

Abstract
Background
Hepatocellular carcinoma (HCC) is the most common liver malignancy worldwide. However, present studies of its multiple gene interaction and cellular pathways still could not explain the initiation and development of HCC perfectly. To find the key genes and miRNAs as well as their potential molecular mechanisms in HCC, microarray data GSE22058, GSE25097, and GSE57958 were analyzed.
Methods
The microarray datasets GSE22058, GSE25097, and GSE57958, including mRNA and miRNA profiles, were downloaded from the GEO database and were analyzed using GEO2R. Functional and pathway enrichment analyses were performed using the DAVID database, and the protein–protein interaction (PPI) network was constructed using the Cytoscape software. Finally, miRDB was applied to predict the targets of the differentially expressed miRNAs (DEMs).
Results
A total of 115 differentially expressed genes (DEGs) were found in HCC, including 52 up-regulated genes and 63 down-regulated genes. The gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses from DAVID showed that up-regulated genes were significantly enriched in chromosome segregation and cell division, while the down-regulated genes were mainly involved in complement activation, protein activation cascades, carboxylic acid metabolic processes, oxoacid metabolic processes, and the immune response. From the PPI network, the 18 nodes with the highest degree were screened as hub genes. Among them, ESR1 was found to have close interactions with FOXO1, CXCL12, and GNAO1. In addition, a total of 64 DEMs were identified, which included 58 up-regulated miRNAs and 6 down-regulated miRNAs. ESR1 was potentially targeted by five miRNAs, including hsa-mir-18a and hsa-mir-221.
Conclusions
The roles of DEMs like hsa-mir-221 in HCC through interactions with DEGs such as ESR1 and CXCL12 may provide new clues for the diagnosis and treatment of HCC patients.
Electronic supplementary material
The online version of this article (doi:10.1186/s12957-017-1127-2) contains supplementary material, which is available to authorized users.
Keywords: Liver cancer, Microarray analysis, Differentially expressed genes, miRNA, Protein–protein interaction network
Background
HCC is the most common primary liver malignancy and is one of the leading causes of cancer-related deaths around the world. In Asia, there are more than 580,000 new cases expected every year [1]. As well as other carcinomas, gene aberrations, cellular context, and environmental influences are believed to be the reason of the occurrence, progression, and metastasis of HCC. Although the study of the multiple genes and cellular pathways that take part in the initiation and development of HCC has been discussed for many years, the therapy for HCC accurately still remains scarcely. Accordingly, it is crucial to investigate the molecular mechanisms involved in the proliferation, apoptosis, and invasion of HCC for the improvement of diagnostic and therapeutic strategies.
In recent years, the microarray, a high-throughput platform for analysis of gene expression, has been extensively conducted as an efficient tool for the identification of general genetic alteration during tumorigenesis [2]. While studies of DEGs and DEMs of HCC have been performed in the past decades and some of their functions in different pathways, biological processes, or molecular functions have been reported, there remain questions about how the DEGs and microRNAs interact through molecular pathways because of limitations on the comparative analysis of the DEGs in independent studies. Currently, due to bioinformatics methods, we can finally deal with the data generated by microarray technology and find the interactions among DEGs and microRNAs, especially the pathways in the interaction network, to conclude their potential mechanisms in HCC.
In this study, we chose three gene expression profiles (GSE22058, GSE25097, and GSE57958), which were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), to obtain DEGs and DEMs between liver cancer tissues and normal tissue samples. Then, functional enrichment and network analyses were applied to identify the DEGs, which were combined with mRNA–microRNA interaction analysis, to describe the key genes and miRNAs as well as their potential molecular mechanisms in HCC.
Methods
Collection and inclusion criteria of studies
We searched the GEO database (https://www.ncbi.nlm.nih.gov/geo/) for publicly available studies from January 1, 2010 to October 30, 2016 using the following keywords: “hepatocellular carcinoma” (study keyword), “Homo sapiens” (organism), “Expression profiling by array” (study type), “70 to 3000” (sample count) and “tissue” (attribute name). After a systematic review, 13 GSE studies were retrieved. The inclusion criteria for studies were as follows: (1) samples diagnosed with HCC tissue samples and normal tissue samples, (2) gene expression profiling of mRNA, (3) sample count of each group are more than 35, and (4) sufficient information to perform the analysis. Then, three gene expression profiles (GSE22058, GSE25097, and GSE57958) were collected for analysis.
Microarray data
Three gene expression profiles (GSE22058, GSE25097, and GSE57958) were downloaded from the GEO database. The array data for GSE25097 included 268 HCC tissue samples and 243 normal tissue samples [3]. The array data for GSE57958 consisted of 39 HCC tissue samples and 39 normal tissue samples [4]. The array data for GSE22058 consisted of one mRNA expression profile (including 100 HCC tissue samples and 97 normal tissue samples) and a miRNA expression profile (including 96 HCC tissue samples and 96 normal tissue samples) [5].
Data processing
GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) is an interactive web tool for comparing two groups of data that can analyze any GEO series [6]. GEO2R was applied to screen differentially expressed mRNAs and miRNAs between HCC and normal tissue samples. The adjusted P values (adj. P) using Benjamini and Hochberg (BH) false discovery rate (FDR) method by default were applied to correct for the occurrence of false positive results. An adj. P < 0.05 and a |logFC| ≥ 1 were set as the cut-off criteria. Heat map of DEGs was generated using the online tool Morpheus (https://software.broadinstitute.org/morpheus/).
