

Abstract
The synthesis of a series of thiolated paclitaxel analogs is described as part of a novel nanomedicine program aimed at developing formulations of paclitaxel that will bind to gold nanoparticles for tumor targeted drug delivery. Preliminary evaluation of the new nanomedicine comprised of 27 nm gold nanoparticles, tumor necrosis factor alpha (TNFα), thiolated polyethylene glycol (PEG-Thiol) and one of several thiolated paclitaxel analogs is presented.
Graphic abstract
INTRODUCTION
Chemotherapy has been and continues to be one of the most effective ways to treat cancer, but many of the chemotherapeutic drugs used are cytotoxic and have significant side effects. Among chemotherapy drugs paclitaxel is one of the most widely used and effective agents, and it is the first-line treatment for breast, ovarian, lung, and colon cancer. Its mechanism of action involves binding to microtubules and stabilizing them, ultimately causing apoptosis.1, 2 It does, however have significant side effects, including alopecia, nausea and vomiting, myelosuppression,3 and peripheral neuropathy.4 These side effects are caused in large part because paclitaxel is given by intravenous infusion, and thus has the potential to be distributed throughout the body and not just to the tumor site. In spite of these problems a 2008 review stated “Paclitaxel and docetaxel are considered fundamental drugs in the treatment of breast cancer,”5 and a 2012 review stated “Today, paclitaxel and docetaxel are widely prescribed antineoplastic agents for a broad range of malignancies including lung cancer, breast cancer, prostate cancer, Kaposi’s sarcoma, squamous cell carcinoma of the head and neck, gastric cancer, esophageal cancer, bladder cancer, and other carcinomas. Although very active clinically, paclitaxel and docetaxel are associated with many serious side effects, which often preclude continued use of these agents in patients. A number of these side effects have been associated with the solvents used for the dilution of these antineoplastic agents.”6 The major problem with paclitaxel is thus a drug delivery problem; if it could be delivered specifically to the tumor, its side effects would be minimized, potentially making it even more effective.
This concept of targeting chemotherapeutic drugs in general and paclitaxel in particular to tumors is not a new one, and numerous publications on different approaches have appeared in the last few years. These include antibody-drug conjugates,7–9 and various nanoparticle-based approaches.10 The advantage of the nanoparticle approach is that a measure of selectivity is assured because of the enhanced permeability and retention (EPR) effect,11 which is primarily due to the preferential extravasation of nanoparticles through the leaky vasculature of tumors over the tight vasculature of normal tissue.
A fundamental issue in nanomedicine development is whether and to what extent the EPR effect exists in human cancers, as over time it may become a self-limiting phenomenon. One such limitation is that the combination of fluid leaking into the tumor coupled with impaired lymphatic vessels, which drain fluid from the tumor, combine to pressurize the tumor. This pressure, known as tumor interstitial fluid pressure (IFP), acts as a physical barrier that prevents or limits the ability of cancer therapies, potentially including nanomedicines, from penetrating the tumor. Thus it is not surprising that on average only 0.7% of the administered dose of nanoparticle drugs is delivered to solid tumors.12 This number is however still ten to one hundred-fold higher than that of naked drug molecules, and drug delivery efficiencies are closer to 10% for nanomedicines in clinical trial.13
Several nanomedicines that utilize the EPR effect to selectively deliver paclitaxel or its analog docetaxel have been proposed. In the case of paclitaxel the albumin-bound formulation Abraxane® and the polymeric micellar nanoparticle formulation Genexol-PM are in clinical use, and the micellar nanoparticle formulation NK105 is in Phase III clinical trials.14, 15 However, these formulations do not completely resolve the toxicity issues associated with paclitaxel, as indicated by the fact that Abraxane® shows significant neuropathy.16
Various studies have also appeared of paclitaxel linked to iron oxide nanoparticles,17 to gold nanoparticles,17–20 or to fullerenes,21 but none of these have resulted in any published clinical or preclinical studies. Various other paclitaxel delivery systems are described in a recent review.22 The use of nanoparticles for the delivery of docetaxel has also been reviewed recently and thus will not be discussed further.23
Given the many potential benefits of selective delivery of paclitaxel to tumors, we initiated a research program to explore its delivery by nanoparticles. We selected gold nanoparticles (AuNP) for this tumor-targeted delivery because they are readily prepared in a range of sizes, they are chemically stable and biocompatible, and they can be functionalized with a variety of ligands.24–26 As stated in a recent review “Because of their facile surface chemistry, gold nanoparticles can act as artificial antibodies whose binding affinity can be precisely tuned by varying the density of binding ligands on their surfaces.”27 In particular, they can be functionalized with tumor necrosis factor (TNF) and polyethylene glycol to generate a potent and biocompatible tumor-targeting drug delivery vehicle.28–30
The cytokine TNF has several effects, including reduction of IFP31 and apoptosis of cancer cells,32 but it is highly toxic, and cannot be used systemically. Its value has been demonstrated by its clinical use in the isolated limb perfusion (ILP) surgical procedure involving TNF and chemotherapy. In this technique, which has been approved in Europe since 1998, TNF followed by a chemotherapy, such as melphalan or doxorubicin, are sequentially perfused via complex surgery into the tumor-burdened limb, resulting in a 60–85% complete local response rate in cancer patients with refractory tumors.33, 34 The localized delivery of TNF induces vascular leak in the tumor, reduction in the tumor IFP,31 and enhanced tumor uptake of follow-on chemotherapy.35 However, ILP is not applicable to most tumors, either primary or metastatic, and the potential benefits of TNF are thus only available to a small subset of cancer patients, but the success of ILP with TNF followed by chemotherapy suggests a strategy for developing tumor-targeting nanomedicines for cancer treatment.
AuNPs provide an attractive alternative to the use of ILP as a means of selective delivery of TNF to tumors. In a key study of this approach, AuNPs were functionalized with TNF, bound through its cysteine residues, and with sulfhydryl-polyethylene glycol (PEG-Thiol) to make them able to target tumors and to be undetectable by the immune system, respectively.36 The resulting nanomedicine, termed CYT-6091, has to date demonstrated many of the anti-tumor effects of TNF without inducing its toxic side effects. For example, by targeting the tumor vasculature, CYT-6091 induces the selective disruption of the tumor vascular bed37 and increases the uptake of contrast MRI contrast agents, a surrogate for the uptake of chemotherapy in solid tumors.38 Following vascular disruption CYT-6091 reduces tumor interstitial fluid pressure in some but not all tumors tested,39 and increases the efficacy of energy based treatments such as radio and thermal based therapies40 as well as chemotherapy.41 Evaluation of CYT-6091 in MC38 tumor-bearing mice indicated that it selectively induced tumor neovascular permeability.37 Finally, in a pivotal Phase I clinical study CYT-6091 selectively accumulated in patient tumors, and increased overall systemic exposure to the cytokine without inducing the dose limiting toxicities of hypotension and hepatotoxicity associated with systemic TNF administration.28 These findings suggest that CYT-6091 may be replicating the first arm of the ILP procedure to effectively deliver the benefits of TNF to a broad spectrum of cancers.
Although these findings are highly significant, both CYT-6091 and the ILP protocol require the use of a separate chemotherapeutic agent, which is not targeted specifically to the tumor. Consequently, linking a chemotherapeutic agent directly to the TNF-PEGylated-AuNP construct would target both TNF and the chemotherapeutic agent to the tumor and thus reduce side effects associated with untargeted agents. We selected paclitaxel as the chemotherapeutic agent of choice because of its premier place among cancer drugs. Portions of this work are the subject of a US Patent.42
The design of a paclitaxel analog for binding to gold nanoparticles for drug delivery must meet three basic requirements: it must contain a thiol or substituted thiol group positioned to bind to the gold nanoparticle, it must be stable in whole blood so that it does not release the drug in the circulation, and it must be capable of release from the gold nanoparticle to regenerate paclitaxel once it reaches the tumor. This last criterion is important if the objective is to deliver native paclitaxel to the tumor. In earlier work we designed paclitaxel analogs such as 1 (Fig. 1) that could be linked to gold nanoparticles,29 but did not readily regenerate paclitaxel from the nanoparticle since the linkage was the relatively stable C-7 ester,43 and their efficacy was thus limited. A similar concern applies to the well-characterized paclitaxel-linked AuNP (2) prepared by Gibson et al.,18 since these were also linked to the nanoparticle through a relatively stable ester linkage.
Figure 1.
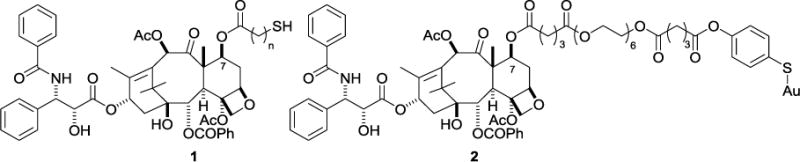
Previous thiolated paclitaxel derivatives.
In this work we describe the synthesis and initial evaluation of four paclitaxel analogs that meet the criteria desired above and are suitable for development as TNF-PEGylated-AuNP-linked drugs for selective delivery to tumors, and provide preliminary biological data on two of them.
