

Abstract
The first step in removing cholesterol from a cell is the ATP-binding cassette transporter 1 (ABCA1)-driven transfer of cholesterol to lipid-free or lipid-poor apolipoprotein A-I (apoA-I), which yields cholesterol-rich nascent high-density lipoprotein (nHDL) that then matures in plasma to spherical, cholesteryl ester-rich HDL. However, lipid-free apoA-I has a three-dimensional (3D) conformation that is significantly different from that of lipidated apoA-I on nHDL. By comparing the lipid-free apoA-I 3D conformation of apoA-I to that of 9–14 nm diameter nHDL, we formulated the hypothetical helical domain transitions that might drive particle formation. To test the hypothesis, ten apoA-I mutants were prepared that contained two strategically placed cysteines several of which could form intramolecular disulfide bonds and others that could not form these bonds. Mass spectrometry was used to identify amino acid sequence and intramolecular disulfide bond formation. Recombinant HDL (rHDL) formation was assessed with this group of apoA-I mutants. ABCA1-driven nHDL formation was measured in four mutants and wild-type apoA-I. The mutants contained cysteine substitutions in one of three regions: the N-terminus, amino acids 34 and 55 (E34C to S55C), central domain amino acids 104 and 162 (F104C to H162C), and the C-terminus, amino acids 200 and 233 (L200C to L233C). Mutants were studied in the locked form, with an intramolecular disulfide bond present, or unlocked form, with the cysteine thiol blocked by alkylation. Only small amounts of rHDL or nHDL were formed upon locking the central domain. We conclude that both the N- and C-terminal ends assist in the initial steps in lipid acquisition, but that opening of the central domain was essential for particle formation.
Graphical Abstract
Plasma HDL levels negatively correlate with coronary heart disease (CHD) risk,1 and therefore, HDL cholesterol concentration was identified several years ago as an ideal target for lowering the incidence of atherosclerosis.2–4 HDL is an integral part of the reverse cholesterol transport pathway and thus plays a pivotal role in transporting excess cholesterol from peripheral tissues to the liver for excretion, thereby promoting cholesterol homeostasis.5,6 However, the atheroprotective properties of HDL may not be directly related to its concentration in plasma,7–10 as recent reports have demonstrated that HDL-related CHD risk reduction in humans more strongly correlated with the efficiency of cholesterol efflux to LDL-depleted plasma, a correlation that was only partially dependent on plasma HDL cholesterol concentration.11–13 Efflux of cholesterol to HDL relies on the interaction of lipid-free or lipid-poor apolipoprotein A-I (apoA-I) with ATP-binding cassette transporter 1 (ABCA1), which generates nHDL14–16 that is subsequently converted to mature HDL through the actions of lecithin-cholesterol acyltransferase (LCAT), lipases, and lipid transfer proteins. An essential aspect of understanding the process through which cholesterol is transferred by ABCA1 is delineating the molecular events taking place with lipid-free apoA-I that facilitate lipidation.
ApoA-I is a 28 kDa protein composed of 243 amino acids. A unique structural property of apoA-I is that the region defined by amino acids 44–243 is composed of 10 amphipathic helices, including two 11-mer and eight 22-mer segments. ApoA-I has six class A amphipathic helices, four class Y amphipathic helices, and one class G* amphipathic helix.17–19 Basically, an amphipathic helix has one hydrophobic face and a second charged face. The hydrophobic face of apoA-I interacts with the hydrophobic portion of glycerophospholipids at the surface of HDL, while the charged face interacts with solvent and the polar residues of the glycerophospholipids.
Biophysical studies have led to a solution structure of apoA-I consisting of an internal helix bundle, helices 2–8, with the N-and C-terminal ends in the proximity of one another.20–26 Structural studies of lipid-free apoA-I in solution have generally confirmed the proposed structure.27–31 Efforts to obtain a crystal structure of full-length lipid-free apoA-I have proven to be challenging. Crystal structures of truncation mutations of apoA-I, Δ1–43 and Δ185–243, N- and C-terminal deletion mutations,32,33 respectively, have revealed that the central region prefers to crystallize in a saddle or semicircular conformation, reminiscent of the conformations deduced for apoA-I on discoidal rHDL.34–37
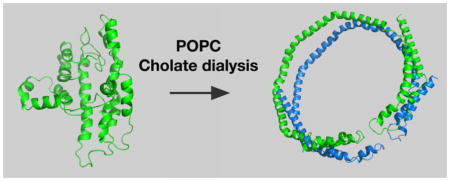
If the solution structure of lipid-free apoA-I is compact and highly associated, how does it form HDLs? The basic question is illustrated in Figure 1A, showing compact, lipid-free apoA-I and the first product of lipidation, which has the apoA-I belting phospholipid. On one hand, cholate dialysis in the presence of palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) yields rHDL, 9.6 diameter lipid discs carrying ~120 POPC molecules where two antiparallel apoA-I molecules act like a belt around the periphery with the 5,5′ amphipathic helices of the two apoA-Is adjacent to one another,34–36,38,39 the LL5/5 rotomer orientation.40 In the presence of ABCA1, in this case human embryonic kidney 293 (HEK) cells expressing human ABCA1, the predominant nHDL was spherical, had a diameter of 9–14 nm, and carried 109 cholesterol molecules with 130 molecules of phospholipid and three antiparallel molecules of apoA-I that seem to have maintained their 5,5′ helix association.16
Figure 1.

“Question”: What are the important factors for a compact, folded lipid-free apoA-I molecule to open up and accept lipid? Panel A shows apoA-I in the compact, in-solution conformation (left) and the conformation that it assumes with lipid (right) in rHDL. Panel B illustrates the unique features of lipidated apoA-I generated by cholate dialysis (top) and ABCA1-catalyzed lipidation (bottom). The top panel shows two antiparallel strands of apoA-I on a POPC lipid disc with coincidence of the 5,5′ helices. The bottom panel shows three antiparallel strands that belt a sphere of lipids composed of glycerophospholipid, sphingomyelin, and cholesterol with the 5,5′,5″ helical regions coincident.
To discuss the opening of lipid-free apoA-I, the molecule was divided into three regions: amino acids 1–65 (N-terminus), amino acids 190–243 (C-terminus), and residues 66–189 (internal). The N-terminal region contains amino acids 1–43 and helix 1, the C-terminal region helices 8–10, while the internal region helices 2–7. Figure 1B shows unique features of lipidated apoA-I generated by cholate dialysis (top panel) and ABCA1-catalyzed lipidation (bottom panel).
The three regions of lipid-free apoA-I are reported to have somewhat different roles in promoting lipidation. Only modest reductions in the level of lipid association and rHDL formation were reported when the N-terminal region, amino acids 1–65 of apoA-I, was deleted,23,41–44 although, when studied in isolation, helices 1 and 10 showed the greatest lipid binding affinities.43 Various combinations of deletions of amino acids 190–243 at the C-terminus suggested that this end contributed significantly to lipid binding, rHDL formation, and lipid clearance.44–46 Double-deletion mutants of the N- and C-termini of apoA-I almost completely abolished lipid binding and rHDL formation.44 In contrast, for cAMP-dependent, ABCA1-mediated cholesterol efflux, the central domain of apoA-I, helices 2–7, was comparable to N-terminal deletions showing an only slightly reduced level of lipidation and HDL particle formation compared to that of wild-type apoA-I.47–49
Several mechanisms have been proposed to account for the composition and conformation of apoA-I during lipidation and formation of nHDL.50–59 With regard to the changes in apoA-I conformation, the Phillips group has proposed that the initial step of a two-step process of nHDL formation involves the C-terminal domain of apoA-I, specifically residues 190–243, with the region of residues 223–243 being essential for HDL formation.53,60 In contrast, Gursky et al. suggested that nHDL forms through the adsorption of lipid-free apoA-I to the plasma membrane followed by “domain swapping” in the helix 5 region promoting dimerization of apoA-I molecules and, finally, the insertion of helices 8–10 assisted by ABCA1.52 The opening of the helix bundle located in the central domain is proposed to follow the initial interactions of first C-terminal and then N-terminal regions with lipid.19,23
Because apoA-I structural reorganization is essential for accepting lipid, this study was designed to delineate how restricting different regions effected the acquisition of lipid by apoA-I. Our published model of human lipid-free apoA-I was used as a template.27 Most models for lipid-free apoA-I are compact; each region has several folds that must open to accommodate lipid, giving the structures shown in Figure 1B. We engineered 10 apoA-I mutants having two cysteines that can potentially “lock” regions together through disulfide bonds when the spatial separation of the sulfhydryl groups is 0.3–0.5 nm. Mutations span the N- and C-termini as well as the central domain of apoA-I. Mutant apoA-Is E34C/S55C, L200C/L233C, and F104C/H162C apoA-I were the most thoroughly studied. Formation of rHDL and nHDL was measured for mutant proteins in both the “locked” and “unlocked” states.