Functional and pathway enrichment analysis
Gene ontology (GO) is a common method for annotating genes, gene products and sequences to underlying biological phenomena [7, 8]; the Kyoto Encyclopedia of Genes and Genomes (KEGG) is an integrated database resource for biological interpretation of genome sequences and other high-throughput data [9]. Both analyses were available in the DAVID database (https://david.ncifcrf.gov/), which is a bioinformatics data resource composed of an integrated biology knowledge base and analysis tools to extract meaningful biological information from large quantities of genes and protein collections [10]. GO and KEGG analyses were performed using the DAVID database to identify DEGs. A P value <0.05 was set as the cut-off criterion.
PPI network construction and analysis of modules
The STRING database (http://string-db.org/) is an online software that aims to provide a critical assessment and integration of protein–protein interactions, including direct (physical) and indirect (functional) associations [11]. Cytoscape is a popular open-source software tool for the visual exploration of biomolecule interaction networks composed of protein, gene, and other types of interactions [12]. The DEGs were mapped to STRING to evaluate the PPI information and then visualized with Cytoscape. A combined score >0.15 was set as the cut-off criterion. To screen the hub genes, node degree ≥10 was set as the cut-off criterion. Then, the Molecular Complex Detection (MCODE) plug-in was used to screen modules of hub genes from the PPI network with degree cut-off = 10, haircut on, node score cut-off = 0.2, k-core = 2, and max. depth = 100. Moreover, the functional and pathway enrichment analyses of DEGs in each module were performed by DAVID. A P value <0.05 was set as the cut-off criterion.
Prediction of miRNA targets
The target genes of the DEMs from GSE22058 were predicted with miRDB (http://mirdb.org/miRDB/), which is an online database for predicting microRNA targets [13]. The target genes were aligned with the DEGs to obtain an intersection for further analysis.
Results
Identification of DEGs
A total of 2021, 1097, and 409 DEGs were identified after the analyses of the GSE22058, GSE25097, and GSE57958 datasets, respectively (Additional files 1, 2, and 3). Among them, 116 genes were found in all three datasets (Fig. 1). Of these, 115 gene expressions were matched, including 52 up-regulated genes and 63 down-regulated genes in HCC tissue samples compared with normal liver tissue samples.
Fig. 1.
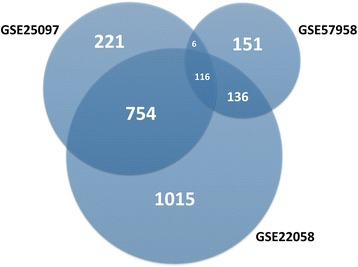
Identification of differentially expressed genes (DEGs) in mRNA expression profiling datasets GSE22058, GSE25097, and GSE57958
Functional and pathway enrichment analyses
To further understand the function and mechanism of the identified DEGs, functional and pathway enrichment analyses, including GO and KEGG, were performed using DAVID. The GO term enrichment analysis showed that in the biological processes-associated category, the up-regulated genes were significantly enriched in chromosome segregation and cell division, while the down-regulated genes were mainly involved in complement activation, protein activation cascades, carboxylic acid metabolic processes, oxoacid metabolic processes, and the immune response (Table 1). In addition, cell component analysis showed that the up-regulated genes were enriched in the chromosome, cytosol, proteinaceous extracellular matrix, and basement membrane collagen trimer, and that the down-regulated genes were mainly found in the collagen trimer, blood microparticles, membrane attack complexes, membrane-bounded vesicles, and the extracellular region part (Table 1). Moreover, for molecular function, the up-regulated genes were enriched in histone deacetylase binding, and the down-regulated genes were enriched in steroid binding, aromatase activity, oxidoreductase activity, monooxygenase activity, and serine-type endopeptidase activity (Table 1). Furthermore, the KEGG pathway analysis showed that the up-regulated genes were significantly enriched in ECM-receptor interaction, while four pathways were overrepresented in the down-regulated genes: chemical carcinogenesis, steroid hormone biosynthesis, retinol metabolism, and drug metabolism—cytochrome P450 (Table 1).
Table 1.