RESULTS
Synthesis of New Thiolated Paclitaxel Analogs
The series of paclitaxel analogs 3 – 6 (Fig. 2) was designed with thiopyridyl or lipoic acid moieties at the C-7 and C-2′ positions as the linkers to the AuNP. By design each of these compounds is converted to paclitaxel at the tumor site by either a reductive or a hydrolytic cleavage mechanism. The C-7 derivatives 3 and 4 were designed with a C-7 acyloxyalkoxy carbonate group, since this group had been shown to be readily hydrolyzed under mildly acidic conditions to regenerate paclitaxel,44 and would thus be expected to hydrolyze in the acidic tumor environment.45 The C-2′ lipoic acid derivative 6 likewise contained an acyloxyalkoxy carbonate linkage which would be expected to hydrolyze to 2′-O-succinylpaclitaxel, which would then be hydrolyzed to paclitaxel.46, 47 The 7-O-thiobenzyl derivative 5 was designed to undergo reductive cleavage from the AuNP by glutathione in the reducing tumor environment and to release native paclitaxel.48 Glutathione is the most abundant intracellular thiol, with concentrations ranging from 0.2–10 mM,49 and with concentration in tumors higher than in normal tissue.50 And, this range of concentrations is far higher than the 2.09 ± 1.14 μM found in human plasma,51 indicating that a compound such as 5 should be reasonably stable in plasma but should undergo reductive cleavage in the tumor.
Figure 2.
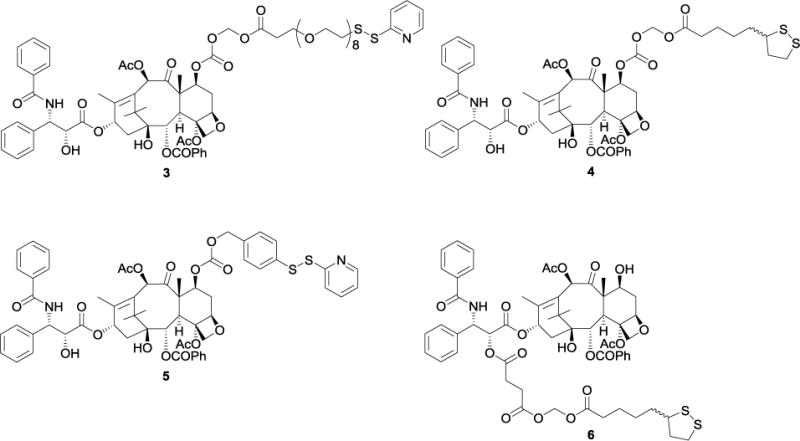
Paclitaxel derivatives 3 – 6.
Compound 3 was prepared from carboxylic acid 8 and paclitaxel derivative 11 (Scheme 1). Compound 11 was prepared from 2′-(O-tbutyldimethylsilyl)paclitaxel46 by the reported procedures.52 Acid 8 was prepared from commercially available acid 7 and 2,2′-dipyridyl disulfide in one step. Compound 11 was then refluxed with NaI in acetone to carry out halogen exchange, followed by subsequent SN2 reaction with acid 8 to produce the desired analog 3 in 60% yield.
Scheme 1. Synthesis of paclitaxel derivative 3a.
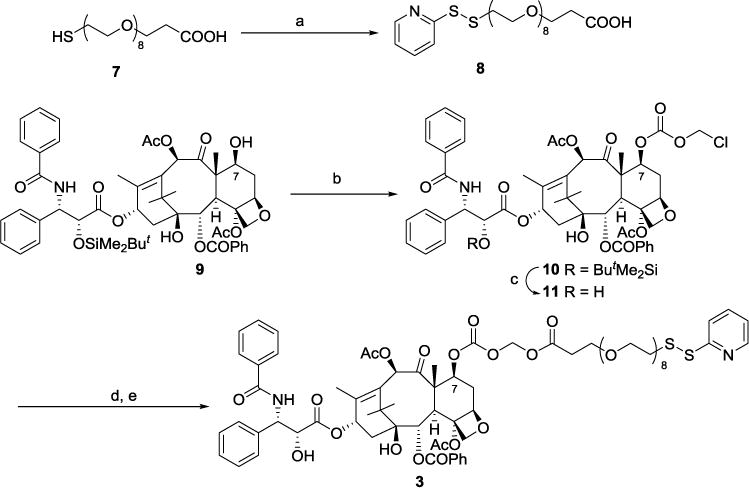
a(a) 2,2′-dipyridyldisulfide, MeOH, AcOH, 55%; (b) chloromethyl chloroformate, pyridine, CH2Cl2, 12 h, 75%; (c) HF-pyridine, THF, rt, 12 h, 90%; (d) NaI, acetone, reflux 10 h; (e) 8, K2CO3, 18-crown-6, benzene, 2 h, 76 °C, 60%.
Compound 4 was prepared by reaction of compound 11 with racemic lipoic acid using a similar procedure to that used for the conversion of 11 to 3 (Scheme 2).
Scheme 2. Synthesis of paclitaxel derivative 4a.
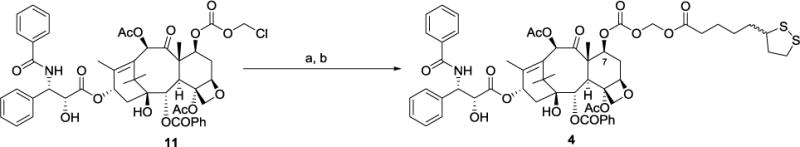
a(a) NaI, acetone, reflux 10 h; (b) Lipoic acid, K2CO3, 18-crown-6, benzene, 3.5 h, 65 °C, 78%.
Paclitaxel analog 5 was prepared (Scheme 3) by coupling precursor 1253 with the corresponding alcohol 13,48 followed by deprotection of the resulting TBDMS ether 14.
Scheme 3. Synthesis of paclitaxel derivative 5a.
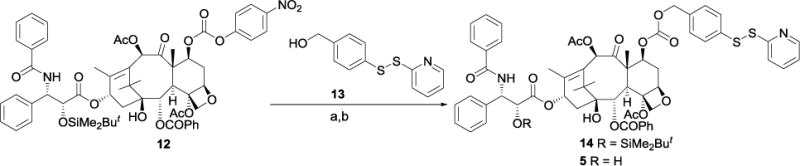
a(a) 13, DMAP, CH2Cl2, 18 h, 88%; (b) HF-pyridine, THF, rt, 16 h, 92%.
Compound 6 is the only analog in this series that possesses a C-2′ linker. Reaction of 2′-succinyl paclitaxel 1546 with chloromethyl chlorosulfate under phase-transfer conditions generated the chloromethyl ester 16. Compound 16 was then treated with lipoic acid as previously described to furnish analog 6 (Scheme 4).52
Scheme 4. Synthesis of paclitaxel derivative 6a.
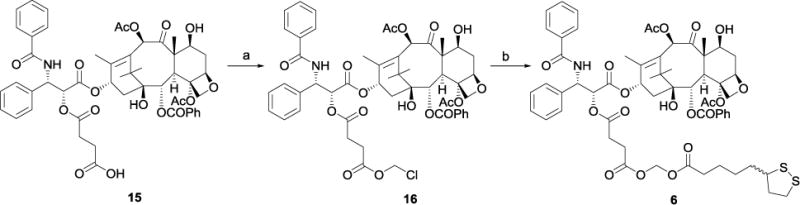
a(a) Chloromethyl chlorosulfate, NaHCO3, TBAH, H2O/CH2Cl2, 46%; (b) Lipoic acid, 18-crown-6, NaI, benzene, reflux, 60%.
The gold nanoparticle-bound version of 5 consisting of compound 5, PEG-Thiol, and TNF bound to gold nanoparticles was designated CYT-21625, and the similar nanoparticle formulation of 3 was designated CYT-20203.
Paclitaxel Release from Analogs in Buffer, Media, and Whole Blood
The stability of the linkers used to connect paclitaxel with the gold surface is a key for the gold nanoparticle-based system to achieve selective delivery of paclitaxel to tumors. Ideally the linkers should be stable as the nanomedicine travels through the circulatory system, but should be cleaved after entering the tumor to release paclitaxel. As noted earlier, analog 4 was designed to release paclitaxel under acidic conditions, so an initial investigation of its stability in buffer solution at the physiological pH 7.4 was carried out. Analog 4 was dissolved in a 3:1 mixture of methanol and PBS buffer (pH 7.4), with the methanol used to increase the solubility of the analog. Aliquots were taken at various time points and the concentrations of the analog and paclitaxel were determined by quantitative HPLC analyses. The analog was gradually hydrolyzed within 24 hours in the buffer solution and paclitaxel was generated slowly as the only product (Figure S1). A linear regression analysis indicated pseudo 1st order kinetics, and a half-life of analog 4 of 3.2 h at pH 7.4 was calculated. This study indicated that analog 4 was too labile at physiological pH to be suitable as an AuNP drug candidate. Analogs 3 and 6 were not evaluated in this study. In the case of analog 6, the relative ease of hydrolysis of 2′ esters of paclitaxel43, 47 combined with the acid-labile diacylmethylene linkage, indicated that this compound would most probably be too labile for use as a drug candidate. Analog 3 was not evaluated for paclitaxel release in this study, but it was evaluated in one experiment as the nanoparticle bound form CYT-20203 (see below).
Analog 5 was designed to release paclitaxel under reductive rather than hydrolytic conditions, and so a time course study with analysis by HPLC was carried out for this compound to assess the release of paclitaxel in the absence and in the presence of a 3-fold excess of dithiothreitol (DTT). In bicarbonate buffer without DTT 5 released less than 10% paclitaxel after 24 h, rising to 50% after 72 h (Table 1 and Figures S2 and S3). Treatment of 5 in buffer with a threefold excess of DTT resulted in 90% conversion to paclitaxel within 60 min, comparable with the results for the release of mitomycin from similar disulfides.48
Table 1.
