
- 3.Costa LJ, Maddocks K, Epperla N, Reddy NM, Karmali R, Umyarova E, et al. Diffuse large B-cell lymphoma with primary treatment failure: ultra-high risk features and benchmarking for experimental therapies. Am J Hematol. 2017;92:161–70. doi: 10.1002/ajh.24615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hitz F, Connors JM, Gascoyne RD, Hoskins P, Moccia A, Savage KJ, et al. Outcome of patients with primary refractory diffuse large B cell lymphoma after R-CHOP treatment. Ann Hematol. 2015;94:1839–43. doi: 10.1007/s00277-015-2467-z. [DOI] [PubMed] [Google Scholar]
- 5.Gisselbrecht C, Glass B, Mounier N, Singh Gill D, Linch DC, Trneny M, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28:4184–90. doi: 10.1200/JCO.2010.28.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.A predictive model for aggressive non-Hodgkin’s lymphoma The International Non-Hodgkin’s Lymphoma Prognostic Factors Project. N Engl J Med. 1993;329:987–94. doi: 10.1056/NEJM199309303291402. [DOI] [PubMed] [Google Scholar]
- 7.Sehn LH, Berry B, Chhanabhai M, Fitzgerald C, Gill K, Hoskins P, et al. The revised International Prognostic Index (R-IPI) is a better predictor of outcome than the standard IPI for patients with diffuse large B-cell lymphoma treated with R-CHOP. Blood. 2007;109:1857–61. doi: 10.1182/blood-2006-08-038257. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Z, Sehn LH, Rademaker AW, Gordon LI, LaCasce AS, Crosby-Thompson A, et al. An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood. 2014;123:837–42. doi: 10.1182/blood-2013-09-524108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tzankov A, Xu-Monette ZY, Gerhard M, Visco C, Dirnhofer S, Gisin N, et al. Rearrangements of MYC gene facilitate risk stratification in diffuse large B-cell lymphoma patients treated with rituximab-CHOP. Mod Pathol. 2014;27:958–71. doi: 10.1038/modpathol.2013.214. [DOI] [PubMed] [Google Scholar]
- 10.Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359:2313–23. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer PN, Fu K, Greiner TC, Smith LM, Delabie J, Gascoyne RD, et al. Immunohistochemical methods for predicting cell of origin and survival in patients with diffuse large B-cell lymphoma treated with rituximab. J Clin Oncol. 2011;29:200–7. doi: 10.1200/JCO.2010.30.0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott DW, Wright GW, Williams PM, Lih C-J, Walsh W, Jaffe ES, et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin embedded tissue. Blood. 2014;123:1214–7. doi: 10.1182/blood-2013-11-536433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tzankov A, Leu N, Muenst S, Juskevicius D, Klingbiel D, Mamot C, et al. Multiparameter analysis of homogeneously R-CHOP-treated diffuse large B cell lymphomas identifies CD5 and FOXP1 as relevant prognostic biomarkers: report of the prospective SAKK 38/07 study. J Hematol Oncol. 2015;8:70. doi: 10.1186/s13045-015-0168-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyazaki K, Yamaguchi M, Suzuki R, Kobayashi Y, Maeshima AM, Niitsu N, et al. CD5-positive diffuse large B-cell lymphoma: a retrospective study in 337 patients treated by chemotherapy with or without rituximab. Ann Oncol Off J Eur Soc Med Oncol. 2011;22:1601–7. doi: 10.1093/annonc/mdq627. [DOI] [PubMed] [Google Scholar]
- 15.Salles G, de Jong D, Xie W, Rosenwald A, Chhanabhai M, Gaulard P, et al. Prognostic significance of immunohistochemical biomarkers in diffuse large B-cell lymphoma: a study from the Lunenburg Lymphoma Biomarker Consortium. Blood. 2011;117:7070–8. doi: 10.1182/blood-2011-04-345256. [DOI] [PubMed] [Google Scholar]
- 16.Winter JN, Li S, Aurora V, Variakojis D, Nelson B, Krajewska M, et al. Expression of p21 protein predicts clinical outcome in DLBCL patients older than 60 years treated with R-CHOP but not CHOP: a prospective ECOG and Southwest Oncology Group correlative study on E4494. Clin Cancer Res. 2010;16:2435–42. doi: 10.1158/1078-0432.CCR-09-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song G, Ni H, Zou L, Wang S, Tian F, Liu H, et al. Expression of CD40 is a positive prognostic factor of diffuse large B-cell lymphoma treated with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) Onco Targets Ther. 2016;9:3799–805. doi: 10.2147/OTT.S96992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43:830–7. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morin RD, Mungall K, Pleasance E, Mungall AJ, Goya R, Huff RD, et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood. 2013;122:1256–65. doi: 10.1182/blood-2013-02-483727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110:1398–403. doi: 10.1073/pnas.1205299110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasqualucci L, Dalla-Favera R. The genetic landscape of diffuse large B-cell lymphoma. Semin Hematol. 