Functional and pathway enrichment analysis of up-regulated and down-regulated genes in hepatocellular carcinoma (HCC) tissue
| Category | Term | Count | % | P Value |
|---|---|---|---|---|
| Up-regulated | ||||
| GOTERM_BP_FAT | GO:0007059 ~ chromosome segregation | 9 | 17.6 | 2.66E-06 |
| GOTERM_BP_FAT | GO:0098813 ~ nuclear chromosome segregation | 8 | 15.7 | 1.06E-05 |
| GOTERM_BP_FAT | GO:0000819 ~ sister chromatid segregation | 7 | 13.7 | 2.91E-05 |
| GOTERM_BP_FAT | GO:0051301 ~ cell division | 9 | 17.6 | 1.31E-04 |
| GOTERM_BP_FAT | GO:0007067 ~ mitotic nuclear division | 8 | 15.7 | 1.44E-04 |
| GOTERM_CC_FAT | GO:0000793 ~ condensed chromosome | 5 | 9.8 | 2.14E-03 |
| GOTERM_CC_FAT | GO:0005829 ~ cytosol | 18 | 35.3 | 3.98E-03 |
| GOTERM_CC_FAT | GO:0005578 ~ proteinaceous extracellular matrix | 5 | 9.8 | 1.51E-02 |
| GOTERM_CC_FAT | GO:0098651 ~ basement membrane collagen trimer | 2 | 3.9 | 2.08E-02 |
| GOTERM_CC_FAT | GO:0005694 ~ chromosome | 7 | 13.7 | 3.38E-02 |
| GOTERM_MF_FAT | GO:0042826 ~ histone deacetylase binding | 3 | 5.9 | 3.03E-02 |
| KEGG_PATHWAY | hsa04512:ECM-receptor interaction | 3 | 5.9 | 1.68E-02 |
| Down-regulated | ||||
| GOTERM_BP_FAT | GO:0006956 ~ complement activation | 6 | 10.0 | 1.83E-05 |
| GOTERM_BP_FAT | GO:0072376 ~ protein activation cascade | 6 | 10.0 | 6.43E-05 |
| GOTERM_BP_FAT | GO:0019752 ~ carboxylic acid metabolic process | 12 | 20.0 | 8.21E-05 |
| GOTERM_BP_FAT | GO:0043436 ~ oxoacid metabolic process | 12 | 20.0 | 8.66E-05 |
| GOTERM_BP_FAT | GO:0006955 ~ immune response | 16 | 26.7 | 1.23E-04 |
| GOTERM_CC_FAT | GO:0005581 ~ collagen trimer | 4 | 6.7 | 7.15E-03 |
| GOTERM_CC_FAT | GO:0072562 ~ blood microparticle | 4 | 6.7 | 1.91E-02 |
| GOTERM_CC_FAT | GO:0005579 ~ membrane attack complex | 2 | 3.3 | 2.67E-02 |
| GOTERM_CC_FAT | GO:0031988 ~ membrane-bounded vesicle | 21 | 35.0 | 4.60E-02 |
| GOTERM_CC_FAT | GO:0044421 ~ extracellular region part | 22 | 36.7 | 5.00E-02 |
| GOTERM_MF_FAT | GO:0005496 ~ steroid binding | 4 | 6.7 | 3.47E-03 |
| GOTERM_MF_FAT | GO:0070330 ~ aromatase activity | 3 | 5.0 | 3.57E-03 |
| GOTERM_MF_FAT | GO:0016712 ~ oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen | 3 | 5.0 | 4.11E-03 |
| GOTERM_MF_FAT | GO:0004497 ~ monooxygenase activity | 4 | 6.7 | 4.77E-03 |
| GOTERM_MF_FAT | GO:0004252 ~ serine-type endopeptidase activity | 5 | 8.3 | 7.38E-03 |
| KEGG_PATHWAY | hsa05204:Chemical carcinogenesis | 6 | 10.0 | 4.53E-05 |
| KEGG_PATHWAY | hsa00140:Steroid hormone biosynthesis | 4 | 6.7 | 3.04E-03 |
| KEGG_PATHWAY | hsa00830:Retinol metabolism | 4 | 6.7 | 4.19E-03 |
| KEGG_PATHWAY | hsa00982:Drug metabolism - cytochrome P450 | 4 | 6.7 | 4.76E-03 |
Count: the number of enriched genes in each term
If there were more than five terms enriched in this category, the top five terms were selected per the P value
PPI network construction and analysis of modules
Ninety-seven nodes and 319 edges were mapped in the PPI network of identified DEGs, including 43 up-regulated genes and 54 down-regulated genes (Fig. 2a). The 18 nodes with the higher degrees were screened as hub genes, including TOP2A, FOS, TK1, CDC20, ESR1, CCNB2, CXCL12, FOXO1, HMMR, VWF, ACSM3, COL4A1, ZIC2, RFC4, TXNRD1, GNAO1, CYP3A4, and RAP2A (Table 2). Hub genes expression heat map in GSE25097 is shown in Fig. 2c. Among these, ESR1 was found to have close interactions with FOS, FOXO1, CXCL12, and GNAO1; CDC20 had interactions with CCNB2, CDCA5, CENPM, and GMNN (combined score > 0.90). A significant module including 16 nodes and 58 edges was obtained using MCODE (Fig. 2b). GO term enrichment analysis showed that in biological processes, the genes in this module were mainly associated with the cell cycle and responses to oxidation reactions and steroid hormones (Table 3). The genes were significantly enriched in the nucleoplasm, cytosol, and chromosome by cell component analysis (Table 3). The molecular function analysis showed that the genes were mainly involved in the binding of macromolecular complexes, chromatin, enzymes, RNA polymerases, and carbohydrate derivatives (Table 3). The KEGG analysis showed that the genes were mainly enriched in ECM-receptor interactions (Table 3).
Fig. 2.
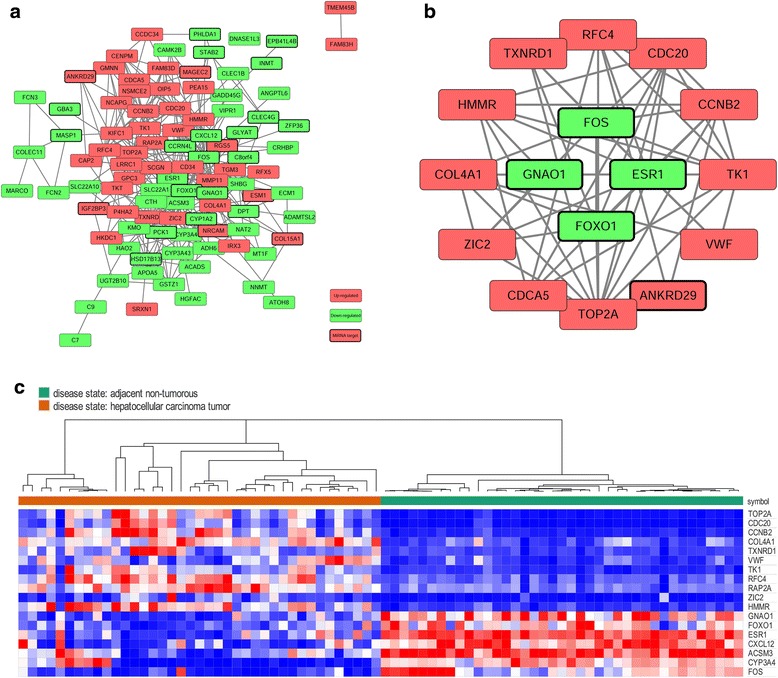
Protein–protein interaction (PPI) network and hub genes. a PPI network of differentially expressed genes (DEGs). b A significant module selected from the PPI network. Red nodes denote up-regulated genes, while green nodes denote down-regulated genes. Black border shows that the gene is a potential target for differentially expressed miRNAs (DEMs). The lines represent an interaction relationship between the nodes. c Hub genes expression heat map (11 up-regulated genes and 7 down-regulated genes) in GSE25097. Red: up-regulation; purple: down-regulation
Table 2.