Percent Conversion of 5 and CYT-21625 to Paclitaxel in Buffer and in Rabbit Whole Blood
| Incubation time (h) | |||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| Analyte | Medium | 0 | 1 | 4 | 24 | 48 | 72 |
| 5 | Buffer | 0.3 | 4.1 | 4 | 4.2 | 14.4 | 50.4 |
| 5 | Buffer + DTT | NDa | 90 | – | – | – | – |
| 5 | Rabbit Whole Blood | 100 | – | – | – | – | – |
| CYT-21625 | Rabbit Whole Bloodb | 0.1 | 0.1 | 0.1 | 1–2 | NDa | NDa |
Not determined.
In rabbit whole blood significant release of paclitaxel was only achieved by pretreating the sample with BME
These results thus indicated that paclitaxel analog 5 was likely to be stable in blood but would release paclitaxel in the reducing tumor environment.
Studies of Gold-Bound Analogs
The nanoparticle drug construct CYT-21625 was prepared by binding analog 5, TNF, and PEG-Thiol to gold nanoparticles as described in the experimental section. The construct CYT-20203 was prepared similarly from analog 3. CYT-21625 was added to rabbit blood, and the paclitaxel released was analyzed by HPLC. No conversion to paclitaxel was observed over a 24 h time period. Only by addition of excess β-mercaptoethanol (BME) to the blood sample was paclitaxel released from the nanomedicine, although the amount was not quantified.
CYT-21625 was also treated in bicarbonate buffer with BME and various concentrations of MeOH to assess the influence of solvent polarity on paclitaxel release. The amount of paclitaxel released from the nanomedicine was measured by quantitative RP-HPLC, while particle agglomeration was measured by determining the decrease in absorbance of the particle preparation at 525 nm. The results are shown in Table 2. Eliminating MeOH from the releasing buffer significantly reduced the ability of BME to release paclitaxel from CYT-21625. These data are consistent with the inability of BME to induce particle agglomeration and precipitation. Adding 50% MeOH to the releasing buffer resulted in increased amounts of paclitaxel released from CYT-21625 as well as MeOH-dependent particle agglomeration (Figure S2).
Table 2.
Effect of MeOH on BME-induced release of compound 5 from CYT-21625 and on BME induced particle agglomeration
| Releasing Buffera | Compound 5 released (μg/mL)b,c |
Percent Loss in OD 525 nm | |
|---|---|---|---|
| MeOH (%) | BME (μM) | ||
| 0 | 0 | 3.82 | 0 |
| 0 | 100 | 20.56 | −30 |
| 3.11 | 100 | ND | −32 |
| 12.5 | 100 | 23.13 | −39 |
| 50 | 100 | 36.52 | −54 |
Base buffer included Na2CO3 and NaHCO3
Concentration of Compound 5 (detected as paclitaxel) following release from the nanoparticle
Concentration of paclitaxel when particles fully precipitated = 40 μg/mL
The data presented in Table 2 are consistent with the hypothesis that the binding of the paclitaxel analogs onto the particle surface orients them so that they form a hydrophobic layer on the particle surface (Figure S3). The absence of MeOH from the releasing buffer is then hypothesized to reduce the ability of the reducing agent BME from accessing the disulfide group on the particle surface, thus preventing the release of the paclitaxel analogs as well as blocking BME-induced particle agglomeration. Since blood is a highly buffered homeostatic environment the hydrophobic layers serve to prevent release of the particle bound analog in the circulation (Table 1). However, we believe that the reductive tumor environment coupled with the potential disruption of the tumor vasculature and tumor necrosis mediated by TNF leads to an eventual breakdown of this layer and release of the particle bound chemotherapeutic.
Biological Evaluation of the Thiolated Analogs
The antiproliferative activities of analogs 3 – 6 were evaluated against the A2780 human ovarian cancer cell line (Table 3). Analogs 3, 4, and 6 were slightly more potent than native paclitaxel, but analog 5 was significantly less potent. These differences are presumably due to the lability of analogs 3, 4, and 6 as compared with the stability of analog 5 in buffer. The antiproliferative activities of nanoconstructs CYT-20203 and CYT-21625 were also determined. As with the unbound analogs, CYT-20203 had similar activity to native paclitaxel, but CYT-21625 was almost four-fold less potent, consistent with its greater stability.
Table 3.
Antiproliferative activities of paclitaxel and analogs against the A2780 cell line
| Compound | 3 | 4 | 5 | 6 | CYT-20203 | CYT-21625 | paclitaxel |
|---|---|---|---|---|---|---|---|
| IC50 (nM) | 2.8 ± 0.3 | 4.5 ± 0.4 | 34 ± 18 | 6.6 ± 1.7 | 6.7 ± 2.2 | 56 ± 30 | 15 ± 1a |
Data from reference54
These results taken together indicate that although analogs 3, 4, and 6 are most probably not suitable for use as AuNP drug candidates, because of their potential lability at physiological pH, analog 5 does have the right combination of stability in buffer and lability in the presence of DTT to serve as a drug candidate. The nanoparticle formulation CYT-21625 was thus investigated in more detail.
Pharmacokinetics and Tumor Accumulation of Paclitaxel, Compound 5 and CYT-21625
The data reported above indicate that compound 5 is rapidly converted to paclitaxel in rabbit whole blood, while CYT-21625 exhibits little to no release and conversion of the particle bound compound 5 to paclitaxel. A similar study was then carried out in mice. B16/F10 tumor burdened mice were injected with 5 μg of compound 5 or CYT-21625 containing the same amount of 5, and whole blood samples were collected at intervals and divided into 2 aliquots. The first aliquot was analyzed directly without any additional treatment while the second aliquot was treated with BME. Both samples were analyzed for paclitaxel content by ELISA. Consistent with the previous data compound 5 was rapidly degraded to the paclitaxel parent in the circulation, but only marginal paclitaxel release from CYT-21625 was observed (Figure 3A). In a second experiment (Figures 3B and 3C) either 50 μg of either paclitaxel or CYT-21625 containing the same amount of paclitaxel were intravenously injected into B16/F10 tumor burdened C57Bl/6 mice. At 1, 3, 6, and 15 h a group of animals (n = 3 per time point) were sacrificed and whole blood and tumors collected. The whole blood was processed as described above while tumors were first homogenized and then treated identically to the whole blood samples. Tumor measurements were normalized for protein content. The data (Figures 3B and 3C) show that CYT-21625 increased both the pharmacokinetic exposure and tumor uptake of paclitaxel as compared to native drug. In addition, CYT-21625, by virtue of being a PEGylated nanomedicine, significantly increased pharmacokinetic exposure, as measured by terminal half-life and area under the curve, when compared to native paclitaxel (Figure 3B).
Figure 3.
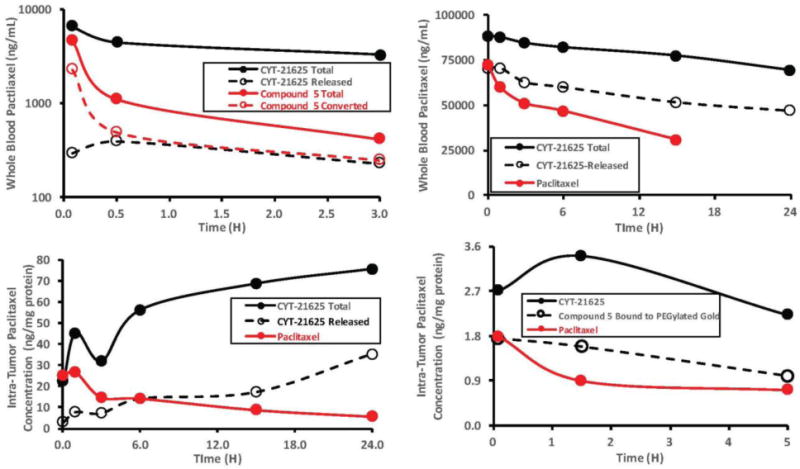
Pharmacokinetics and tumor accumulation profiles of compound 5, CYT-21625 and paclitaxel in tumor bearing mice. Figure 3A: levels of CYT-21625 and 5 in B16/F10 tumor burdened mice after injection with 5 μg of compound 5 or the same dose as CYT-21625. Figure 3B: Blood levels of CYT-21625, of paclitaxel released from CYT-21625 after treatment with DTT, and of native paclitaxel after treatment of tumor burdened C57BL/6 mice with 50 μg of either native paclitaxel or the equivalent paclitaxel dose on CYT-21625. Figure 3C: Tumor levels of CYT-21625, of paclitaxel released from CYT-21625 after treatment with DTT, and of native paclitaxel from a similar experiment as described for Figure 3B. Figure 3D: Tumor levels of CYT-21625, compound 5 bound to PEGylated gold nanoparticles, and native paclitaxel; 5 μg of paclitaxel or its equivalent dose on CYT-21625 were used.
Consistent with previous results with CYT-609128, 36 we observed that TNF mediated targeting of CYT-21625 to B16/F10 solid tumors. Figure 3C demonstrates that CYT-21625 delivered significantly more paclitaxel (both free and bound) to B16/F10 tumors when compared to the native drug. Initially we observed a rapid uptake of CYT-21625 into B16/F10 solid tumors and a clear latency between its arrival at the tumor and the release of compound 5 from the particle and its conversion to paclitaxel.