2015;52:67–76. doi: 10.1053/j.seminhematol.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu W, et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood. 2012;120:3986–96. doi: 10.1182/blood-2012-05-433334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernández-Rodríguez C, Bellosillo B, García-García M, Sánchez-González B, Gimeno E, Vela MC, et al. MYD88 (L265P) mutation is an independent prognostic factor for outcome in patients with diffuse large B-cell lymphoma. Leukemia. 2014;28:2104–6. doi: 10.1038/leu.2014.184. [DOI] [PubMed] [Google Scholar]
- 24.Trinh DL, Scott DW, Morin RD, Mendez-Lago M, An J, Jones SJM, et al. Analysis of FOXO1 mutations in diffuse large B-cell lymphoma. Blood. 2013;121:3666–74. doi: 10.1182/blood-2013-01-479865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mamot C, Klingbiel D, Hitz F, Renner C, Pabst T, Driessen C, et al. Final results of a prospective evaluation of the predictive value of interim positron emission tomography in patients with diffuse large B-cell lymphoma treated with R-CHOP-14 (SAKK 38/07) J Clin Oncol. 2015;33:2523–9. doi: 10.1200/JCO.2014.58.9846. [DOI] [PubMed] [Google Scholar]
- 26.Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–86. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 27.Juskevicius D, Lorber T, Gsponer J, Perrina V, Ruiz C, Stenner-Liewen F, et al. Distinct genetic evolution patterns of relapsing diffuse large B-cell lymphoma revealed by genome-wide copy number aberration and targeted sequencing analysis. Leukemia. 2016;30:2385–95. doi: 10.1038/leu.2016.135. [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Mutat. 2016;37:235–41. doi: 10.1002/humu.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24:2125–37. doi: 10.1093/hmg/ddu733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCall CM, Mosier S, Thiess M, Debeljak M, Pallavajjala A, Beierl K, et al. False positives in multiplex PCR-based next-generation sequencing have unique signatures. J Mol Diagn. 2014;16:541–9. doi: 10.1016/j.jmoldx.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–95. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–9. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morin RD, Assouline S, Alcaide M, Mohajeri A, Johnston RL, Chong L, et al. Genetic landscapes of relapsed and refractory diffuse large B-cell lymphomas. Clin Cancer Res. 2016;22:2290–300. doi: 10.1158/1078-0432.CCR-15-2123. [DOI] [PubMed] [Google Scholar]
- 34.Inthal A, Zeitlhofer P, Zeginigg M, Morak M, Grausenburger R, Fronkova E, et al. CREBBP HAT domain mutations prevail in relapse cases of high hyperdiploid childhood acute lymphoblastic leukemia. Leukemia. 2012;26:1797–803. doi: 10.1038/leu.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamizono S, Hanada T, Yasukawa H, Minoguchi S, Kato R, Minoguchi M, et al. The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. J Biol Chem. 2001;276:12530–8. doi: 10.1074/jbc.M010074200. [DOI] [PubMed] [Google Scholar]
- 36.Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921–4. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- 37.Schif B, Lennerz JK, Kohler CW, Bentink S, Kreuz M, Melzner I, et al. SOCS1 mutation subtypes predict divergent outcomes in diffuse large B-cell lymphoma (DLBCL) patients. Oncotarget. 2013;4:35–47. doi: 10.18632/oncotarget.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steidl C, Gascoyne RD. The molecular pathogenesis of primary mediastinal large B-cell lymphoma. Blood. 2011;118:2659–69. doi: 10.1182/blood-2011-05-326538. [DOI] [PubMed] [Google Scholar]
- 39.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–11. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gascoyne DM, Banham AH. The significance of FOXP1 in diffuse large B-cell lymphoma. Leuk Lymphoma. 2016;58:1037–51. doi: 10.1080/10428194.2016.1228932. [DOI] [PubMed] [Google Scholar]
- 41.Wong KK, Gascoyne DM, Soilleux EJ, Lyne L, Spearman H, Roncador G, et al. FOXP2-positive diffuse large B-cell lymphomas exhibit a poor response to R-CHOP therapy and distinct biological signatures. Oncotarget. 2016;7:52940–56. doi: 10.18632/oncotarget.9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pastore A, Jurinovic V, Kridel R, Hoster E, Staiger AM, Szczepanowski M, et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015;16:1111–22. doi: 10.1016/S1470-2045(15)00169-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Targeted high-throughput sequencing data was deposited in the Sequence Read Archive (SRA) Ref Nr.: SRP098666.