Hub genes and their related DEMs
| Gene symbol | Node degree | Related DEMs |
|---|---|---|
| TOP2A | 30 | none |
| FOS | 27 | hsa-miR-221 |
| TK1 | 24 | none |
| CDC20 | 23 | none |
| ESR1 | 21 | hsa-miR-148b, hsa-miR-221, hsa-miR-18a, hsa-miR-181b, hsa-miR-19a |
| CCNB2 | 18 | none |
| CXCL12 | 17 | hsa-miR-221 |
| FOXO1 | 16 | hsa-miR-135b, hsa-miR-324-3p, hsa-miR-369-3p |
| HMMR | 15 | none |
| VWF | 15 | none |
| ACSM3 | 14 | none |
| COL4A1 | 14 | none |
| ZIC2 | 14 | none |
| RFC4 | 13 | none |
| TXNRD1 | 13 | none |
| GNAO1 | 12 | hsa-miR-193a |
| CYP3A4 | 10 | none |
| RAP2A | 10 | none |
Only top 18 DEGs with higher node degrees were showed in this table. Node degree: the number of edges incident to the node; Related miRNAs: miRNAs may potentially target the gene
Table 3.
Functional and pathway enrichment analysis of the genes in the module
| Category | Term | Count | % | P Value |
|---|---|---|---|---|
| GOTERM_BP_FAT | GO:0000302 ~ response to reactive oxygen species | 4 | 26.7 | 8.25E-04 |
| GOTERM_BP_FAT | GO:1901700 ~ response to oxygen-containing compound | 7 | 46.7 | 1.06E-03 |
| GOTERM_BP_FAT | GO:0006979 ~ response to oxidative stress | 4 | 26.7 | 4.59E-03 |
| GOTERM_BP_FAT | GO:0048545 ~ response to steroid hormone | 4 | 26.7 | 4.78E-03 |
| GOTERM_BP_FAT | GO:1903047 ~ mitotic cell cycle process | 5 | 33.3 | 5.21E-03 |
| GOTERM_CC_FAT | GO:0005654 ~ nucleoplasm | 9 | 60.0 | 1.54E-03 |
| GOTERM_CC_FAT | GO:0005829 ~ cytosol | 8 | 53.3 | 1.94E-02 |
| GOTERM_CC_FAT | GO:0005694 ~ chromosome | 4 | 26.7 | 4.75E-02 |
| GOTERM_MF_FAT | GO:0044877 ~ macromolecular complex binding | 9 | 60.0 | 4.65E-06 |
| GOTERM_MF_FAT | GO:0003682 ~ chromatin binding | 6 | 40.0 | 5.72E-05 |
| GOTERM_MF_FAT | GO:0019899 ~ enzyme binding | 6 | 40.0 | 1.67E-02 |
| GOTERM_MF_FAT | GO:0000982 ~ transcription factor activity, RNA polymer | 3 | 20.0 | 3.72E-02 |
| GOTERM_MF_FAT | GO:0097367 ~ carbohydrate derivative binding | 6 | 40.0 | 4.13E-02 |
| KEGG_PATHWAY | hsa04512:ECM-receptor interaction | 3 | 20.0 | 9.53E-03 |
Count: the number of enriched genes in each term
If there were more than five terms enriched in this category, the top five terms were selected per the P value
miRNA-DEG pairs
A total number of 64 DEMs between HCC tissue samples and normal tissue samples were identified after the analyses of the GSE22058 datasets (Additional file 4), including 58 up-regulated miRNAs and 6 down-regulated miRNAs (Fig. 3). The miRDB database was used to predict target genes of the identified DEMs (Table 4). By comparing the target gene to the DEGs, we screened the genes with a consistent expression trend for further analysis. miRNA-221, one of the most significantly up-regulated miRNAs, was found to target ESR1, FOS, and CXCL12. In addition, miRNA-142-5p, one of the main down-regulated miRNAs, potentially targeted ANKRD29, IGF2BP3, and IGSF3. Moreover, we found that ESR1 was potentially targeted by five miRNAs, including hsa-mir-148b, hsa-mir-181b, hsa-mir-18a, hsa-mir-19a, and hsa-mir-221 (Table 2). In addition, FOXO1 was the potential target of hsa-mir-135b, hsa-mir-324, and hsa-mir-369.
Fig. 3.