This observation supports the theory that CYT-21625 mimics the entire surgical procedure of ILP on a single gold nanoparticle. As shown in Figure 3D, a control preparation of 5 bound to pegylated gold but without the TNF targeting molecule exhibits very similar uptake into B16/F10 tumors as native paclitaxel. These data are consistent with those reported by Paciotti et.al.36 who demonstrated that passive targeting of MC38 colon tumors, for example by the EPR effect, is ineffective and active targeting was only achieved by incorporating TNF into the gold nanoconstructs. Since CYT-21625 was engineered to deliver therapeutic doses of both TNF and paclitaxel it is possible that TNF is not only targeting its receptor on the vascular endothelium but also inducing many of mechanisms demonstrated for CYT-6091.28, 36
Treatment of tumor-burdened mice with CYT-21625 and CYT-20203
The efficacy of CYT-21625 was then tested on tumor-burdened mice using significantly lower doses of paclitaxel, based on the significant increases in overall system exposure to paclitaxel observed for this construct (Figure 3B). B16/F10 melanoma was established in C57BL/66 mice, and the mice were treated with either paclitaxel (2.5 or 40 mg/kg), CYT-21625 (2.5 mg/kg), or CYT-20203 (2.5 mg/kg) and tumor volumes were measured over time. In this initial experiment a 50% reduction in tumor volume after 9 days was selected as a significant anti-tumor response. Consistent with the targeted delivery leading to increased uptake of paclitaxel in the B16F/10 tumors, CYT-21625 and CYT-20203 induced similar anti-tumor responses as paclitaxel but at 16 fold lower doses of drug (Table 4).
Table 4.
Anti-Tumor Responses of B16/F10 Tumor Burdened C57BL/6 Mice to Paclitaxel and CYT-21625
| Treatment Group | Paclitaxel Dose (mg/kg) | Response: Decrease in Tumor Volume vs Control: (%) |
|---|---|---|
| PBS control | 0 | 0 |
| Paclitaxel | 2.5 | −24 |
| Paclitaxel | 40 | −68 |
| CYT-21625 | 2.5 | −62 |
| CYT-20203 | 2.5 | −60 |
DISCUSSION
As part of the effort to develop a colloidal-gold-nanoparticle-based drug delivery system for the targeted delivery of paclitaxel to tumors, paclitaxel derivatives 3 – 6 with various sulfur containing linkers were synthesized. In studies of the hydrolytic stabilities of the analogs, analog 4 was converted to paclitaxel in buffer solution with a half-life of 3.2 hours, but analog 5 was stable in the absence of a reducing agent such as DTT or BME. Gold bound analog Au-3 released paclitaxel much more slowly than the corresponding native analog 3.
Analogs 3 and 5 were converted to the nanoparticle constructs CYT-20203 and CYT-21625, respectively, containing the analog, TNF, and PEG-Thiol in predetermined ratios. CYT-21625 was shown to deliver paclitaxel selectively to tumors and to serve as a slow-release reservoir of paclitaxel, and both constructs were effective at reducing the volume of B16/F10 melanomas at doses of 2.5 mg/Kg, with CYT-21625 showing slightly better efficacy.
CYT-21625 represents a completely novel nanomedicine with the capabilities of not only targeting solid tumors (via particle bound TNF) but also providing a two-pronged attack on the architecture of the solid tumor. Since the nanomedicine platform delivers therapeutically relevant doses of TNF and paclitaxel we predict that the particle-bound TNF will induce vascular leakage, similar to that observed with CYT-6091,38, 55 and a reduction in tumor interstitial fluid pressure.39 As these events unfold CYT-21625 is expected to release paclitaxel that can then attack the cancer cells.
Since the TNF-mediated events of targeting and creation of vascular leakage are early stage events, coupled with the latency of the release of paclitaxel, it is reasonable to expect that CYT-21625 could successfully mimic the ILP paradigm as a systemically deliverable nanomedicine that can attack not only primary but also metastatic tumors. The full promise of this approach to cancer treatment requires additional routine pharmacologic studies and a study of the general applicability of TNF, since both Farma55 and Koonce39 report differential sensitivities of solid tumors to TNF-mediated vascular effects. However, in tumors where TNF does induce vascular disruption CYT-21625 may provide a strong and synergistic attack on solid tumors.
EXPERIMENTAL SECTION
The following standard conditions apply unless otherwise stated. All reactions were performed under argon or nitrogen in oven-dried glassware using dry solvents and standard syringe techniques. Tetrahydrofuran (THF) was distilled from the sodium benzophenone ketyl radical ion. DCM was distilled from calcium hydride. HPLC grade methanol and water were purchased from Fisher Scientific. All reagents were of commercial quality and used as received. 1-Mercapto-3,6,9,12,15,18,21,24-octaoxaheptacosan-27-oic acid was purchased from Polypure AS, Gaustadalleen 21, NO-0349 Oslo, Norway. PEG-Thiol (MW 20,000) was purchased from SunBio, Inc., Seoul, South Korea, and TNF was purchased from Boehringer Ingelheim, GmbH, Vienna, Austria. After workup, partitioned organic layers were washed with water and brine and dried over Na2SO4. Reaction progress was monitored using aluminum-backed thin layer chromatography (TLC) plates pre-coated with silica UV254. Purification by preparative thin layer chromatography (PTLC) was performed using glass-backed plates pre-coated with silica UV254. HPLC was conducted on a Shimadzu SCL-10AVP system using a column purchased from Phenomenex (Luna 5μ C18 (2) 25 × 4.6 mm). 1H and 13C NMR spectra were recorded on a 400 MHz (400 MHz for 1H and 100 MHz for 13C) spectrometer or a 500 MHz (500 MHz for 1H and 126 MHz for 13C) spectrometer in CDCl3 unless otherwise stated. All chemical shifts (δ) were referenced to the solvent peaks of CDCl3 (7.26 ppm for 1H, 77.0 ppm for 13C).
1-(Pyridin-2-yldisulfanyl)-3,6,9,12,15,18,21,24-octaoxaheptacosan-27-oic acid (8)
A 25 mL round bottom flask was dried in the oven overnight, cooled to room temperature (rt) and equipped with a magnetic stirring bar and a rubber septum. The flask was evacuated for 5 min and flushed with argon. A solution of 1-mercapto-3,6,9,12, 15,18,21,24-octaoxaheptacosan-27-oic acid 7 (194 mg, 0.423 mmol) in dry tetrahydrofuran (2 mL) and excess 2,2′-dithio-dipyridine56 was added into the flask via syringe in the presence of an argon atmosphere (balloon). The solution was stirred vigorously at rt and 30 mL of glacial acetic acid was added via syringe. The resulting solution was stirred at rt for 48 h. The resulting light yellow solution was concentrated by rotary evaporation (35 °C) to yield a yellow oil. This sample was applied to column chromatography (silica gel, 50 g) eluting with a mixture of hexane and EtOAc (1:3, 60 mL) and then MeOH. The fraction eluted by MeOH was concentrated by rotary evaporation (35 °C) to give a yellow oil, which was then purified by preparative TLC (developed with CH2Cl2:MeOH = 10:1) to produce 8 (200 mg, 0.35 mmol) as a colorless oil (82 %).
(2′-tert-Butyldimethylsilyloxy)-7-chloromethyloxycarbonylpaclitaxel 10
To a 25 mL round bottom flask containing a magnetic stirring bar was charged compound 957 (399 mg, 0.41 mmol). The flask was then equipped with a rubber septum, and evacuated and flushed with argon. Dry CH2Cl2 (5 mL) was added to dissolve the solid and the resulting solution was cooled to 0 °C in an ice bath. To the mixture was added chloromethyl chloroformate (40.3 μL, 0.45 mmol) dropwise via syringe. After the solution was stirred for 5 min at 0 °C, pyridine (40.3 μL, 0.49 mmol) was added over a period of 5 min via syringe. The resulting mixture was allowed to warm to rt and stir for 15 h under an argon atmosphere (balloon). The reaction was diluted with EtOAc (100 mL), washed with water (2 × 5 mL) and brine (2 × 5 mL) and dried with anhydrous Na2SO4. Rotary evaporation (35 °C) of the organic extract gave 10 (445 mg, crude) as a white powder, which was used in the next step without further purification. 1H NMR (CDCl3, 400 MHz) δ 8.12 (2H, d, J = 8.5 Hz), 7.74 (2H, d, J = 8.5 Hz), 7.25–7.65 (11H, m), 7.08 (1H, d, J = 9.0 Hz), 6.32 (1H, s), 6.26 (1H, t, J = 8.5 Hz), 5.98 (1H, d, J = 6.5 Hz), 5.73 (1H, d, J = 8.5 Hz), 5.69 (1H, d, J = 6.5 Hz), 5.57, 5.51 (1H, d, J = 6.5 Hz), 4.98 (1H, d, J = 9.5 Hz), 4.67 (1H, d, J = 2.5 Hz), 4.35 (1H, d, J = 8.0 Hz), 4.20 (1H, d, J = 8.5 Hz), 3.96 (1H, d, J = 7.0 Hz), 2.66 (1H, m), 2.58 (3H, s), 2.41 (1H, dd, J = 15, 8.5 Hz), 2.14 (3H, s), 2.04 (3H, s), 1.99 (3H, s), 1.82 (3H, s), 1.21 (3H, s), 1.15 (3H, s), 0.80 (9H, s), −0.03 (3H, s), −0.30 (3H, s); HRFABMS m/z 1060.3853 [M + H] + (calcd for C55H67ClNO16Si, 1060.3918).