MATERIALS AND METHODS
Materials
Human apoA-I variants were expressed using the IMPACT protein expression system, including Escherichia coli strain ER2566, plasmid vector pTYB11, and chitin beads purchased from New England Biolabs. International DNA Technologies synthesized polymerase chain reaction (PCR) primers. DNase I was from Worthington Biochemical. Custom DNA Constructs prepared mutant apoA-I cDNA constructs. 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) was from Avanti Polar Lipids Inc. Solvents used in mass spectrometry (MS) and liquid chromatography and mass spectrometry (LC–MS) were “B&J GC2” grade from Burdick and Jackson. All other solvents and routine reagents were of the highest commercial grade available.
Cloning, Expression, and Purification of ApoA-I
The coding sequence for wild-type and mutant apoA-I was cloned from the CMV5 vector by Custom DNA Constructs and amplified by PCR as described and inserted into the pTYB11 vector. Expression and purification of the double-cysteine-containing apoA-I mutants from inclusion bodies were conducted as previously described.61–63 Mutant apoA-Is released from a chitin affinity column with dithiothreitol (DTT) are monomeric and essentially pure as determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). Protein purities and molecular weights were determined by mass spectrometry of the intact proteins and are listed in Table 1. The position of cysteine substitution for each mutant was confirmed by mass spectrometry after the addition of 500 mM 1,4-dithiothreitol, treatment with 1 M iodoacetamide to alkylate the cysteine residues, exhaustive dialysis against 10 mM ammonium bicarbonate, and in-gel trypsin digestion as previously reported.27 These constructs have an alanine added at the N-terminal end so that the average mass of the expressed, wild-type apoA-I is 28150 Da. Human plasma apoA-I has an average mass of 28079 Da.
Table 1.
List of ApoA-I Molecules Used in This Study
| apoA-I helical domain amino acid | formation of intramolecular disulfide | formation of rHDL particles | mass of mutant apoA-I from mass spectrometry/disulfide ionn+ | ||||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| predicted | actual | apoA-I modification | size (nm) | POPC/apoA-I ± SD | |||
| ND13C | H2V67C | no | no | unlocked | 9.6 | 54:1 ± 6 | 28142 |
| alkylated | 9.8/9.6 | 44:1 ± 4 | 28143 | ||||
| NG26C | H1K59C | no | no | unlocked | 9.6/8.0 | 54:1 ± 6 | 28171 |
| alkylated | 9.8 | 56:1 ± 5 | 28170 | ||||
| NE34C | H1S55C | yes | yes | locked | 9.8 | 36:1 ± 5 | 28140/751.344+ |
| alkylated | 9.6 | 58:1 ± 5 | 28140 | ||||
| H1V53C | H5R123C | yes | yes | locked | NPFa | b | 28119/758.354+ |
| alkylated | 9.6 | 53:1 ± 14 | 28119 | ||||
| H4D103C | H7R177C | yes | yes | locked | 9.4 | b | 28137/733.043+ |
| alkylated | 9.6 | 56:1 ± 10 | 28137 | ||||
| H4F104C | H6M148C | no | no | unlocked | 9.6 | 69:1 ± 1 | 28078 |
| alkylated | 9.6 | 50:1 ± 4 | 28078 | ||||
| H4F104C | H6R160C | yes | yes | locked | NPFa | b | 28053/804.884+ |
| alkylated | 9.6 | 64:1 ± 5 | 28053 | ||||
| H4F104C | H6H162C | yes | yes | locked | NPFa | 28:1 ± 3 | 27998/820.023+ |
| alkylated | 9.6 | 64:1 ± 10 | 27998 | ||||
| H6D157C | H7L178C | yes | yes | unlocked | 8.0 | b | 28128/665.342+ |
| alkylated | 9.6 | 60:1 ± 8 | 28129 | ||||
| H9L200C | H10L233C | yes | yes | locked | 9.6 | 54:1 ± 2 | 28130/645.554+ |
| alkylated | 9.8 | 64:1 ± 4 | 28130 | ||||
| wild-type apoA-I | not applicable | none | 9.6 | 52:1 ± 4 | 28150 | ||
No particle found by nondenaturing gel electrophoresis.
No lipid phosphorus was detected.
Formation of Intramolecular Disulfide Bonds
Intramolecular disulfide bond formation between specific residues within apoA-I was promoted under optimized conditions as previously described.27 Briefly, double-cysteine-containing apoA-I mutants were denatured with 6 M guanidine hydrochloride in the presence of 500 mM dithiothreitol and then diluted to 0.02 μg/μL. The proteins were extensively dialyzed against 10 mM ammonium bicarbonate. Mass spectrometry of intact mutant apoA-Is was used to verify that a preparation contained monomeric protein. Disulfide coupling was verified by in-gel trypsin digestion of the refolded protein followed by mass spectrometry. The m/z values of disulfide-linked peptides are included in Table 1.
rHDL Particle Preparation and Purification
Reconstituted HDL particles were prepared by adding 1 mol of apoA-I to 80 mol of POPC as previously described.34 Briefly, POPC dissolved in chloroform was first dried in a stream of nitrogen on the side of a glass tube. The last traces of solvent were removed under vacuum for 18 h. POPC was added to a solution containing sodium deoxycholate at a deoxycholate:-POPC molar ratio of 2:1 by incubation at 37 °C with intermittent shaking for 1 h and then vigorously shaken on a standard multitube vortexer (VWR Scientific) for 1 h at room temperature. The crude rHDL was purified by application to two Superdex 200 HR 10/30 columns linked in tandem and eluted at 0.5 mL/min as previously described.61,64 The purified rHDL was then analyzed on 4 to 30% nondenaturing gels (CBS Scientific) to assess yield and particle size.34,36
Cell Culture and Purification of Nascent HDL
HEK293 cells expressing ABCA1 were a generous gift from M. Hayden (University of British Columbia, Vancouver, BC) and supplied by J. Parks. HEK293 Flp-In cells from Invitrogen that do not express ABCA1 were used for controls.16 All cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L glucose, 50 μg/mL hygromycin, 100 μg/mL streptomycin, 100 units/mL penicillin, 2 mM L-glutamine, and 10% fetal bovine serum (FBS). Cells were maintained at 37 °C in an atmosphere of 5% CO2. Cells were plated on 100 mm dishes, incubated until the cells reached ~90–100% confluence, then washed twice with 1 mL of balance salt solution, and incubated for 2 h with serum-free DMEM. Cells were then incubated for 24–48 h with 10 μg/mL lipid-free WT or cysteine containing mutant apoA-I with a [125I]apoA-I tracer in serum-free DMEM and harvested for analysis as described previously.16
Recombinant HDL Composition
Lipids were extracted from rHDL FPLC fractions 59–71 according to the method of Bligh and Dyer.16,65 For lipid phosphorus analysis,16,66 extracts were evaporated with a stream of nitrogen and redissolved in 1 mL of chloroform and methanol (1:1), and the solutions were transferred to screw-cap glass tubes. All traces of chloroform were removed under a vacuum; 150 μL of perchloric acid was added, and the mixture was incubated at 180 °C. After 18 h, the samples were cooled and treated with 900 μL of distilled water, 176 μL of 2.6% aqueous Na2MoO4, and 176 μL of aqueous ascorbic acid. After being incubated at 50 °C for 15 min, the samples were cooled, and the level of lipid phosphorus was determined from the absorbance at 820 nm. The protein content of rHDL was measured by the method of Lowry et al.67 using bovine serum albumin as the standard.
SDS–PAGE and In-Gel Trypsin Digest
The center cut fractions of rHDL from the Superdex FPLC system were separated via 12% SDS–PAGE and visualized using SimplyBlue SafeStain. The protein band was cut from the gel and used for in-gel digestion followed by peptide analysis using MS/MS techniques. Tryptic digestion of apoA-I was performed as described previously.16,34,35,68 Briefly, protein bands from monomeric apoA-I were excised from the gel, minced, and repeatedly dehydrated with acetonitrile. Next gel pieces were rehydrated with a cold, freshly prepared solution containing 20 ng/μL trypsin, 0.1% (w/v) RapiGest SF, and 1 mM CaCl2 dissolved in 10 mM ammonium bicarbonate (pH 7.8). The final trypsin:apoA-I mass ratio was 1:20. After mixing, digests sat on ice for 10 min and were then incubated for 18 h at 37 °C.