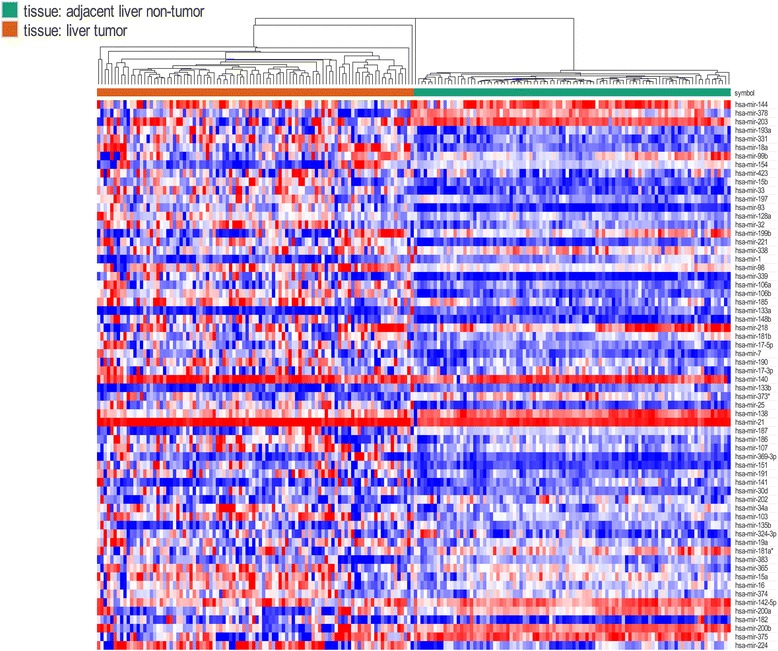
Heat map of the differentially expressed miRNAs (DEMs) (58 up-regulated genes and 6 down-regulated genes). Red: up-regulation; purple: down-regulation
Table 4.
DEMs in HCC tissue and their potential target genes
| miRNA | Adj. P | logFC | Target genes |
|---|---|---|---|
| hsa-mir-106b | 1.94E-41 | 1.49 | ANKRD29, IL1RAP, ADARB1, ARID4B, EPHA4, FBXL5, PDCD1LG2, PKD2, PTPN4, SLC40A1 |
| hsa-mir-148b | 9.59E-22 | 1.37 | ESR1, ABCB7, B4GALT6, CDK19, LDLR, MXD1, NPTN, RPS6KA5, SOS2, SZRD1 |
| hsa-mir-151 | 2.61E-21 | 1.14 | AGO2, ZFAND5, CLK1, AQP4, CASD1, RPS6KA5, FAM104A, NAA15, YTHDF3, HIF1A |
| hsa-mir-221 | 7.37E-21 | 1.66 | ESR1, FOS, CXCL12, CDKN1B, GABRA1, PANK3, TCF12, HECTD2, RFX7, TMCC1 |
| hsa-mir-18a | 2.16E-20 | 2.48 | ESR1, NEDD9, BBX, INADL, MAP7D1, PHF19, RORA, ZBTB47, CDK19, DICER1 |
| hsa-mir-200b | 4.50E-17 | −3.15 | MAGEC2, ESM1, TCEB1, TRIM33, LHFP, PTPN21, ARHGAP6, VASH2, HIPK3, NR5A2 |
| hsa-mir-224 | 4.85E-15 | −71.38 | NRCAM, CPNE8, ZNF207, ACSL4, RNF144B, SH3KBP1, RNF38, SLC4A4, GPR158, GGNBP2 |
| hsa-mir-200a | 5.03E-14 | −2.29 | COL15A1, MYBL1, ZEB2, ATP8A1, DCP2, TMEM170B, ZBTB34, DUSP3, TRHDE, RSAD2 |
| hsa-mir-182 | 1.41E-06 | −2.69 | NRCAM, ABHD13, MFAP3, PCNX, NADK2, CAMSAP2, SLC39A9, NCALD, ANK3, HOXA9 |
| hsa-mir-142-5p | 6.59E-06 | −1.49 | ANKRD29, IGF2BP3, IGSF3, ZFP36, ZFPM2, BAI3, AFF4, DIAPH2, AHR, ARID4B |
Adj. P: adjusted P value, FC: fold change
Positive logFC values denote up-regulated miRNAs, while negative logFC values denote down-regulated miRNAs. If there were more than ten genes predicted by miRDB, only ten genes were listed in the table
Discussion
Although people have continuously studied HCC, the early diagnosis and treatment of HCC is still a large problem due to the lack of understanding of the molecular mechanisms that drive the occurrence and development of HCC. Therefore, in-depth research into the factors and mechanisms of HCC progression are necessary for HCC diagnosis and treatment. Due to well-developed microarray technology, it is easier to determine the general genetic alterations in the progression of diseases, which can allow for the identification of gene targets for diagnosis, therapy, and prognosis of tumors.
In our study, a total of 115 DEGs were screened, including 52 up-regulated genes and 63 down-regulated genes. The up-regulated genes were enriched in chromosome segregation and cell division, while the down-regulated genes were mainly involved in complement activation, protein activation cascades, carboxylic acid metabolic processes, oxoacid metabolic processes, and the immune response. Moreover, by constructing the PPI, we identified high degree genes including ESR1, which was found to have close interactions with FOXO1, FOS, CXCL12, and GNAO1.