7-Chloromethyloxycarbonyl paclitaxel 11
A plastic vial (25 mL) equipped with a magnetic stirring bar was charged with a solution of 10 (445 mg, crude) in tetrahydrofuran (10 mL). To the solution was added HF-pyridine (0.6 mL) dropwise at 0 °C. The resulting mixture was allowed to warm up to rt and stir for 10 h. The reaction mixture was diluted with EtOAc (100 mL), washed with saturated aqueous sodium bicarbonate (2 × 5 mL), water (2 × 5 mL) and brine (2 × 5 mL) and dried with anhydrous Na2SO4. Rotary evaporation (35 °C) gave a light brown residue, which was then purified by column chromatography (silica gel, 150 g, eluted with hexane:EtOAc, 3:2~2:3) to yield 11 (298 mg, 0.32 mmol) as a white powder (77 % for two steps from 10. 1H NMR (500 MHz) δ 8.11 (d, J = 7.4 Hz, 2H), 7.76 (d, J = 7.4 Hz, 2H), 7.62 (t, J = 7.4 Hz, 1H), 7.55 – 7.46 (m, 6H), 7.41 (m, 4H), 7.35 (t, J = 7.3 Hz, 1H), 7.04 (d, J = 8.9 Hz, 1H), 6.28 (s, 1H), 6.18 (t, J = 8.5 Hz, 1H), 5.97 (d, J = 6.5 Hz, 1H), 5.80 (dd, J = 8.9, 2.4 Hz, 1H), 5.67 (d, J = 6.9 Hz, 1H), 5.51 (q, J = 5.7 Hz, 2H), 4.95 (d, J = 8.4 Hz, 1H), 4.80 (dd, J = 4.7, 2.5 Hz, 1H), 4.32 (d, J = 8.5 Hz, 1H), 4.18 (d, J = 8.5 Hz, 1H), 3.92 (d, J = 6.9 Hz, 1H), 3.60 (d, J = 4.9 Hz, 1H), 2.65 (ddd, J = 14.5, 9.5, 7.3 Hz, 1H), 2.38 (s, 3H), 2.36 – 2.30 (m, 2H), 2.15 (s, 3H), 2.04 – 1.97 (m, 1H), 1.85 (s, 3H), 1.81 (s, 3H), 1.21 (s, 3H), 1.15 (s, 3H); 13C NMR (100 MHz) δ 201.54, 172.59, 170.53, 169.29, 167.12, 166.92, 152.71, 140.80, 138.10, 133.91, 133.74, 132.96, 132.04, 130.26, 129.09, 128.84, 128.80, 128.43, 127.16, 127.15, 83.78, 80.92, 78.62, 76.64, 76.47, 75.49, 74.26, 73.29, 73.06, 72.23, 56.25, 55.04, 47.00, 43.29, 35.63, 33.32, 26.61, 22.62, 20.98, 20.86, 14.72, 10.72.
Paclitaxel analog 3
A dry 25 mL round bottom flask was charged with a magnetic stirring bar, compound 11 (140 mg, 0.15 mmol) and sodium iodide (33.3 mg, 0.22 mmol). Acetone (5 mL) was added via syringe to dissolve the solid. The flask was then equipped with a water condenser and the reaction mixture was allowed to reflux (~62 °C, oil bath) for 10 h. The resulting yellow solution was concentrated by rotary evaporation (35 °C) to give a yellow residue. To the flask was introduced a solution of 8 (128 mg, 0.22 mmol) in benzene (5 mL). After K2CO3 (61.3 mg, 0.44 mmol) and 18-crown-6 (235 mg, 0.89 mmol) were added, the flask was capped with a water condenser, warmed up to 75 °C and stirred for 2 h. The reaction solution was allowed to cool to rt and diluted with EtOAc (100 mL), washed with saturated aqueous sodium bicarbonate (2 × 5 mL), water (2 × 5 mL) and brine (2 × 5 mL) and dried with anhydrous Na2SO4. Rotary evaporation (35 °C) gave a yellowish oil. This was applied to preparative TLC (developed with 5 % MeOH in CH2Cl2) to yield 3 (136 mg, 0.092 mmol, 62 %). 1H NMR (500 MHz) δ 8.44 (1H, d, J = 4.8 Hz), 8.11 (2H, d, J = 7.6 Hz), 7.25–7.80 (15H, m), 7.10 (2H, m), 6.30 (1H, s), 6.18 (1H, dd, J = 8.0, 8.0 Hz), 5.90 (1H, d, J = 6.0 Hz), 5.79 (1H, dd, J = 9.2, 2.4 Hz), 5.71 (1H, d, J = 6.0 Hz), 5.67 (1H, d, J = 6.8 Hz), 5.48 (1H, dd, J = 10.8, 6.8 Hz), 5.30 (1H, s), 4.94 (1H, d, J = 8.4 Hz), 4.80 (1H, dd, J = 6.4, 4.0 Hz), 4.31 (1H, d, J = 8.4 Hz), 4.19 (1H, d, J = 7.6 Hz), 3.91 (1H, d, J = 6.8 Hz), 3.55–3.80 (33H, m), 2.99 (2H, dd, J = 6.4, 6.4 Hz), 2.67 (2H, dd, J = 6.4, 6.4 Hz), 2.64 (1H, m), 2.38 (3H, s), 2.32 (2H, d, J = 9.2 Hz), 2.16 (3H, s), 2.01 (1H, m), 1.85 (3H, s), 1.80 (3H, s), 1.21 (3H, s), 1.16 (3H, s); 13C NMR (126 MHz) δ 201.4, 172.6, 170.5, 170.1, 169.1, 167.0, 166.8, 153.3, 149.6, 140.7, 138.1, 137.2, 133.9, 133.0, 132.0, 130.3, 129.1, 129.1, 128.8, 128.8, 128.4, 127.1, 127.1, 120.7, 119.7, 84.5, 83.8, 82.6, 81.2, 81.0, 79.2, 78.7, 76.5, 76.2, 75.7, 75.4, 74.3, 73.2, 72.5, 72.2, 70.6, 70.5, 69.0, 66.1, 65.5, 56.2, 54.9, 47.0, 43.3, 38.5, 35.6, 34.8, 33.4, 29.8, 27.0, 26.6, 22.6, 21.0, 20.8, 14.8, 10.7; HRFABMS m/z 1477.5421 [M + H] + (calcd for C73H93N2O26S2, 1477.5458).
(7-O-Paclitaxelcarbonyloxy)methyl 5-(1,2-dithiolan-3-yl)pentanoate 4
A dry 25 mL round bottom flask was charged with a magnetic stirring bar, compound 11 (40 mg, 42.3 μmol) and sodium iodide (9.5 mg, 63.5 μmol). Acetone (2 mL) was added via syringe to dissolve the solid. The flask was then equipped with a water condenser and the reaction mixture was allowed to reflux (~62 °C, oil bath) for 10 h. The resulting yellow solution was concentrated by rotary evaporation (35 °C) to give a yellow residue. To the flask was added lipoic acid (17.0 mg, 82.6 μmol) and benzene (2 mL). After K2CO3 (17.5 mg, 126.9 μmol) and 18-crown-6 (67.1 mg, 253.8 μmol) was added, the flask was capped with a water condenser, warmed to 65 °C and stirred for 3.5 h. TLC was used to examine the reaction progress. Decomposed product could be detected with prolonged reaction times. The reaction solution was allowed to cool down to rt and diluted with EtOAc (75 mL), washed with saturated aqueous sodium bicarbonate (2 × 4 mL), water (2 × 4 mL) and brine (2 × 4 mL) and dried with anhydrous Na2SO4. Rotary evaporation (35 °C) gave a yellowish oily residue, which was purified by preparative TLC (developed with hexane:EtOAc, 1:1) to yield 4 (36 mg, 32.2 μmol, 76 %). 1H NMR (400 MHz) δ 8.10 (2H, dd, J = 7.5, 1.5 Hz), 7.76 (2H, d, J = 7.0, 1.5 Hz), 7.62 (1H, m), 7.47–7.52 (5H, m), 7.33–7.43 (4H, m), 7.04 (1H, d, J = 9.0 Hz), 6.30 (1H, s), 6.18 (1H, dd, J = 8.0, 8.0 Hz), 5.90 (1H, d, J = 6.0 Hz), 5.79 (1H, dd, J = 9.0, 2.0 Hz), 5.70 (1H, d, J = 6.0 Hz), 5.66 (1H, d, J = 7.0 Hz), 5.48 (1H, dd, J = 10.0, 7.0 Hz), 4.94 (1H, d, J = 8.0 Hz), 4.79 (1H, d, J = 2.5 Hz), 4.31 (1H, d, J = 8.5 Hz), 4.17 (1H, d, J = 8.5 Hz), 3.91 (1H, d, J = 7.5 Hz), 3.56 (2H, m), 3.08–3.18 (2H, m), 2.62 (1H, m), 2.45 (1H, m), 2.38 (3H, s), 2.31 (2H, dd, J = 9.0, 4.0 Hz), 2.15 (3H, s), 1.96 (1H, m), 1.89 (1H, m), 1.84 (3H, s), 1.80 (3H, s), 1.67 (6H, m), 1.46 (2H, m), 1.21 (3H, s), 1.18 (3H, s); 13C NMR (126 MHz) δ 201.44, 172.59, 172.00, 170.52, 169.11, 167.10, 166.92, 153.28, 140.66, 138.08, 133.90, 133.73, 133.05, 132.06, 130.26, 129.10, 128.84, 128.81, 128.44, 127.17, 127.14, 83.81, 82.65, 80.97, 78.62, 76.49, 76.22, 75.40, 74.27, 73.27, 72.29, 56.35, 56.34, 56.19, 55.01, 47.02, 43.31, 40.28, 38.57, 35.61, 34.64, 33.74, 33.38, 28.71, 28.70, 26.64, 24.29, 22.64, 20.98, 20.87, 14.75, 10.73; HRESIMS (m/z): [M + H]+ calcd for C57H66NO18S2 1116.3716; found, 1116.3721.