Q-TOF Mass Spectrometry
Extraction of tryptic peptides was accomplished as described previously.16,34,35,68 Briefly, the digestion solution was removed, and then 200 μL of an acetonitrile/water/formic acid mixture [50:45:5 (v/v/v)] was added to cover gel pieces. After sitting for 10 min, the solvent was transferred to a fresh tube. The extraction was repeated, and the combined aliquots were acidified to an HCl:apoA-I ratio of 1:10 (v/v) using 500 mM HCl. After the acidified solution had been incubated for 35 min at 37 °C, the sample was centrifuged for 10 min at 13000 rpm. The supernatant was transferred to a fresh tube before mass spectrometry. Survey scans were performed on each peptide mixture using a Waters Q-TOF API-US mass spectrometer equipped with a Waters CapLC and Advion Nanomate source. Acquisition was controlled by Mass-Lynx version 4.0. Peptides were loaded onto a PLRP-S trapping column before elution onto the analytical PLRP-S column as previously described.27 Initially, peptides of interest were identified using a tolerance of ±0.05 m/z and their identities confirmed by mass spectrometric sequence analysis.
Analysis of intact peptides was accomplished using direct infusion through the Nanomate source into the Waters Q-TOF API-US mass spectrometer. Approximately 10 μg of apoA-I in 100 μL of 10 mM ammonium bicarbonate buffer was diluted with 100 μL of acetonitrile. Approximately 2 μL of the protein solution was slowly infused into the mass spectrometer. The electrospray spectrum was deconvoluted using Mass-Lynx version 4.0, giving the uncharged mass of the protein.
RESULTS
Selection, Expression, and Isolation of Cysteine-Containing ApoA-I Variants
Two cysteines will form a disulfide bond, a process called locking, if they are 3–5 Å apart and no steric hindrance is present to impede bond formation. To study lipidation and the role of helix opening, two cysteines were added to apoA-I using standard techniques61–63 to create mutant proteins that could undergo intramolecular locking, which prevents regions from moving apart. Several double-cysteine-containing mutants were prepared that, based on our model, would not form intramolecular disulfide bonds. Table 1 lists 10 mutant apoA-I proteins prepared for these studies along with predicted and observed intramolecular disulfide bond formation. ApoA-I mutant protein purity was assessed by SDS–PAGE and MS analysis. To achieve lipid-free, refolded, locked protein samples, mutant apoA-Is were dissolved in 6 M guanidine hydrochloride, reduced with DTT, diluted to ≤0.02 μg/μL, and then dialyzed against ammonium bicarbonate to allow the intramolecular disulfide bonds to predominate. This procedure is similar to that reported by Lu et al.69 Refolding at a low protein concentration encouraged intramolecular disulfide bond formation over intermolecular disulfide cross-linking. Direct infusion mass spectrometry of each refolded mutant protein was run to verify if the protein was indeed monomeric, because mass spectrometry is much more precise than SDS–PAGE.35 An additional mass spectrometric analysis was used to confirm that we had only intramolecular bonds in our recombinant HDL complexes. For each mutant protein containing rHDL, we performed trypsin digestion and then subjected the digests to LC–MS/MS analysis as was done in our chemical cross-linking studies.27,35 A peptide with a mass corresponding to that of the intended intramolecular disulfide bond formation was sequenced by MS/MS to confirm that it was the correct sequence. The samples were searched for potential intermolecular disulfide bonds to ensure that there was no intermolecular disulfide bond formation.
Contribution of ApoA-I Domains to Recombinant HDL Particle Assembly and Composition
Lipidation of lipid-free cysteine-containing apoA-I was investigated by incubating them with POPC at an 80:1 molar ratio to form rHDL. Two different preparations were used, one in which cysteines were allowed to form an intramolecular disulfide bond and a second in which the cysteine thiols were alkylated with iodoacetamide. S-Alkylated forms of each mutant are monomeric, should readily form rHDL, and were used as controls. Figure 2 shows a representative group of rHDL particles separated on a 4 to 30% nondenaturing gel and stained with Coomassie Blue. Table 1 lists rHDL particle compositions and diameters for the rHDL particles. All alkylated, double-cysteine-containing apoA-I mutants formed rHDL particles having diameters between 9.6 and 9.8 nm, like wild-type apoA-I, and on average gave rHDL that carried slightly more lipid than wild-type apoA-I (Table 1), the two exceptions being D13C/V67C and F104C/M148C. Three double-cysteine-containing mutants with cysteines located toward the ends of the molecule, E34C/S55C, L200C/L233C, and D13C/V67C, formed rHDL particles having diameters of 9.6–9.8 nm. The first two were locked before lipidation, while D13C/V67C was locked during the process of lipidation. E34C/S55C and L200C/L233C acquired fewer POPC molecules than the alkylated forms did, but D13C/V67C acquired more POPC molecules, suggesting that the valine at position 67 may be important for synthetic lipidation of apoA-I. In contrast, the three locked, double-cysteine-containing mutants, F104C/R160C, F104C/H162C, and D157C/L178C, which prevent the central helical regions 4–6 from opening to accept lipid, did not form discrete rHDL particles and are denoted NPF in Table 1. Lipid extracts from V53C/R123C, F104C/R160C, and D157C/L178C did not have sufficient lipid phosphorus signal over background to report and are marked with footnote b. The lipid composition of F104C/H162C apoA-I reported in Tables 1 and 2 was generously sampled from those FPLC fractions around and including 9.6–9.8 nm diameter particles.
Figure 2.

Coomassie Blue-stained 4 to 30% NDGGE of rHDL particles formed from cholate dialysis of (80:1 molar ratio) POPC and apoA-I. Locked indicates the presence (+) or absence (−) of a disulfide bond. Alkylated indicates the presence (+) or absence (−) of carbamidomethylated cysteines. The top part of the figure shows apoA-I divided into its 10 repeats with the first amino acid in the repeat displayed. The intramolecular disulfide bonds are indicated above with the position of the first and last cysteine.
Table 2.
Comparison of the Formation Nascent HDL and Recombinant HDLa
| apoA-I | % nHDL peak 1 to wild-type peak 1 | % nHDL peak 1 to total | % molar rHDL POPC:ApoA-I to wild-type control |
|---|---|---|---|
| wild-type | 100 | 32 | 100 |
| alkyated | |||
| E34C/S55C | 81 | 26 | 112 |
| F104C/H162C | 57 | 18 | 123 |
| L200C/L233C | 76 | 24 | 123 |
| locked | |||
| E34C/S55C | 40 | 13 | 69 |
| F104C/H162C | 13 | 4 | 54 |
| L200C/L233C | 46 | 15 | 104 |
nHDL traces from Figure 3 were used to calculate the percent of peak 1 relative to peak 1 from wild-type apoA-I. Molar ratios of rHDL POPC:ApoA-I were determined from analysis of protein content and lipid content as described in Materials and Methods.
ApoA-I Domains That Contribute to Nascent HDL Particle Assembly, Size, and Composition
Four 125I-labeled, double-cysteine-containing apoA-I mutants, E34C/S55C, D13C/V67C, F104C/H162C, and L200C/L233C, were each incubated with ABCA1-expressing HEK293 cells in serum-free medium for 24 h. nHDL particles were separated by FPLC, and the results are plotted in Figure 3 as total [125I]apoA-I radioactivity versus elution fraction. Wild-type apoA-I formed three different nHDLs with the largest particles, which carry the most cholesterol, having diameters of 9–14 nm,16,70 called peak 1. Alkylation of N- and C-terminal mutants E34C/S55C and L200C/L233C, which without alkylation spontaneously lock to form disulfide bonds, gave ~22% less peak 1 nHDL upon incubation with HEK293 cells expressing ABCA1 compared to wild-type apoA-I (Table 2). The level of peak 1 formation for the alkylated, internal mutant, F104C/H162C, was reduced 43%. Upon locking, there was an average 45% reduction in the level of peak 1 formation from N- and C-terminal mutants compared to the corresponding alkylated mutants, but a 77% reduction for the F104C/H162C locked mutant.
Figure 3.

FPLC analysis of nascent HDL particles formed after incubating [125I]apoA-I with ABCA1-expressing HEK293 cells. (A) Locked and alkylated D13C/V67C apoA-I. (B) Locked and alkylated E34C/S55C apoA-I. (C) Locked and alkylated F104C/H162C apoA-I. (D) Locked and alkylated L200C/L233C apoA-I. Wild-type apoA-I is shown in all plots for comparison.