ESR1 could encode the transcription factor that enhances the response to various stimuli such as estrogen and growth factors in different kinds of tissue types [14]. Some researchers noticed that it may play a potential tumor suppressor features in HCC with its novel associations among the hepatocyte-specific pathways [15]. Moreover, there are also some reports claiming that ESR1 could suppress the inflammatory process mediated by interleukin-6 and reduce hepatic injury and the compensatory proliferation of hepatocytes [16]. Recent research has shown that cancers such as non-small cell lung cancer (NSCLC) also have very low-expression level of ESR1 [17]. FOXO, which has four members including FOXO1, is one of the 19 kinds of forkhead box transcription factors and is mainly express in mammals [18, 19]. Reports showed that the overexpression of FOXO could inhibit the growth and size of tumors [18, 20]. Some research on breast cancer also claimed that when FOXO proteins accumulate in the nucleus, they could suspend cell cycle progression and promote apoptosis [20, 21]. Recent research on FOXO proteins provides a new viewpoint that they may possess antitumor properties in HCC, inducing the expression of pro-apoptotic genes and interfering with signaling cascades, such as the Wnt/β-catenin, PI3K/AKT/mTOR, or MAPK pathways that are commonly changed in HCC [21, 22]. Meanwhile, FOS is reported as an oncogene in several kinds of cancers such as bladder cancer and HCC [23, 24]. There is also a recent research that shows that FOS has a high expression in HCC cell lines [25]. Yet in this work, we found it as a low-expression hub gene through the Microarray data analysis. Therefore it may need a further test in the future experiments and is excluded from the work this time. However, the molecular mechanism of ESR1 for HCC and the relationship of ESR1 and FOXO1 were rarely explored. In our study, we found that ESR1 and FOXO1 both had low-expression levels and had an interaction, indicating a joint function in HCC.
Stromal-derived factor 1 alpha, also known as CXCL12, is a specific ligand for CXCR4 and CXCR7. These three proteins together drive the migration of progenitor cells in embryonic development. Some invasion-related or metastasis-related pathways, such as cytokine-cytokine receptor interaction and axon guidance, contain CXCL12 as an important role in the regulation of themselves [26]. Several studies have shown that CXCL12 has a lower expression level in HCC tissue than in normal liver tissue [27]. There is also a study showing that CXCL12 and CXR4 may play significant roles in the metastasis of HCC by promoting the migration of tumor cells [28]. GNAO1 is a member of the subunit family of Gα proteins. As a molecular switch that controls signal transduction, the deregulation of GNAO1 can facilitate oncogenesis [29]. Kan et al. found that the role of mutant GNAO1 in oncogenesis might be to act as a tumor suppressor gene [30]. Jia et al. combined an integrated CNA (chromosomal copy number alteration) analysis with gene expression data to demonstrate that GNAO1 may play a key role in the pathogenesis of HCC [31]. Meanwhile, some research has shown that GNAO1 also plays a significant role in breast cancer and hepatocellular carcinoma [32]. Above all, these results suggest that ESR1, FOXO1, CXCL12, and GNAO1 are involved in the pathogenesis of carcinoma by affecting cell division, complement activation, and protein activation cascades, which support our findings.
Increasing evidence has shown that the dysregulation of miRNAs is an important part of the pathogenesis of multiple cancer types, including HCC. In our study, we identified 64 DEMs, including 58 up-regulated and 6 down-regulated miRNAs in HCC. miR-221 is one of the most significantly up-regulated miRNAs and was found to target ESR1, FOS, and CXCL12. Additionally, miRNA-142-5p is the main down-regulated miRNA and potentially targets ANKRD29, IGF2BP3, and IGSF3. A recent report shows that miRNA-142-5p could regulate the expression of IGF2BP3, which is strongly associated with an advanced tumor stage and is a predictor of poor prognosis among patients with HCC, as with IGF2BP1 [33]. miR-221, which is one of the most frequently and consistently up-regulated microRNAs (miRNAs) in human cancer, has been suggested to act as a tumor promoter. A recent study presents that miR-221 overexpression could accelerate hepatocyte proliferation and contribute to liver tumorigenesis [34]. Additionally, miR-221 also had a higher expression level in NSCLC [35]. Further researches give a conclusion that through the stimulation from staphylococcal nuclease domain-containing 1 (SND1), miR-221 could result in the overexpression of angiogenic factors like angiogenin and CXCL16 [36]. By using parallel measurement, a change in CDKN1B, CDKN1C, paralemmin-2, and CXCL12 levels was suggested as consistent with increased miR-221 activity in the same group. Meanwhile, Yau et al. found that miR-221 and miR-18a levels were also significantly higher in colorectal carcinoma tissues compared with their respective adjacent normal tissues [37]. miR-18a and miR-19a, regulated by E2F-MYC signaling pathways, are reported playing a crucial role in inducing cell proliferation [38]. As we found that ESR1 was potentially targeted by hsa-mir-148b, hsa-mir-181b, hsa-mir-18a, hsa-mir-19a, and hsa-mir-221, it indicates that these miRNAs may play a key role in HCC by mediating ESR1.
Conclusions
In conclusion, our study tried to identify DEGs using comprehensive bioinformatics analyses and found potential biomarkers to predict the progression of diseases. After analysis, a total of 115 DEGs and 64 DEMs were screened including ESR1, FOXO1, CXCL12, GNAO1, and several miRNAs such as miR-221 and miR-142-5p. Significantly, the roles of hsa-mir-148b, hsa-mir-181b, hsa-mir-18a, hsa-mir-19a, and hsa-mir-221 in HCC through interactions with ESR1 and CXCL12 may provide new clues for the diagnosis and treatment of HCC patients. However, lacking of experimental verification is a limitation of this study. In the future, these predicted results obtained from bioinformatics analysis can be verified by further experimental researches, such as qRT-PCR and Western Blot.
Acknowledgements
Not applicable.