Synthesis of 2΄-(tert-butyldimethylsiloxy)-7-(((4-(pyridin-2-yldisulfanyl)-benzyloxy)-carbonyloxy)-paclitaxel 14
To a stirred solution of compounds 1253 (48.5 mg, 0.043 mmol) and 1348 (16 mg, 0.064 mmol) in dry DCM (2 mL) was added 4-(dimethylamino) pyridine (15.6 mg, 0.128 mmol), and the mixture was stirred at rt for 18 h. The resulting mixture was diluted with DCM (40 mL) and washed with water (2 × 2 mL) and brine (2 × 2 mL). The solution was dried with anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by preparative TLC (50% EtOAc in hexanes) to afford compound 14 (47 mg, 88%): 1H NMR (500 MHz) δ 8.46 (1H, d, J = 4.9 Hz), 8.11 (2H, d, J = 7.3 Hz), 7.76 (2H, d, J = 7.6 Hz), 7.25–7.55 (17 H, m), 7.10 (1H, d, J = 8.7 Hz), 6.39 (1H, s), 6.26 (1H, dd, J = 9.2, 8.1 Hz), 5.73 (1H, d, J = 8.7 Hz), 5.70 (1H, br d, J = 6.7 Hz), 5.54 (1H, dd, J = 10.1, 6.9), 5.16 (2H, br s), 4.97 (1H, d, J = 9.2 Hz), 4.67 (1H, br s), 4.34 (1H, d, J = 8.5 Hz), 4.20 (1H, d, J = 8.5 Hz), 3.97 (1H, d, J = 6.7 Hz), 2.60 (1H, m), 2.58 (3H, s), 2.42 (1H, dd, J = 15.1, 9.4 Hz), 2.15 (3H, s), 2.15 (1H, dd, J = 15.4, 8.9 Hz), 2.00 (1H, m), 2.00 (3H, s), 1.80 (3H, s), 1.22 (3H, s), 1.16 (3H, s), 0.80 (9H, s), −0.03 (3H, s), −0.30 (3H, s); 13C NMR (126 MHz) δ 201.6, 171.4, 170.0, 169.1, 167.1, 166.9, 159.0, 154.0, 149.0, 141.1, 138.2, 136.6, 135.7, 134.0, 133.7, 132.7, 131.8, 130.2, 129.7, 129.0, 128.8, 128.3, 128.0, 127.0, 126.3, 126.0, 121.5, 120.5, 115.7, 83.8, 80.9, 78.7, 76.3, 75.5, 75.3, 75.0, 74.4, 71.3, 69.2, 56.0, 55.7, 46.8, 43.3, 35.5, 33.3, 26.4, 25.5, 23.0, 21.4, 20.8, 18.1, 14.6, 10.7, −5.2, −5.8; HRFABMS m/z 1243.4370 [M + H]+ (calcd for C66H75N2O16S2Si, 1243.4327).
Synthesis of 7-(((4-(pyridin-2-yldisulfanyl)benzyloxy)carbonyloxy)-paclitaxel 5
A plastic vial was charged with a solution of compound 14 (45 mg, 0.036 mmol) in 4.5 mL of dried THF and cooled to 0 °C. To this solution was added 0.15 mL of HF-pyridine. The mixture was allowed to warm to rt and stirred for 16 h. The reaction was quenched by careful addition of saturated aqueous NaHCO3 until no bubbles were formed. The resulting solution was extracted with EtOAc (3 × 15 mL). The organic solution was washed with water (2 × 2 mL) and brine (2 × 2 mL), dried with anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by preparative TLC (50% EtOAc/hexane) to give 5 (35 mg, 92 % based on unreacted starting material), together with 3 mg unreacted compound 14. 1H NMR (500 MHz) δ 8.46 (1H, d, J = 4.9 Hz), 8.11 (2H, d, J = 7.2 Hz), 7.76 (2H, d, J = 8.0 Hz), 7.25–7.65 (17H, m), 7.10 (1H, br dd, J = 7.0, 4.1 Hz), 7.04 (1H, d, J = 8.8 Hz), 6.36 (1H, s), 6.19 (1H, t, J = 6.9 Hz), 5.80 (1H, dd, J = 8.8, 2.5 Hz), 5.67 (1H, d, J = 6.8 Hz), 5.47 (1H, dd, J = 10.7, 7.5), 5.17 (1H, d, J = 12.3 Hz), 5.13 (1H, d, J = 12.3 Hz), 4.93 (1H, d, J = 8.8 Hz), 4.80 (1H, dd, J = 5.0, 2.5 Hz), 4.31 (1H, d, J = 8.7 Hz), 4.18 (1H, d, J = 8.7 Hz), 3.93 (1H, d, J = 6.8 Hz), 3.85 (1H, d, J = 5.0 Hz), 2.58 (1H, m), 2.38 (3H, s), 2.33 (2H, d, J = 6.9 Hz), 2.15 (3H, s), 1.95 (1H, m), 1.85 (3H, s), 1.80 (3H, s), 1.22 (3H, s), 1.17 (3H, s); 13C NMR (126 MHz) δ 201.5, 172.5, 170.4, 169.0, 167.0, 166.8, 159.0, 153.8, 149.0, 140.5, 138.0, 137.8, 133.8, 133.8, 133.6, 133.0, 131.9, 130.1, 129.6, 129.0, 128.7, 128.7, 128.4, 127.7, 127.0, 127.0, 127.0, 121.5, 120.5, 83.8, 80.9, 78.5, 76.4, 75.6, 75.3, 74.2, 73.1, 72.2, 69.3, 56.1, 54.9, 46.9, 43.2, 35.5, 33.4, 26.5, 22.5, 20.9, 20.8, 14.6, 10.6; HRFABMS m/z 1129.3464 [M + H]+ (calcd for C60H61N2O16S2, 1129.3463).
Synthesis of paclitaxel-2′-yl (chloromethyl)succinate (16)
A mixture of CH2Cl2 (2 mL) and 2 mL of an aqueous solution containing compound 1546 (20.8 mg, 0.022 mmol), NaHCO3 (9 mg, 0.11 mmol), and tetrabutylammonium bisulfate (1 mg, 0.003 mmol) was stirred for 10 min at rt. Chloromethyl chlorosulfate (4.6 mg, 0.028 mmol) was then added and the reaction mixture vigorously stirred at rt for 2 h. The reaction mixture was diluted with 10 mL DCM, washed with brine (2 × 1 mL), dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure. The residue was separated by PTLC with hexane and EtOAc (1:2) as developing solvent to yield compound 19 (10 mg, 46% yield). 1H NMR (500 MHz):1.12 (s, 3H), 1.23 (s, 3H), 1.67 (s, 3H), 1.87 (m, 1H), 1.92 (s, 3H), 2.15 (m, 1H), 2.22 (s, 3H), 2.37 (m, 1H), 2.44 (s, 3H), 2.55 (m, 1H), 2.68 (m, 2H), 2.78 (m, 2H), 3.80 (d, J = 7.0 Hz, 1H), 4.19 (d, J = 8.5 Hz, 1H), 4.31 (d, J = 8.5 Hz, 1H), 4.43 (m, 1H), 4.96 (d, J = 7.5 Hz, 1H) 5.50 (d, J = 3.0 Hz), 5.57 (s, 2H), 5.68 (d, J = 7.0 Hz, 1H), 5.98 (dd, J = 6.5, 3.0 Hz, 1H), 6.23 (t, 9.0 Hz, 1H) 6.28 (s, 1H), 6.94 (d, J = 9.0 Hz, 1H), 7.34–7.45 (m, 7H), 7.52 (m, 3H), 7.60 (m 1H), 7.77 (m, 2H), 8.14 (m, 2H); 13C NMR (126 MHz) δ 203.91, 171.37, 170.83, 170.38, 169.91, 169.17, 167.89, 167.16, 167.15, 153.29, 142.85, 138.91, 136.93, 133.79, 133.59, 132.85, 132.15, 130.33, 129.24, 129.21, 128.84, 128.80, 128.60, 127.28, 127.27, 126.59, 84.52, 81.15, 79.27, 77.30, 76.80, 75.68, 75.16, 74.45, 72.24, 72.00, 68.94, 62.97, 58.61, 52.74, 45.64, 43.25, 35.58, 29.09, 28.71, 26.89, 22.78, 22.20, 20.91, 14.91, 9.67.
Synthesis of ((5-(1,2-dithiolan-3-yl)pentanoyl)oxy)methyl paclitaxel-2′-yl succinate (6)
A mixture of 16 (10 mg, 0.01 mmol), lipoic acid (4.4 mg, 0.02 mmol), K2CO3 (4.4 mg, 0.03 mmol), 18-crown-6 (16.8 mg, 0.06 mmol) and NaI (cat.) was refluxed in benzene for 5 hr. The reaction mixture was diluted with ether (10 mL) and washed with water (2 × 1 mL) and brine (2 × 1 mL). The resulting crude product was purified by PTLC (1:2 hexane/EtOAc) to give compound 6 as a white solid (7 mg, 60% yield). 1H NMR (500 MHz):1.12 (s, 3H), 1.23 (s, 3H), 1.40–1.50 (m, 2H), 1.64 (m, 2H), 1.67 (s, 3H), 1.87 (m, 1H), 1.92 (s, 3H), 2.15 (m, 1H), 2.22 (s, 3H), 2.30–2.40 (m, 3H), 2.44 (s, 3H), 2.45 (m, 2H), 2.55 (m, 1H), 2.68 (m, 2H), 2.78 (m, 2H), 3.06–3.18 (m, 2H), 3.54 (m, 1H), 3.80 (d, J = 7.0 Hz, 1H), 4.19 (d, J = 8.5 Hz, 1H), 4.31 (d, J = 8.5 Hz, 1H), 4.43 (m, 1H), 4.96 (d, J = 7.5 Hz, 1H) 5.50 (d, J = 3.0 Hz), 5.64 (m, 2H), 5.68 (d, J = 7.0 Hz, 1H), 5.98 (dd, J = 6.5, 3.0 Hz, 1H), 6.23 (t, 9.0 Hz, 1H) 6.28 (s, 1H), 6.98 (d, J = 9.0 Hz, 1H), 7.34–7.45 (m, 7H), 7.52 (m, 3H), 7.60 (m 1H), 7.77 (m, 2H), 8.14 (m, 2H); HRESIMS (m/z): [M + H]+ calcd for C60H70NO19S2 1172.3978, found 1172.3849.