Conformation of ApoA-I on Recombinant HDL Particles
We measured the intramolecular disulfide bond prior to and after rHDL particle formation in a subset of mutant apoA-Is. Table 3 shows that mutant apoA-Is E34C/S55C, F104C/H162C, and L200C/L233C carried the correct disulfide cross-link before and after rHDL formation. Alkylated double-cysteine-containing mutants did not have detectable disulfide cross-links. However, D13C/V67C apoA-I was unique in that it did not form an intramolecular disulfide bond in the lipid-free state, but an intramolecular disulfide bond did form upon lipidation. The tryptic fragment of residues T3–T9 (790.144+, m/zcharge+), which corresponds to the fragment containing the disulfide bridge in D13C/V67C apoA-I, was absent in the lipid-free state, indicating the N-terminus through helix 1, specifically residues 13 and 67, is not within 3–5 Å in the absence of lipid. Our model of lipidated apoA-I was adjusted to account for this difference.
Table 3.
List of Unique Disulfide Mutants and Disulfide States upon Lipidationa
| apoA-I | disulfide tryptic fragment [(m/z)charge+] | lipid-free locked | lipid-bound locked |
|---|---|---|---|
| D13C/V67C | T3–T9 [(790.14)4+] | no | yes |
| E34C/S55C | T5–T7 [(751.34)4+] | yes | yes |
| F104C/H162C | T14–T26 [(820.05)3+] | yes | yes |
| L200C/L233C | T32–T35 [(645.44)4+] | yes | yes |
Trypsin digest and MS analysis of mutant apoA-I proteins in the lipid-free or lipid-bound conformation yielded the set of ions listed above. Proteins were examined for the presence of the disulfide tryptic fragment in the lipid-free state or after formation of rHDL.
DISCUSSION
Choice of Model
To assess the contributions of the various amphipathic helices to lipid binding, we used our recently published structure of human lipid-free apoA-I as a model to engineer double-cysteine-containing apoA-I.27 This model is similar to several other published models.28,30 To identify the conformation of lipid-free apoA-I, the Pollard study27 used mixtures of d0-BS3 and d4-BS3 to cross-link lysines. Chemically cross-linked peptides were identified by MS and verified by MS/MS sequencing. Using the cross-linking restraints, several models of lipid-free apoA-I were examined to determine if the BS3 cross-links were consistent with the model. The structure of Silva et al.28 generally fit the cross-link results generated in our laboratory and so was used as the template and then modified as required to more accurately reflect BS3 cross-linking results. Conformations for semicircular, truncated apoA-Is deduced from X-ray analysis by Bohani et al.32 and by Mei and Atkinson33 did not fit the biophysical studies that suggested lipid-free apoA-I was a compact protein, and neither of these models fit the chemical cross-linking results for lipid-free apoA-I. The unique feature of the two X-ray structures is that the folding of the N- and C-terminal regions appears to be present in the lipidated disc and in the lipid-free apoA-I, both of which were apparent from the cross-linking studies of Bhat et al.34,35 and Pollard et al.27 Two models that appeared after we started this work were those of Lagerstedt et al.71 and Segrest et al.30 The first did not fit our chemical cross-linking results; however, the second is similar to that of Pollard et al.27
The structure of Pollard et al.27 was used to identify which regions of the backbone needed to move apart to accept lipid. For each region, using visual inspection, several sites in which a cross-link would prevent the protein backbone from moving apart were chosen. Amino acids were considered for mutation if they faced one another on the backbone in the appropriate regions. A series of in silico replacements of cysteine for these amino acids were made, and if the substituted thiol moieties were separated by 0.5–0.7 nm, these sites were chosen for substitution. Other proposed non-cross-linking substitutions were prepared by choosing two amino acids that did not face one another on the opposing regions of the backbone (e.g., they were on opposite faces of the backbone) or by choosing two sites that were on the same face of the backbone, but separated by several nanometers. It was gratifying that the Pollard model correctly predicted whether double-cysteine-containing mutants would form intramolecular disulfide bonds.
A priori, it cannot be inferred that a structure derived from chemical cross-linking has no predictive power. Each cross-link is a high-resolution site with well-defined distance constraints. The regions between these points may have lower resolution, but steric constraints often force intervening amino acids to follow certain rules that lead to accurate predictions at a lower level of confidence. A number of studies have compared structural information derived from chemical cross-linking in solution with a protein structure derived by X-ray crystallography. The mean separation found for chemical cross-linking is a bit larger than that from X-ray analysis, but observed cross-links were consistent with the published X-ray results.72–79 The use of cross-linking in structural studies has been the subject of several papers.80–82 Nuclear magnetic resonance (NMR) is also used to ascertain solution structures for smaller proteins; however, in many cases, loop regions and the C- and N-terminal regions seem to be more flexible as was reported for truncated mouse apoA-I.29 A recent comparison of NMR and X-ray crystal structures pointed out where the two techniques provide similar results and where they differ.83 It may be argued that structural methods applied to proteins in solution are more affected by intramolecular motions.
ApoA-I Opening and rHDL Formation
The use of cysteine substitutions to monitor apolipoprotein helical lipid binding activity was first reported by Narayanaswami et al. to assess the conformational opening of apolipophorin III (apoLp-III), an exchangeable amphipathic protein from the Sphinx moth.84 Results of this study provided a mechanism for the opening of hinge domains as phospholipids are acquired by apoLp-III. The conformational reorganization of the N-terminal four-helix bundle within apoE was also examined using this approach,69 as was the helix registry of apoA-I on lipid discs.40
Ten apoA-I mutants each having two cysteine residues were employed to study rHDL formation. The mutations covered the principal domains of apoA-I (Table 1). The first step in the analysis was to determine if double-cysteine-containing apoA-I mutants would form rHDL with POPC using the cholate dialysis method when the thiol moiety of cysteine was alkylated, preventing disulfide formation. All alkylated double-cysteine-containing apoA-I mutants formed 9.6–9.8 nm diameter rHDL with compositions that were similar to that obtained with wild-type apoA-I, showing that alkylation had little effect on the product distribution generated by cholate dialysis. Locked double-cysteine-containing apoA-I mutants had very different outcomes upon cholate dialysis (Figure 2 and Table 1). Preventing the opening of the C-terminal domain, L200C and L233C, helices 8–10, gave particles that were similar to wild-type apoA-I in size and composition. Locking the N-terminal region of apoA-I, E34C/S55C, N-terminus to helix 1, reduced the extent of lipid accumulation by ~20% compared to that of wild-type rHDL, but the rHDL particles were the same size. However, when the central domain was locked, none of these double-cysteine-containing mutants, including F104C/H162C, which connects helices 4–6, yielded discrete particles (Figure 2 and Table 1). The locked F104C/H162C mutant showed a smear in Figure 2, not a discrete particle, but extraction of FPLC fractions used for NDGGE yielded protein and lipid. The locked, double-cysteine-containing apoA-Is were subject to rigorous analysis by tandem mass spectroscopy before being used in these experiments. Therefore, it is unlikely that more than trace amounts of unlocked protein were in the preparation. We speculate that the disulfide-locked mutant protein can still bind small amounts of lipid, a process that yields a continuum of mutant apoA-I/lipid particles/aggregates.
These results suggest that the unfolding of N-terminal amino acids 1–43 through helix 1 or C-terminal helices 8–10 does not play a critical role in the synthesis of POPC-containing rHDL particles. Previous studies of lipidated rHDL particles34,35,37 suggested that both ends are folded back, and therefore, consistent with those reports, these new results suggest that the N- and C-terminal regions do not need to become mobile for lipidation to proceed.
Previous studies employing N-terminally truncated apoA-I suggested that the N-terminal domain of apoA-I plays a major role in the stability of the lipid-free conformation;85 however, several studies suggested that the N-terminal region, amino acids 1–65, may not have a rate-limiting role in rHDL formation or lipid association. Brouillette et al. observed a slight reduction in the association of POPC with Δ1–43 apoA-I and Δ1–65 apoA-I,23 and the reduced clearance of dimyristoyl-phosphatidylcholine liposomes by N-terminal deletion mutants Δ1–41 and Δ1–59 are consistent with a lower lipid binding affinity.44,86 There is general consensus that the C-terminal domain has the highest affinity for lipid, and it has been suggested that this region initiates the biogenesis of HDL.60,87 Studies comparing deletion mutants Δ190–243, Δ212–233, and Δ213–243 to wild-type apoA-I showed a significant reduction in the extent of DMPC clearance and rHDL formation.44–46 This work indicates that residues 200–233 within the C-terminal domain do not need to be free and flexible but can work in concert to associate with lipid. However, it is evident that reorganization of helices 4–6 is essential to the formation of discrete rHDL particles.