Funding
This study was funded by grants from the National Natural Science Foundation of China (No. 81672959), the Natural Science Foundation of Chongqing, China (No. cstc2015shmszx120019), and the Graduate Student Research Innovation Project of Chongqing, China (No. CYS15127).
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article.
Authors’ contributions
TM and DZ participated in the design of this study, and they both performed the statistical analysis and drafted the manuscript. TTL, ZG, and JLP carried out the study and collected important background information and data. XFW, DFZ, and ZJW helped to draft the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- adj. P
Adjusted P values
- BH
Benjamini and Hochberg
- DEGs
Differentially expressed genes
- DEMs
Differentially expressed miRNAs
- FDR
False discovery rate
- GO
Gene ontology
- HCC
Hepatocellular carcinoma
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- PPI
Protein–protein interaction
Additional files
Complete list of differentially expressed genes (DEGs) in GSE22058. (DOCX 183 kb)
Complete list of differentially expressed genes (DEGs) in GSE25097. (DOCX 103 kb)
Complete list of differentially expressed genes (DEGs) in GSE57958. (DOCX 50 kb)
Complete list of differentially expressed miRNAs (DEMs) in GSE22058. (DOCX 19 kb)
Contributor Information
Tong Mou, Email: mutong_9@hotmail.com.
Di Zhu, Email: hnxyzhudi@126.com.
Xufu Wei, Email: wws2010@163.com.
Tingting Li, Email: ltttytt@163.com.
Daofeng Zheng, Email: daofeng90@126.com.
Junliang Pu, Email: junliangpjl@126.com.
Zhen Guo, Email: sunnyguoz@163.com.
Zhongjun Wu, Phone: (86)18983212119, Email: wzjtcy@126.com.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Andreozzi M, Quintavalle C, Benz D, Quagliata L, Matter M, Calabrese D, Tosti N, Ruiz C, Trapani F, Tornillo L, et al. HMGA1 expression in human hepatocellular carcinoma correlates with poor prognosis and promotes tumor growth and migration in in vitro models. Neoplasia. 2016;18:724–31. doi: 10.1016/j.neo.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tung EK, Mak CK, Fatima S, Lo RC, Zhao H, Zhang C, Dai H, Poon RT, Yuen MF, Lai CL, et al. Clinicopathological and prognostic significance of serum and tissue Dickkopf-1 levels in human hepatocellular carcinoma. Liver Int. 2011;31:1494–504. doi: 10.1111/j.1478-3231.2011.02597.x. [DOI] [PubMed] [Google Scholar]
- 4.Mah WC, Thurnherr T, Chow PK, Chung AY, Ooi LL, Toh HC, Teh BT, Saunthararajah Y, Lee CG. Methylation profiles reveal distinct subgroup of hepatocellular carcinoma patients with poor prognosis. PLoS One. 2014;9:e104158. doi: 10.1371/journal.pone.0104158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burchard J, Zhang C, Liu AM, Poon RT, Lee NP, Wong KF, Sham PC, Lam BY, Ferguson MD, Tokiwa G, et al. microRNA-122 as a regulator of mitochondrial metabolic gene network in hepatocellular carcinoma. Mol Syst Biol. 2010;6:402. doi: 10.1038/msb.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2013;41:D991–5. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bastos HP, Tavares B, Pesquita C, Faria D, Couto FM. Application of gene ontology to gene identification. Methods Mol Biol. 2011;760:141–57. doi: 10.1007/978-1-61779-176-5_9. [DOI] [PubMed] [Google Scholar]
- 8.Gene Ontology C. The Gene Ontology (GO) project in 2006. Nucleic Acids Res. 2006;34:D322–6. doi: 10.1093/nar/gkj021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–62. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 11.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–52. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su G, Morris JH, Demchak B, Bader GD. Biological network exploration with Cytoscape 3. Curr Protoc Bioinformatics. 2014;47:8 13 11–24. doi: 10.1002/0471250953.bi0813s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43:D146–52. doi: 10.1093/nar/gku1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magnani L, Lupien M. Chromatin and epigenetic determinants of estrogen receptor alpha (ESR1) signaling. Mol Cell Endocrinol. 2014;382:633–41. doi: 10.1016/j.mce.2013.04.026. [DOI] [PubMed] [Google Scholar]
- 15.Ramesh V, Ganesan K. Integrative functional genomic delineation of the cascades of transcriptional changes involved in hepatocellular carcinoma progression. Int J Cancer. 2016;139:1586–97. doi: 10.1002/ijc.30195. [DOI] [PubMed] [Google Scholar]
- 16.Baldissera VD, Alves AF, Almeida S, Porawski M, Giovenardi M. Hepatocellular carcinoma and estrogen receptors: polymorphisms and isoforms relations and implications. Med Hypotheses. 2016;86:67–70. doi: 10.1016/j.mehy.2015.11.030. [DOI] [PubMed] [Google Scholar]