Preparation of CYT-21625 and CYT-20203
CYT-21625 is the designation of gold nanoparticles bound to TNF, PEG-Thiol, and compound 5, and CYT-20203 is the corresponding construct formed using analog 3 in place of analog 5. The procedure used was adapted from that described previously.36 For both constructs the gold nanoparticles were made by bringing deionized H2O (16 L) to a boil under reflux, and then adding 32 mL of a high purity 4% gold chloride solution to the rapidly mixed boiling water. Nanoparticle initiation was induced by the rapid addition of 600 mL of a 1% sodium citrate solution. The resulting gold nanoparticles were filtered through a 0.22 μ sterilization filter and stored until needed. Two glass containers equipped with bottom ports were connected via Tygon tubing to the side ports of a single T-connector. One of the containers was filled with 800 mL of colloidal gold nanoparticles while the second container was filled with 800 mL of a solution containing TNF, PEG-Thiol and paclitaxel analog 5 in the concentrations of 0.1, 15, and 2.5 μg/mL, respectively. The exit port of the T-connector was similarly fitted with tubing containing in line mixers. This section of tubing was threaded through a single peristaltic pump which when turned on drew both the gold particles and the solution of TNF, PEG-Thiol, and 5 into the T-Connector, through the pump and into a central collection vessel. After binding, a series of stabilizers were added to the solution, which was then concentrated by ultrafiltration and lyophilized to yield CYT-21625. The same procedure using analog 3 in place of analog 5 gave CYT-20203. Each batch of nanodrug produced was interrogated for TNF content using a cross antibody ELISA as previously described.29 Paclitaxel content was determined by diluting the final drug product to a 50% MeOH solution containing BME and NaHCO3 and Na2CO3 (10 mM each). The solution was then incubated at 37°C until particle precipitation was evident (~ 4h). The precipitated material was centrifuged and the supernatant containing paclitaxel released from compound 5 was analyzed by quantitative HPLC using authentic paclitaxel as a standard. To determine the amount of free paclitaxel an aliquot of the final drug product was diluted in 1% PEG 1450 and centrifuged. The resultant supernatant was diluted and analyzed as described above. The composition of CYT-21625 could be tuned by varying the amounts of each active pharmaceutical ingredient that was bound to the nanoparticle surface. Since both TNF and the thiolated paclitaxel analogs were bound at the same time we observed that as the concentration of one API (i.e., the paclitaxel analog) in the binding vessel increased as the amount of the second API (i.e., TNF) detected on the particle decreased. These data support the concept that both TNF and the thiolated paclitaxel analogs shared the surface of the nanoparticles. These conditions yielded nanoparticles with up to 5000 molecules of paclitaxel analog and 25 molecules of TNF bound to each nanoparticle, as calculated from the weights of the starting materials and confirmed by the lack of significant amounts of free analog in the final product.
Hydrolytic release of paclitaxel by compound 4 in buffer
Concentrations of 4 and paclitaxel were determined by quantitative HPLC with a Shimadzu SCL-10AVP system. Reverse phase C18 column was purchased from Phenomenex (Luna 5μ C18 (2) 25 × 4.6 mm). HPLC analyses were run with a linear gradient elution from MeOH:H2O 1:1 to 100% MeOH in 20 min, followed by 100% MeOH for 10 min, at a flow rate of 1 mL/min. A fresh stock solution of 4 was prepared by dissolving 5.20 mg 4 in 25 mL MeOH. A small vial equipped with a cap and magnetic stirring bar was charged with 3 mL of the above solution. To this solution was added 1 mL 1 M PBS buffer (pH 7.4) quickly in one portion. A 50 μL aliquot of this solution immediately injected onto the HPLC system. The vial was then capped and sealed with parafilm. Aliquots (50 μL) were taken at 30 min, 60 min, 120 min, 180 min, 240 min, 360 min, 540 min, 690 min and 1440 min, and injected onto the HPLC system. Concentrations of paclitaxel and 4 in each aliquot were determined based on previously prepared standard curves of both compounds. The resulting concentration vs time plot is shown in Figure S1.
Release of paclitaxel by compound 5 and by CYT-21625 in bicarbonate buffer
Analog 5 (0.25 mg) was dissolved in 10 mL 90% CH3OH containing 5 mg NaHCO3, and the solution was analyzed by HPLC. Less than 5% of paclitaxel was released over 24 h, but this rose to 50% release after 72 h. A repeat of this study using CYT-21625 showed that paclitaxel release was reduced, with less than 10% of paclitaxel released over 72 h.
Release of paclitaxel from compound 5 in bicarbonate buffer plus dithiothreitol
The previous experiment was repeated to assess the release of paclitaxel in the presence of a 3-fold excess of dithiothreitol (DTT). It was found that 90% of 5 was converted to paclitaxel within 60 min under these conditions.
Generation of paclitaxel from compound 5 in rabbit whole blood
Analog 5 or CYT-21625 were spiked into rabbit whole blood. At t = 0, 1, 2, and 24 h samples were collected and processed. Rabbit whole blood (1 mL) was extracted with 2 mL of ice cold acetonitrile (ACN), and the samples were centrifuged at 3500 rpm to pellet the precipitated protein extract. In the case of 5 the supernatant was analyzed directly by HPLC as described below. In the case of CYT-21625 the colloidal gold particles did not completely pellet out, and the particles that remained in the supernatant were re-centrifuged at 14,000 rpm, reconstituted with H2O and treated with β-mercaptoethanol (BME) prior to analysis by HPLC. The resultant supernatant samples (~3 mL), which contained analog/paclitaxel released from CYT-21625, were diluted to a final volume of 7 mL with deionized H2O and loaded onto a low pressure C-18 resin which was pre-equilibrated with 30% ACN. Once loaded the column was washed with 6 mL of 30% CAN, and the column bound material was eluted with 1 mL of 100% ACN. The eluted material was then diluted to 3 mL with deionized H2O and analyzed by RP-HPLC. Controls included compound 5 spiked into running buffer alone or treated with BME, paclitaxel spiked into whole blood and CYT-21625 spiked into blood and treated with BME. The latter 2 samples were extracted with ACN and purified via C-18 chromatography as described above.
Compound 5 was completely converted to paclitaxel essentially immediately under these conditions. However, CYT-21625 was not converted to paclitaxel over a 2 h time period, but did release paclitaxel on treatment with BME.
Antiproliferative assay
Measurements of antiproliferative activity were performed at Virginia Tech against the A2780 ovarian cancer cell line as previously described.58 The A2780 cell line is a drug - sensitive human ovarian cancer cell line.59
Accumulation of paclitaxel in solid tumors after treatment with CYT-21625
B16/F10 tumor-bearing C57BL/6 mice received an intravenous injection of either native paclitaxel or CYT-21625 at the same dose level of drug (2.5 mg/Kg). At selected time points tumors were harvested, flash frozen and subsequently homogenized in PBS using a Polytron tissue disrupter. Debris was removed by allowing the homogenate to stand on ice for 20 minutes. For samples from mice treated with CYT-21625 the resultant supernatant was split into two aliquots. One aliquot was analyzed directly without further treatment, while the other was incubated in hydrolysis buffer containing DTT and NaHCO3 prior to analysis. The treated samples thus represented total paclitaxel while the untreated samples represented the amount of paclitaxel generated by hydrolysis. In the case of samples from mice treated with paclitaxel the sample was analyzed directly without further treatment. The samples were analyzed for paclitaxel concentration by ELISA, and for total protein using a commercial protein assay (BioRad, Hercules, CA, USA), and the intra-tumor paclitaxel concentration was calculated as ng paclitaxel/mg protein. Tumors from mice treated with native paclitaxel showed an initial intra-tumor paclitaxel concentration of 25 ng/mL, decreasing over 24 h to 5 ng/mL. In contrast, tumors from mice treated with CYT-21625 showed native paclitaxel levels increasing over 24 h from initial levels of 5 ng/mL to 30 ng/mL at 24 h. However, treatment of the homogenate with hydrolysis buffer revealed initial intra-tumor levels of native paclitaxel of 25 ng/mL increasing to 75 ng/mL after 24 h post treatment.
Treatment of tumor-burdened mice with CYT-21625 and CYT-20203
C57BL/6 mice were implanted with B16/F10 melanoma cells. Once the cells formed tumors with an approximate volume of 500 mm3 the mice (n=4/group/formulation) received an injection of either native paclitaxel, CYT-21625 or CYT-20203. Paclitaxel was given at a dose of 40 mg/kg, whereas the CYT-20000 series drugs were administered at a dose of 2.5 mg/Kg. The mice were treated four times, on days 0, 2, 5 and 7, and tumor responses were determined by measuring tumor volume during the course of the study.
Supplementary Material
Acknowledgments
This work was supported in part by SBIR grant R43 CA119399 from the National Cancer Institute and a grant from the Maryland Technology Development Corporation (TEDCO) “Development and Pilot Manufacturing for Multi-functional Tumor Targeting Nanomedicine”.
Footnotes
Current Addresses: G.F.P.: Senior Scientific, LLC, 800 Bradbury Drive SE, Suite 213, Albuquerque, NM 87106; J.Z.: The Procter & Gamble Company, Mason Business Center, DS2-7D5, 8700 S. Mason Montgomery Rd, Mason, OH 45040; S.C.: Department of Pharmaceutical Sciences, Daniel K Inouye College of Pharmacy, University of Hawaii at Hilo, 924 Stainback Hwy, Hilo, Hawaii 96720; L.D.M.: Champions Oncology, 855 N. Wolfe Street, Suite 602, Baltimore, MD 21205; J.F.; M.H.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:10.1021/acs.bioconjchem.6b00211.