ApoA-I Opening and nHDL Particle Formation
On the basis of our results generating rHDL with double-cysteine-containing mutant apoA-Is, we chose one mutant from each of the three regions of lipid-free apoA-I to incubate with ABCA1-expressing HEK293 cells. A fourth mutant was included when it was discovered that an intramolecular disulfide bond formed only after lipidation. The effect of replacing specific amino acid pairs with cysteines on ABCA1-driven lipidation was evaluated by incubating ABCA1-expressing HEK cells with wild-type apoA-I and four of the double-cysteine mutant apoA-Is having the cysteine moieties blocked by carboxyamidomethylation. Whereas the rHDL lipid content increased for alkylated N- and C-terminal mutants (Table 2), the mass of 9–14 nm diameter nHDL particles was reduced by around 20%. While the level of rHDL formation by the alkylated, internal mutant F104C/H162C, helices 4–6, was similar to those of the other mutants, the level of nHDL formation by carboxyamidomethyl-modified protein was reduced by ~40% (Figure 3 and Table 2). Disulfide formation reduced the extent of rHDL lipidation of N- and C-terminal mutants by an average of 25% compared to those of their carboxyamidomethyl derivatives. The same comparison for nHDL had an average reduction of 45%. However, the mutant protein having an internal locking disulfide, F104C/H162C, suffered an 80% reduction in its level of lipidation. Therefore, preventing helix 4 and helix 6 from moving apart effectively stopped ABCA1-mediated lipidation.
Recombinant HDL particles from cholate dialysis are primarily discoidal, carrying two antiparallel molecules of apoA-I; while our previous studies have shown that ABCA1-driven lipidation yields mostly spherical nHDL particles carrying three molecules of apoA-I,16 these observations suggest that the thermodynamic end points for the two processes are very different. By and large, rHDL molecules from mutant apoA-I have slightly more POPC than wild-type apoA-I molecules do, suggesting that replacing a few amino acids with alkylated cysteines does not interfere with POPC–apoA-I binding. However, ABCA1-mediated efflux of cholesterol depends on protein–protein interactions to keep apoA-I and ABCA1 associated at the plasma membrane. Because cysteine replaced charged amino acids or hydrophobic amino acids in these mutants, a reduced level of lipidation of the alkylated mutants strongly suggests that alkylation disrupted interactions between apoA-I and ABCA1 and/or the apoA-I molecules forming nHDL. Locking disulfides had a stronger effect on lipidation than did alkylation, showing that all parts of apoA-I influenced nHDL formation; however, opening of the internal helix bundle, helices 4–6, is absolutely essential for ABCA1-mediated lipidation. These studies are consistent with an earlier report that showed cAMP-dependent, ABCA1-mediated cholesterol efflux by a dual C- and N-terminal deletion mutation of apoA-I, having only the central domain of apoA-I helices 2–7, suffered at most a slight reduction in its level of lipidation and particle formation compared to that of wild-type apoA-I.47
Proposal for ApoA-I Opening
Helical repeat 5 is roughly in the middle of the apoA-I chain. In the lipid-free form, repeat 5 is folded in the middle of what becomes the nearly linear amphipathic helix 5 in nHDL. Models of human and mouse lipid-free apoA-I also show a break or bend in helix 5.27–30 Lipid-free apoA-I can be readily opened to the belt form of circular apoA-I if region 5 of lipid-free apoA-I is first converted to an amphipathic helix (Figure 4). Sequence 4a interprets the opening of lipid-free apoA-I in four basic steps. In lipid-free apoA-I, 4ai, helical repeat 5 is folded in the middle, and converting this repeat to an α-helix opens apoA-I to 4aii. The two arms of the N- and C-terminal regions open further, 4aiii, and then complete the process, giving the belt conformation reported for nHDL,16 4aiv. This figure shows that the lipid-free C-terminal conformation was similar to the lipidated C-terminal conformation. When lipid-free mutant F104C/H162C, 4bi, was locked by intramolecular disulfide bond formation, opening yielded a final conformation, 4bii, that was significantly different from 4aiv and was considerably less able to bind lipid. Mutant E34C/S55C, 4ci, when locked does not permit the N-terminal end to fold back over helical repeats 1 and 2, 4ciii, but can accommodate lipid. Mutant L200C/L233C, 4di, has little or not effect on the final conformation of the C-terminal region, 4div, or lipidation. A reduced level of formation of rHDL and nHDL in mutant F104C/H162C was consistent with the internal intramolecular disulfide bond causing a major disruption to lipidation.
Figure 4.

Hypothetical lipid binding mechanism for ApoA-I. Lipid-free, wild-type apoA-I, 4ai, is compact and bundled such that the N- and C-terminal ends are folded and lie close to one another.24 Upon initial interaction of lipid with apoA-I, helical region 5 undergoes a transition to an extended conformation, 4aii. The hydrophobic core of apoA-I is now exposed, allowing central helices 4–6 to acquire lipid and form HDL particles, 4aiii and 4aiv. The resulting three-apoA-I particle carries up to 115 molecules of free cholesterol. Mutant apoA-Is are shown below wild-type apoA-I, 4ai. The circles indicate where the intramolecular disulfide bond is located in each of the mutants: F104C/H162C, 4bi, E34C/S55C, 4ci, and L200C/L233C, 4di. Note the unusual conformation forced upon the protein by the disulfide cross-link at F104C/H162C prevents helical regions 4–6 from opening to associate with hydrophobic lipids.
For these studies, we also analyzed the presence or absence of intramolecular disulfide bonds in rHDL prepared with double-cysteine-containing apoA-I mutants that should not have cross-linked. Three of the four mutants did not form intramolecular disulfide bonds upon refolding, nor did they form disulfide bonds during cholate dialysis. However, D13C/V67C was unique in that the lipidated particles carried mutant apoA-I having an intramolecular disulfide cross-link, suggesting that during the reorganization required for lipidation, cysteines 13 and 67 were sufficiently close and formed an intramolecular disulfide bond. Reexamining an earlier model for discoidal rHDL showed that the coordinates for these positions were already relatively close to one another.16,35 A recently published X-ray structure for C-terminally truncated apoA-I also indicated these positions were relatively close together.33 We have revised the model for discoidal rHDL to account for the small amount of rotation required to achieve bond formation. The updated apoA-I conformation is labeled 4aiv in Figure 4.
Conclusions
ApoA-I is essential for atheroprotection because of its roles in lipid metabolism and cholesterol homeostasis, and how it achieves these ends is under rigorous investigation in several laboratories. One of the essential questions is how the conformation of lipid-free apoA-I rearranges to accept lipid and whether some steps through which apoA-I opens to accept lipid are more important than others. We have addressed the question by preparing a series of double-cysteine mutants and measuring the ability of these mutants to form rHDL by cholate dialysis and nHDL by ABCA1-promoted lipidation. Our results show that opening of the central region was essential for the synthetic process that yields 9.6–9.8 nm diameter discoidal rHDL. Locking either the N-terminus or C-terminus slowed, but did not prevent, rHDL formation. Therefore, the C- and N-terminal regions participate in the process of lipidation but are not necessarily essential for lipidation. However, nHDL formation promoted by HEK293 cells expressing ABCA1 had a more complicated outcome. Alkylation of the cysteines caused an overall reduction in the level of nHDL formation. Locking the two cysteines caused a further reduction in the level of nHDL formation with the internal locked mutant yielding the least nHDL, consistent with what was found for rHDL formation. These studies indicated that the various steric and bonding interactions that associate apoA-I and ABCA1 were exquisitely sensitive to minor modifications of apoA-I amino acids.
Acknowledgments
We thank Abraham K. Gebre for his help in separating nHDL classes.
Funding
These studies were supported by grants from the National Institutes of Health (NIH) (National Heart, Lung and Blood Institute), HL-49373, HL-64163, and HL-112270 (M.G.S.-T.), and the American Heart Association, 09GRNT2280053 and 14GRNT20500029 (M.J.T.). R.D.P. was supported by NIH Diversity Supplement HL112270A1S1. The Waters Q-TOF mass spectrometer was obtained from NIH Shared Instrumentation Grant 1S10RR17846 (M.J.T.). MS analyses were performed in the Mass Spectrometer Facility of the Comprehensive Cancer Center of the Wake Forest School of Medicine supported in part by National Cancer Institute Center Grant 5P30CA12197.
ABBREVIATIONS
- ABCA1
ATP-binding cassette transporter 1
- apoLp-III
apolipophorin III
- CHD
coronary heart disease
- FBS
fetal bovine serum
- HDL
high-density lipoprotein
- HEK
human embryonic kidney
- LCAT
lecithin cholesterol acyltransferase
- nHDL
nascent HDL
- POPC
palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- rHDL
recombinant HDL
Footnotes
The authors declare no competing financial interest.