- 17.Li W, Tse LA, Wang F. Prognostic value of estrogen receptors mRNA expression in non-small cell lung cancer: a systematic review and meta-analysis. Steroids. 2015;104:129–36. doi: 10.1016/j.steroids.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 18.Yu W, Wang J, Ma L, Tang X, Qiao Y, Pan Q, Yu Y, Sun F. CD166 plays a pro-carcinogenic role in liver cancer cells via inhibition of FOXO proteins through AKT. Oncol Rep. 2014;32:677–83. doi: 10.3892/or.2014.3226. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 1813;2011:1978–86. doi: 10.1016/j.bbamcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 20.Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–7. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Wang Y, Wang J, Sun F. Protein-protein interactions among signaling pathways may become new therapeutic targets in liver cancer (review) Oncol Rep. 2016;35:625–38. doi: 10.3892/or.2015.4464. [DOI] [PubMed] [Google Scholar]
- 22.Carbajo-Pescador S, Mauriz JL, Garcia-Palomo A, Gonzalez-Gallego J. FoxO proteins: regulation and molecular targets in liver cancer. Curr Med Chem. 2014;21:1231–46. doi: 10.2174/0929867321666131228205703. [DOI] [PubMed] [Google Scholar]
- 23.Li X, Zhang X, Xie W, Zhou C, Li Y, Zhang X. Alterations in transcription and protein expressions of HCC-related genes in HepG2 cells caused by microcystin-LR. Toxicol In Vitro. 2017;40:115–23. doi: 10.1016/j.tiv.2016.12.016. [DOI] [PubMed] [Google Scholar]
- 24.Li S, Xu X, Xu X, Hu Z, Wu J, Zhu Y, Chen H, Mao Y, Lin Y, Luo J, et al. MicroRNA-490-5p inhibits proliferation of bladder cancer by targeting c-Fos. Biochem Biophys Res Commun. 2013;441:976–81. doi: 10.1016/j.bbrc.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Jiang T, Li Z, Lu L, Zhang R, Zhang D, Wang X, Tan J. Analysis of differentially co-expressed genes based on microarray data of hepatocellular carcinoma. Neoplasma. 2017;64:216–21. [DOI] [PubMed]
- 26.Ding M, Li J, Yu Y, Liu H, Yan Z, Wang J, Qian Q. Integrated analysis of miRNA, gene, and pathway regulatory networks in hepatic cancer stem cells. J Transl Med. 2015;13:259. doi: 10.1186/s12967-015-0609-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghanem I, Riveiro ME, Paradis V, Faivre S, de Parga PM, Raymond E. Insights on the CXCL12-CXCR4 axis in hepatocellular carcinoma carcinogenesis. Am J Transl Res. 2014;6:340–52. [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H, Pan Z, Li A, Fu S, Lei Y, Sun H, Wu M, Zhou W. Roles of chemokine receptor 4 (CXCR4) and chemokine ligand 12 (CXCL12) in metastasis of hepatocellular carcinoma cells. Cell Mol Immunol. 2008;5:373–8. doi: 10.1038/cmi.2008.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pei X, Zhang J, Wu L, Lu B, Zhang X, Yang D, Liu J. The down-regulation of GNAO1 and its promoting role in hepatocellular carcinoma. Biosci Rep. 2013;33:755–60. [DOI] [PMC free article] [PubMed]
- 30.Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–73. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 31.Jia D, Wei L, Guo W, Zha R, Bao M, Chen Z, Zhao Y, Ge C, Zhao F, Chen T, et al. Genome-wide copy number analyses identified novel cancer genes in hepatocellular carcinoma. Hepatology. 2011;54:1227–36. doi: 10.1002/hep.24495. [DOI] [PubMed] [Google Scholar]
- 32.Liu Z, Zhang J, Wu L, Liu J, Zhang M. Overexpression of GNAO1 correlates with poor prognosis in patients with gastric cancer and plays a role in gastric cancer cell proliferation and apoptosis. Int J Mol Med. 2014;33:589–96. doi: 10.3892/ijmm.2013.1598. [DOI] [PubMed] [Google Scholar]
- 33.Liu X, Sempere LF, Galimberti F, Freemantle SJ, Black C, Dragnev KH, Ma Y, Fiering S, Memoli V, Li H, et al. Uncovering growth-suppressive MicroRNAs in lung cancer. Clin Cancer Res. 2009;15:1177–83. doi: 10.1158/1078-0432.CCR-08-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan Q, Loya K, Rani B, Mobus S, Balakrishnan A, Lamle J, Cathomen T, Vogel A, Manns MP, Ott M, et al. MicroRNA-221 overexpression accelerates hepatocyte proliferation during liver regeneration. Hepatology. 2013;57:299–310. doi: 10.1002/hep.25984. [DOI] [PubMed] [Google Scholar]
- 35.Duncavage E, Goodgame B, Sezhiyan A, Govindan R, Pfeifer J. Use of microRNA expression levels to predict outcomes in resected stage I non-small cell lung cancer. J Thorac Oncol. 2010;5:1755–63. doi: 10.1097/JTO.0b013e3181f3909d. [DOI] [PubMed] [Google Scholar]
- 36.Bronte F, Bronte G, Fanale D, Caruso S, Bronte E, Bavetta MG, Fiorentino E, Rolfo C, Bazan V, Di Marco V, Russo A. HepatomiRNoma: the proposal of a new network of targets for diagnosis, prognosis and therapy in hepatocellular carcinoma. Crit Rev Oncol Hematol. 2016;97:312–21. doi: 10.1016/j.critrevonc.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 37.Yau TO, Wu CW, Dong Y, Tang CM, Ng SS, Chan FK, Sung JJ, Yu J. microRNA-221 and microRNA-18a identification in stool as potential biomarkers for the non-invasive diagnosis of colorectal carcinoma. Br J Cancer. 2014;111:1765–71. doi: 10.1038/bjc.2014.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramesh V, Ganesan K. Integrative analysis of transcriptome and miRNome unveils the key regulatory connections involved in different stages of hepatocellular carcinoma. Genes Cells. 2016;21:949–65. doi: 10.1111/gtc.12396. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article.