Contributor Information
Ricquita D. Pollard, Section on Molecular Medicine, Department of Internal Medicine, Wake Forest School of Medicine, Winston-Salem, North Carolina 27101, United States
Brian Fulp, Department of Biochemistry, Wake Forest School of Medicine, Winston-Salem, North Carolina 27101, United States.
Mary G. Sorci-Thomas, Departments of Medicine, Division of Endocrinology, Pharmacology and Toxicology, and Blood Research Institute, BloodCenter of Wisconsin, Medical College of Wisconsin, Milwaukee, Wisconsin 53226, United States
Michael J. Thomas, Department of Pharmacology and Toxicology, Medical College of Wisconsin, 8701 Watertown Plank Road, Milwaukee, Wisconsin 53226, United States.
References
- 1.Castelli WP, Doyle JT, Gordon T, Hames CG, Hjortland MC, Hulley SB, Kagan A, Zukel WJ. HDL cholesterol and other lipids in coronary heart disease. The cooperative lipoprotein phenotyping study. Circulation. 1977;55:767–772. doi: 10.1161/01.cir.55.5.767. [DOI] [PubMed] [Google Scholar]
- 2.Barter PJ, Brewer HB, Jr, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler, Thromb, Vasc Biol. 2003;23:160–167. doi: 10.1161/01.atv.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- 3.de Grooth GJ, Klerkx AH, Stroes ES, Stalenhoef AF, Kastelein JJ, Kuivenhoven JA. A review of CETP and its relation to atherosclerosis. J Lipid Res. 2004;45:1967–1974. doi: 10.1194/jlr.R400007-JLR200. [DOI] [PubMed] [Google Scholar]
- 4.Kuvin JT, Alsheikh-Ali AA, Karas RH. High-density lipoprotein cholesterol-raising strategies. J Cardiovasc Pharmacol. 2006;47:196–204. doi: 10.1097/01.fjc.0000199684.20578.7c. [DOI] [PubMed] [Google Scholar]
- 5.Glomset JA. The plasma lecithins:cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–167. [PubMed] [Google Scholar]
- 6.Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. J Lipid Res. 1995;36:211–228. [PubMed] [Google Scholar]
- 7.Joy TR, Hegele RA. The failure of torcetrapib: what have we learned? Br J Pharmacol. 2008;154:1379–1381. doi: 10.1038/bjp.2008.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tall AR. CETP inhibitors to increase HDL cholesterol levels. N Engl J Med. 2007;356:1364–1366. doi: 10.1056/NEJMe078029. [DOI] [PubMed] [Google Scholar]
- 9.Wright RS. Recent clinical trials evaluating benefit of drug therapy for modification of HDL cholesterol. Curr Opin Cardiol. 2013;28:389–398. doi: 10.1097/HCO.0b013e328362059d. [DOI] [PubMed] [Google Scholar]
- 10.Sorci-Thomas MG, Thomas MJ. Why targeting HDL should work as a therapeutic tool, but has not. J Cardiovasc Pharmacol. 2013;62:239–246. doi: 10.1097/FJC.0b013e31829d48a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de la Llera-Moya M, Drazul-Schrader D, Asztalos BF, Cuchel M, Rader DJ, Rothblat GH. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler, Thromb, Vasc Biol. 2010;30:796–801. doi: 10.1161/ATVBAHA.109.199158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, Shaul PW. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–2393. doi: 10.1056/NEJMoa1409065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duong PT, Collins HL, Nickel M, Lund-Katz S, Rothblat GH, Phillips MC. Characterization of nascent HDL particles and microparticles formed by ABCA1-mediated efflux of cellular lipids to apoA-I. J Lipid Res. 2006;47:832–843. doi: 10.1194/jlr.M500531-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Ji A, Wroblewski JM, Cai L, de Beer MC, Webb NR, van der Westhuyzen DR. Nascent HDL formation in hepatocytes and role of ABCA1, ABCG1, and SR-BI. J Lipid Res. 2012;53:446–455. doi: 10.1194/jlr.M017079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sorci-Thomas MG, Owen JS, Fulp B, Bhat S, Zhu X, Parks JS, Shah D, Jerome WG, Gerelus M, Zabalawi M, Thomas MJ. Nascent high density lipoproteins formed by ABCA1 resemble lipid rafts and are structurally organized by three apoA-I monomers. J Lipid Res. 2012;53:1890–1909. doi: 10.1194/jlr.M026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segrest JP, Li L, Anantharamaiah GM, Harvey SC, Liadaki KN, Zannis V. Structure and function of apolipoprotein A-I and high-density lipoprotein. Curr Opin Lipidol. 2000;11:105–115. doi: 10.1097/00041433-200004000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Segrest JP, Garber DW, Brouillette CG, Harvey SC, Anantharamaiah GM. The amphipathic alpha helix: a multifunctional structural motif in plasma apolipoproteins. Adv Protein Chem. 1994;45:303–369. doi: 10.1016/s0065-3233(08)60643-9. [DOI] [PubMed] [Google Scholar]
- 19.Brouillette CG, Anantharamaiah GM, Engler JA, Borhani DW. Structural models of human apolipoprotein AI: a critical analysis and review. Biochim Biophys Acta, Mol Cell Biol Lipids. 2001;1531:4–46. doi: 10.1016/s1388-1981(01)00081-6. [DOI] [PubMed] [Google Scholar]
- 20.Stone WL, Reynolds JA. The self-association of the apo-Gln-I and apo-Gln-II polypeptides of human high density serum lipoproteins. J Biol Chem. 1975;250:8045–8048. [PubMed] [Google Scholar]
- 21.Barbeau DL, Jonas A, Teng T, Scanu AM. Asymmetry of apolipoprotein A-I in solution as assessed from ultracentrifugal, viscometric, and fluorescence polarization studies. Biochemistry. 1979;18:362–369. doi: 10.1021/bi00569a021. [DOI] [PubMed] [Google Scholar]
- 22.Roberts LM, Ray MJ, Shih TW, Hayden E, Reader MM, Brouillette CG. Structural analysis of apolipoprotein A-I: limited proteolysis of methionine-reduced and -oxidized lipid-free and lipid-bound human apo A-I. Biochemistry. 1997;36:7615–7624. doi: 10.1021/bi962952g. [DOI] [PubMed] [Google Scholar]
- 23.Rogers DP, Roberts LM, Lebowitz J, Datta G, Anantharamaiah GM, Engler JA, Brouillette CG. The lipid-free structure of apolipoprotein A-I: effects of amino-terminal deletions. Biochemistry. 1998;37:11714–11725. doi: 10.1021/bi973112k. [DOI] [PubMed] [Google Scholar]
- 24.Rogers DP, Roberts LM, Lebowitz J, Engler JA, Brouillette CG. Structural analysis of apolipoprotein A-I: effects of amino- and carboxy-terminal deletions on the lipid-free structure. Biochemistry. 1998;37:945–955. doi: 10.1021/bi9713512. [DOI] [PubMed] [Google Scholar]
- 25.Brouillette CG, Dong WJ, Yang ZW, Ray MJ, Protasevich II, Cheung HC, Engler JA. Forster resonance energy transfer measurements are consistent with a helical bundle model for lipid-free apolipoprotein A-I. Biochemistry. 2005;44:16413–16425. doi: 10.1021/bi051018v. [DOI] [PubMed] [Google Scholar]
- 26.Koyama M, Tanaka M, Dhanasekaran P, Lund-Katz S, Phillips MC, Saito H. Interaction between the N- and C-terminal domains modulates the stability and lipid binding of apolipoprotein A-I. Biochemistry. 2009;48:2529–2537. doi: 10.1021/bi802317v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pollard RD, Fulp B, Samuel MP, Sorci-Thomas MG, Thomas MJ. The conformation of lipid-free human apolipoprotein A-I in solution. Biochemistry. 2013;52:9470–9481. doi: 10.1021/bi401080k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silva RA, Hilliard GM, Fang J, Macha S, Davidson WS. A three-dimensional molecular model of lipid-free apolipoprotein A-I determined by cross-linking/mass spectrometry and sequence threading. Biochemistry. 2005;44:2759–2769. doi: 10.1021/bi047717+. [DOI] [PubMed] [Google Scholar]
- 29.Yang Y, Hoyt D, Wang J. A complete NMR spectral assignment of the lipid-free mouse apolipoprotein A-I (apoAI) C-terminal truncation mutant, apoAI(1–216) Biomol NMR Assignments. 2007;1:109–111. doi: 10.1007/s12104-007-9031-2. [DOI] [PubMed] [Google Scholar]
- 30.Segrest JP, Jones MK, Shao B, Heinecke JW. An experimentally robust model of monomeric apolipoprotein A-I created from a chimera of two X-ray structures and molecular dynamics simulations. Biochemistry. 2014;53:7625–7640. doi: 10.1021/bi501111j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melchior JT, Walker RG, Morris J, Jones MK, Segrest JP, Lima DB, Carvalho PC, Gozzo FC, Castleberry M, Thompson TB, Davidson WS. An Evaluation of the Crystal Structure of C-terminal Truncated Apolipoprotein A-I in Solution Reveals Structural Dynamics Related to Lipid Binding. J Biol Chem. 2016;291:5439–5451. doi: 10.1074/jbc.M115.706093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borhani DW, Rogers DP, Engler JA, Brouillette CG. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc Natl Acad Sci U S A. 1997;94:12291–12296. doi: 10.1073/pnas.94.23.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mei X, Atkinson D. Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J Biol Chem. 2011;286:38570–38582. doi: 10.1074/jbc.M111.260422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhat S, Sorci-Thomas MG, Alexander ET, Samuel MP, Thomas MJ. Intermolecular contact between globular N-terminal fold and C-terminal domain of ApoA-I stabilizes its lipid-bound conformation: studies employing chemical cross-linking and mass spectrometry. J Biol Chem. 2005;280:33015–33025. doi: 10.1074/jbc.M505081200. [DOI] [PubMed] [Google Scholar]
- 35.Bhat S, Sorci-Thomas MG, Tuladhar R, Samuel MP, Thomas MJ. Conformational adaptation of apolipoprotein AI to discretely sized phospholipid complexes. Biochemistry. 2007;46:7811–7821. doi: 10.1021/bi700384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Lyles DS, Thomas MJ, Pan W, Sorci-Thomas MG. Structural determination of lipid-bound ApoA-I using fluorescence resonance energy transfer. J Biol Chem. 2000;275:37048–37054. doi: 10.1074/jbc.M005336200. [DOI] [PubMed] [Google Scholar]
- 37.Thomas MJ, Bhat S, Sorci-Thomas MG. Three-dimensional models of HDL apoA-I: implications for its assembly and function. J Lipid Res. 2008;49:1875–1883. doi: 10.1194/jlr.R800010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silva RA, Hilliard GM, Li L, Segrest JP, Davidson WS. A mass spectrometric determination of the conformation of dimeric apolipoprotein A-I in discoidal high density lipoproteins. Biochemistry. 2005;44:8600–8607. doi: 10.1021/bi050421z. [DOI] [PubMed] [Google Scholar]
- 39.Wu Z, Wagner MA, Zheng L, Parks JS, Shy JM, 3rd, Smith JD, Gogonea V, Hazen SL. The refined structure of nascent HDL reveals a key functional domain for particle maturation and dysfunction. Nat Struct Mol Biol. 2007;14:861–868. doi: 10.1038/nsmb1284. [DOI] [PubMed] [Google Scholar]
- 40.Li L, Li S, Jones MK, Segrest JP. Rotational and hinge dynamics of discoidal high density lipoproteins probed by interchain disulfide bond formation. Biochim Biophys Acta, Mol Cell Biol Lipids. 2012;1821:481–489. doi: 10.1016/j.bbalip.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka M, Tanaka T, Ohta S, Kawakami T, Konno H, Akaji K, Aimoto S, Saito H. Evaluation of lipid-binding properties of the N-terminal helical segments in human apolipoprotein A-I using fragment peptides. J Pept Sci. 2009;15:36–42. doi: 10.1002/psc.1092. [DOI] [PubMed] [Google Scholar]
- 42.Gillotte KL, Zaiou M, Lund-Katz S, Anantharamaiah GM, Holvoet P, Dhoest A, Palgunachari MN, Segrest JP, Weisgraber KH, Rothblat GH, Phillips MC. Apolipoprotein-mediated plasma membrane microsolubilization. Role of lipid affinity and membrane penetration in the efflux of cellular cholesterol and phospholipid. J Biol Chem. 1999;274:2021–2028. doi: 10.1074/jbc.274.4.2021. [DOI] [PubMed] [Google Scholar]
- 43.Palgunachari MN, Mishra VK, Lund-Katz S, Phillips MC, Adeyeye SO, Alluri S, Anantharamaiah GM, Segrest JP. Only the two end helixes of eight tandem amphipathic helical domains of human apo A-I have significant lipid affinity. Implications for HDL assembly. Arterioscler, Thromb, Vasc Biol. 1996;16:328–338. doi: 10.1161/01.atv.16.2.328. [DOI] [PubMed] [Google Scholar]
- 44.Fang Y, Gursky O, Atkinson D. Lipid-binding studies of human apolipoprotein A-I and its terminally truncated mutants. Biochemistry. 2003;42:13260–13268. doi: 10.1021/bi0354031. [DOI] [PubMed] [Google Scholar]
- 45.Minnich A, Collet X, Roghani A, Cladaras C, Hamilton RL, Fielding CJ, Zannis VI. Site-directed mutagenesis and structure-function analysis of the human apolipoprotein A-I. Relation between lecithin-cholesterol acyltransferase activation and lipid binding. J Biol Chem. 1992;267:16553–16560. [PubMed] [Google Scholar]
- 46.Holvoet P, Zhao Z, Vanloo B, Vos R, Deridder E, Dhoest A, Taveirne J, Brouwers E, Demarsin E, Engelborghs Y. Phospholipid binding and lecithin-cholesterol acyltransferase activation properties of apolipoprotein A-I mutants. Biochemistry. 1995;34:13334–13342. doi: 10.1021/bi00041a009. [DOI] [PubMed] [Google Scholar]
- 47.Chroni A, Liu T, Gorshkova I, Kan HY, Uehara Y, Von Eckardstein A, Zannis VI. The central helices of ApoA-I can promote ATP-binding cassette transporter A1 (ABCA1)-mediated lipid efflux. Amino acid residues 220–231 of the wild-type ApoA-I are required for lipid efflux in vitro and high density lipoprotein formation in vivo. J Biol Chem. 2003;278:6719–6730. doi: 10.1074/jbc.M205232200. [DOI] [PubMed] [Google Scholar]
- 48.Scott BR, McManus DC, Franklin V, McKenzie AG, Neville T, Sparks DL, Marcel YL. The N-terminal globular domain and the first class A amphipathic helix of apolipoprotein A-I are important for lecithin:cholesterol acyltransferase activation and the maturation of high density lipoprotein in vivo. J Biol Chem. 2001;276:48716–48724. doi: 10.1074/jbc.M106265200. [DOI] [PubMed] [Google Scholar]
- 49.Panagotopulos SE, Witting SR, Horace EM, Hui DY, Maiorano JN, Davidson WS. The role of apolipoprotein A-I helix 10 in apolipoprotein-mediated cholesterol efflux via the ATP-binding cassette transporter ABCA1. J Biol Chem. 2002;277:39477–39484. doi: 10.1074/jbc.M207005200. [DOI] [PubMed] [Google Scholar]
- 50.Wang S, Smith JD. ABCA1 and nascent HDL biogenesis. Biofactors. 2014;40:547–554. doi: 10.1002/biof.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang S, Gulshan K, Brubaker G, Hazen SL, Smith JD. ABCA1 mediates unfolding of apolipoprotein AI N terminus on the cell surface before lipidation and release of nascent high-density lipoprotein. Arterioscler, Thromb, Vasc Biol. 2013;33:1197–1205. doi: 10.1161/ATVBAHA.112.301195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gursky O. Crystal structure of Delta(185–243)ApoA-I suggests a mechanistic framework for the protein adaptation to the changing lipid load in good cholesterol: from flatland to sphereland via double belt, belt buckle, double hairpin and trefoil/tetrafoil. J Mol Biol. 2013;425:1–16. doi: 10.1016/j.jmb.2012.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem. 2014;289:24020–24029. doi: 10.1074/jbc.R114.583658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fielding PE, Nagao K, Hakamata H, Chimini G, Fielding CJ. A two-step mechanism for free cholesterol and phospholipid efflux from human vascular cells to apolipoprotein A-1. Biochemistry. 2000;39:14113–14120. doi: 10.1021/bi0004192. [DOI] [PubMed] [Google Scholar]
- 55.Wang N, Lan D, Gerbod-Giannone M, Linsel-Nitschke P, Jehle AW, Chen W, Martinez LO, Tall AR. ATP-binding cassette transporter A7 (ABCA7) binds apolipoprotein A-I and mediates cellular phospholipid but not cholesterol efflux. J Biol Chem. 2003;278:42906–42912. doi: 10.1074/jbc.M307831200. [DOI] [PubMed] [Google Scholar]
- 56.Smith JD, Le Goff W, Settle M, Brubaker G, Waelde C, Horwitz A, Oda MN. ABCA1 mediates concurrent cholesterol and phospholipid efflux to apolipoprotein A-I. J Lipid Res. 2004;45:635–644. doi: 10.1194/jlr.M300336-JLR200. [DOI] [PubMed] [Google Scholar]
- 57.Rigot V, Hamon Y, Chambenoit O, Alibert M, Duverger N, Chimini G. Distinct sites on ABCA1 control distinct steps required for cellular release of phospholipids. J Lipid Res. 2002;43:2077–2086. doi: 10.1194/jlr.m200279-jlr200. [DOI] [PubMed] [Google Scholar]
- 58.Landry YD, Denis M, Nandi S, Bell S, Vaughan AM, Zha X. ATP-binding cassette transporter A1 expression disrupts raft membrane microdomains through its ATPase-related functions. J Biol Chem. 2006;281:36091–36101. doi: 10.1074/jbc.M602247200. [DOI] [PubMed] [Google Scholar]
- 59.Nagao K, Kimura Y, Mastuo M, Ueda K. Lipid outward translocation by ABC proteins. FEBS Lett. 2010;584:2717–2723. doi: 10.1016/j.febslet.2010.04.036. [DOI] [PubMed] [Google Scholar]
- 60.Nagao K, Hata M, Tanaka K, Takechi Y, Nguyen D, Dhanasekaran P, Lund-Katz S, Phillips MC, Saito H. The roles of C-terminal helices of human apolipoprotein A-I in formation of high-density lipoprotein particles. Biochim Biophys Acta, Mol Cell Biol Lipids. 2014;1841:80–87. doi: 10.1016/j.bbalip.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li HH, Thomas MJ, Pan W, Alexander E, Samuel M, Sorci-Thomas MG. Preparation and incorporation of probe-labeled apoA-I for fluorescence resonance energy transfer studies of rHDL. J Lipid Res. 2001;42:2084–2091. [PubMed] [Google Scholar]
- 62.Bhat S, Zabalawi M, Willingham MC, Shelness GS, Thomas MJ, Sorci-Thomas MG. Quality control in the apoA-I secretory pathway: deletion of apoA-I helix 6 leads to the formation of cytosolic phospholipid inclusions. J Lipid Res. 2004;45:1207–1220. doi: 10.1194/jlr.M300498-JLR200. [DOI] [PubMed] [Google Scholar]
- 63.Owen JS, Bharadwaj MS, Thomas MJ, Bhat S, Samuel MP, Sorci-Thomas MG. Ratio determination of plasma wild-type and L159R apoA-I using mass spectrometry: tools for studying apoA-IFin. J Lipid Res. 2007;48:226–234. doi: 10.1194/jlr.D600031-JLR200. [DOI] [PubMed] [Google Scholar]
- 64.Li HH, Lyles DS, Pan W, Alexander E, Thomas MJ, Sorci-Thomas MG. ApoA-I structure on discs and spheres. Variable helix registry and conformational states. J Biol Chem. 2002;277:39093–39101. doi: 10.1074/jbc.M206770200. [DOI] [PubMed] [Google Scholar]
- 65.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 66.Rouser G, Siakotos AN, Fleischer S. Quantitative analysis of phospholipids by thin-layer chromatography and phosphorus analysis of spots. Lipids. 1966;1:85–86. doi: 10.1007/BF02668129. [DOI] [PubMed] [Google Scholar]
- 67.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 68.Bhat S, Sorci-Thomas MG, Calabresi L, Samuel MP, Thomas MJ. Conformation of dimeric apolipoprotein A-I milano on recombinant lipoprotein particles. Biochemistry. 2010;49:5213–5224. doi: 10.1021/bi1003734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu B, Morrow JA, Weisgraber KH. Conformational reorganization of the four-helix bundle of human apolipoprotein E in binding to phospholipid. J Biol Chem. 2000;275:20775–20781. doi: 10.1074/jbc.M003508200. [DOI] [PubMed] [Google Scholar]
- 70.Mulya A, Lee JY, Gebre AK, Thomas MJ, Colvin PL, Parks JS. Minimal lipidation of pre-beta HDL by ABCA1 results in reduced ability to interact with ABCA1. Arterioscler, Thromb, Vasc Biol. 2007;27:1828–1836. doi: 10.1161/ATVBAHA.107.142455. [DOI] [PubMed] [Google Scholar]
- 71.Lagerstedt JO, Budamagunta MS, Liu GS, DeValle NC, Voss JC, Oda MN. The “beta-clasp” model of apolipoprotein A-I–a lipid-free solution structure determined by electron paramagnetic resonance spectroscopy. Biochim Biophys Acta, Mol Cell Biol Lipids. 2012;1821:448–455. doi: 10.1016/j.bbalip.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leitner A, Reischl R, Walzthoeni T, Herzog F, Bohn S, Forster F, Aebersold R. Expanding the chemical cross-linking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Mol Cell Proteomics. 2012;11:M111014126. doi: 10.1074/mcp.M111.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Young MM, Tang N, Hempel JC, Oshiro CM, Taylor EW, Kuntz ID, Gibson BW, Dollinger G. High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc Natl Acad Sci U S A. 2000;97:5802–5806. doi: 10.1073/pnas.090099097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jacobsen RB, Sale KL, Ayson MJ, Novak P, Hong J, Lane P, Wood NL, Kruppa GH, Young MM, Schoeniger JS. Structure and dynamics of dark-state bovine rhodopsin revealed by chemical cross-linking and high-resolution mass spectrometry. Protein Sci. 2006;15:1303–1317. doi: 10.1110/ps.052040406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walzthoeni T, Leitner A, Stengel F, Aebersold R. Mass spectrometry supported determination of protein complex structure. Curr Opin Struct Biol. 2013;23:252–260. doi: 10.1016/j.sbi.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 76.Leitner A, Walzthoeni T, Kahraman A, Herzog F, Rinner O, Beck M, Aebersold R. Probing native protein structures by chemical cross-linking, mass spectrometry, and bioinformatics. Mol Cell Proteomics. 2010;9:1634–1649. doi: 10.1074/mcp.R000001-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang BX, Dass C, Kim HY. Probing conformational changes of human serum albumin due to unsaturated fatty acid binding by chemical cross-linking and mass spectrometry. Biochem J. 2005;387:695–702. doi: 10.1042/BJ20041624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang BX, Kim HY, Dass C. Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry. J Am Soc Mass Spectrom. 2004;15:1237–1247. doi: 10.1016/j.jasms.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 79.Leitner A. Cross-linking and other structural proteomics techniques: how chemistry is enabling mass spectrometry applications in structural biology. Chemical Science. 2016;7:4792–4803. doi: 10.1039/c5sc04196a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hofmann T, Fischer AW, Meiler J, Kalkhof S. Protein structure prediction guided by crosslinking restraints–A systematic evaluation of the impact of the crosslinking spacer length. Methods. 2015;89:79–90. doi: 10.1016/j.ymeth.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leitner A, Faini M, Stengel F, Aebersold R. Crosslinking and Mass Spectrometry: An Integrated Technology to Understand the Structure and Function of Molecular Machines. Trends Biochem Sci. 2016;41:20–32. doi: 10.1016/j.tibs.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 82.Luo J, Fishburn J, Hahn S, Ranish J. An integrated chemical cross-linking and mass spectrometry approach to study protein complex architecture and function. Mol Cell Proteomics. 2012;11:M111.008318. doi: 10.1074/mcp.M111.008318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sikic K, Tomic S, Carugo O. Systematic comparison of crystal and NMR protein structures deposited in the protein data bank. Open Biochem J. 2010;4:83–95. doi: 10.2174/1874091X01004010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Narayanaswami V, Wang J, Kay CM, Scraba DG, Ryan RO. Disulfide bond engineering to monitor conformational opening of apolipophorin III during lipid binding. J Biol Chem. 1996;271:26855–26862. doi: 10.1074/jbc.271.43.26855. [DOI] [PubMed] [Google Scholar]
- 85.Davidson WS, Hazlett T, Mantulin WW, Jonas A. The role of apolipoprotein AI domains in lipid binding. Proc Natl Acad Sci U S A. 1996;93:13605–13610. doi: 10.1073/pnas.93.24.13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fang Y, Gursky O, Atkinson D. Structural studies of N- and C-terminally truncated human apolipoprotein A-I. Biochemistry. 2003;42:6881–6890. doi: 10.1021/bi034152t. [DOI] [PubMed] [Google Scholar]
- 87.Oda MN, Forte TM, Ryan RO, Voss JC. The C-terminal domain of apolipoprotein A-I contains a lipid-sensitive conformational trigger. Nat Struct Biol. 2003;10:455–460. doi: 10.1038/nsb931. [DOI] [PubMed] [Google Scholar]