1H and 13C NMR spectra of compounds 3 – 6, and Figures S1–S3
Notes
The authors declare the following competing financial interests: L.T. and G.F.P. own stock in CytImmune Sciences, Inc., and D.G.I.K. holds stock options in CytImmune Sciences, Inc.
References
- 1.Orr GA, Verdier-Pinard P, McDaid H, Horwitz SB. Mechanisms of Taxol resistance related to microtubules. Oncogene. 2003;22:7280–7295. doi: 10.1038/sj.onc.1206934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by taxol. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- 3.Chiorazzi A. Paclitaxel: chemotherapy and neurotoxicity - the two sides of the coin. Horiz Cancer Res. 2011;45:1–38. [Google Scholar]
- 4.Rivera E, Cianfrocca M. Overview of neuropathy associated with taxanes for the treatment of metastatic breast cancer. Cancer Chemother Pharmacol. 2015;75:659–670. doi: 10.1007/s00280-014-2607-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saloustros E, Mavroudis D, Georgoulias V. Paclitaxel and docetaxel in the treatment of breast cancer. Expert Opin Pharmacother. 2008;9:2603–2616. doi: 10.1517/14656566.9.15.2603. [DOI] [PubMed] [Google Scholar]
- 6.Yared JA, Tkaczuk KHR. Update on Taxane Development: New Analogs and new Formulations. Drug Des, Dev Ther. 2012;6:371–384. doi: 10.2147/DDDT.S28997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez HL, Cardarelli PM, Deshpande S, Gangwar S, Schroeder GM, Vite GD, Borzilleri RM. Antibody-drug conjugates: current status and future directions. Drug Disc Today. 2014;19:869–881. doi: 10.1016/j.drudis.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Chari RVJ, Miller ML, Widdison WC. Antibody–drug conjugates: An emerging concept in cancer therapy. Angew Chem Int Ed. 2014;53:3796–3827. doi: 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- 9.Bouchard H, Viskov C, Garcia-Echeverria C. Antibody–drug conjugates—A new wave of cancer drugs. Bioorg & Med Chem. 2014;24:5357–5363. doi: 10.1016/j.bmcl.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 10.Swami A, Shi J, Gadde S, Votruba AR, Kolishetti N, Farokhzad OC. Nanoparticles for Targeted and Temporally Controlled Drug Delivery. In: Svenson S, Prud’homme RK, editors. Multifunctional Nanoparticles for Drug Delivery Applications: Imaging, Targeting, and Delivery. Springer Science+Business Media, LLC; 2012. [Google Scholar]
- 11.Maeda H. Tumor-Selective Delivery of Macromolecular Drugs via the EPR Effect: Background and Future Prospects. Bioconjugate Chem. 2010;21:797–802. doi: 10.1021/bc100070g. [DOI] [PubMed] [Google Scholar]
- 12.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1:16014. [Google Scholar]
- 13.Torrice M. Does nanomedicine have a delivery problem? ACS Cent Sci. 2016;2:434–437. doi: 10.1021/acscentsci.6b00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun T, Zhang YS, Pang B, Hyun DC, Yang M, Xia Y. Engineered nanoparticles for drug delivery in cancer therapy. Angew Chem Int Ed. 2014;53:12320–12364. doi: 10.1002/anie.201403036. [DOI] [PubMed] [Google Scholar]
- 15.Luo C, Wang Y, Chen Q, Han X, Liu X, Sun J, He Z. Advances of paclitaxel formulations based on nanosystem delivery technology. Mini-Rev Med Chem. 2012;12:434–444. doi: 10.2174/138955712800493924. [DOI] [PubMed] [Google Scholar]
- 16.Gluck S. nab-Paclitaxel for the treatment of aggressive metastatic breast cancer. Clin Breast Cancer. 2014;14:221–227. doi: 10.1016/j.clbc.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 17.Hwu JR, Lin YS, Josephrajan T, Hsu M-H, Cheng F-Y, Yeh C-S, Su W-C, Shieh D-B. Targeted paclitaxel by conjugation to iron oxide and gold nanoparticles. J Am Chem Soc. 2009;131:66–68. doi: 10.1021/ja804947u. [DOI] [PubMed] [Google Scholar]
- 18.Gibson JD, Khanal BP, Zubarev ER. Paclitaxel-Functionalized Gold Nanoparticles. J Am Chem Soc. 2007;129:11653–11661. doi: 10.1021/ja075181k. [DOI] [PubMed] [Google Scholar]
- 19.Heo DN, Yang DH, Moon HJ, Lee JB, Bae MS, Lee SC, Lee WJ, Sun IC, Kwon IK. Gold Nanoparticles Surface-functionalized with Paclitaxel Drug and Biotin Receptor as Theranostic Agents for Cancer Therapy. Biomaterials. 2012;33:856–866. doi: 10.1016/j.biomaterials.2011.09.064. [DOI] [PubMed] [Google Scholar]
- 20.Ren F, Bhana S, Norman DD, Johnson J, Xu L, Baker BR, Parrill AL, Huang X. Gold nanorods carrying paclitaxel for photothermal-chemotherapy of cancer. Bioconjugate Chem. 2013;24:376–386. doi: 10.1021/bc300442d. [DOI] [PubMed] [Google Scholar]
- 21.Zakharian TY, Seryshev A, Sitharaman B, Gilbert BE, Knight V, Wilson LJ. A Fullerene - Paclitaxel Chemotherapeutic: Synthesis, Characterization, and Study of Biological Activity in Tissue Culture. J Am Chem Soc. 2005;127:12508–12509. doi: 10.1021/ja0546525. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z, Mei L, Feng S-S. Paclitaxel Drug Delivery Systems. Expert Opin Drug Deliv. 2013;10:325–340. doi: 10.1517/17425247.2013.752354. [DOI] [PubMed] [Google Scholar]
- 23.Zhao P, Astruc D. Docetaxel Nanotechnology in Anticancer Therapy. ChemMedChem. 2012;7:952–972. doi: 10.1002/cmdc.201200052. [DOI] [PubMed] [Google Scholar]
- 24.Duncan B, Kim C, Rotello VM. Gold nanoparticle platforms as drug and biomacromolecule delivery systems. J Controlled Release. 2010;148:122–127. doi: 10.1016/j.jconrel.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosh P, Han G, De M, Kim CK, Rotello VM. Gold nanoparticles in delivery applications. Adv Drug Deliv Rev. 2008;60:1307–1315. doi: 10.1016/j.addr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 26.Rana S, Bajaj A, Mout R, Rotello VM. Monolayer coated gold nanoparticles for delivery applications. Adv Drug Deliv Rev. 2012;64:200–216. doi: 10.1016/j.addr.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dreaden EC, Alkilany AM, Huang X, Murphy CJ, El-Sayed MA. The golden age: gold nanoparticles for biomedicine. Chem Soc Rev. 2012;41:2740–2779. doi: 10.1039/c1cs15237h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Libutti SK, Paciotti GF, Byrnes AA, Alexander HR, Gannon WE, Walker M, Seidel GD, Yuldasheva N, Tamarkin L. Phase I and pharmacokinetic studies of CYT-6091, a novel PEGylated colloidal gold-rhTNF nanomedicine. Clin Cancer Res. 2010;16:6139–6149. doi: 10.1158/1078-0432.CCR-10-0978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paciotti GF, Kingston DGI, Tamarkin L. Colloidal gold nanoparticles: A novel nanoparticle platform for developing multifunctional tumor-targeted drug delivery vectors. Drug Dev Res. 2006;67:47–54. [Google Scholar]
- 30.Powell AC, Paciotti GF, Libutti SK. Colloidal gold: A novel nanoparticle for targeted cancer therapeutics. Methods Mol Biol. 2010;624:375–384. doi: 10.1007/978-1-60761-609-2_25. [DOI] [PubMed] [Google Scholar]
- 31.Kristensen CA, Nozue M, Boucher Y, Jain RK. Reduction of interstitial fluid pressure after TNF-α treatment of three human melanoma xenografts. Br J Cancer. 1996;74:533–536. doi: 10.1038/bjc.1996.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goodsell DS. The molecular perspective: Tumor necrosis factor. The Oncologist. 2006;11:83–84. doi: 10.1634/theoncologist.11-1-83. [DOI] [PubMed] [Google Scholar]
- 33.Alexander HR, Bartlett DL, Libutti SK, Fraker DL, Moser T, Rosenberg SA. Isolated hepatic perfusion with tumor necrosis factor and melphalan for unresectable cancers confined to the liver. J Clin Oncol. 1998;16:1479–1489. doi: 10.1200/JCO.1998.16.4.1479. [DOI] [PubMed] [Google Scholar]
- 34.Lejeune FJ. High dose recombinant tumor necrosis factor (rTNFα) administered by isolation perfusion for advanced tumors of the limbs: a model for biochemotherapy of cancer. Eur J Cancer. 1995;31:1009–1016. doi: 10.1016/0959-8049(94)00512-5. [DOI] [PubMed] [Google Scholar]
- 35.de Wilt JH, ten Hagen TL, de Boeck G, van Tiel ST, de Bruijn EA, Eggermont AMM. Tumour necrosis factor alpha increases melphalan concentration in tumour tissue after isolated limb perfusion. Br J Cancer. 2000;82:1000–1003. doi: 10.1054/bjoc.1999.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paciotti GF, Myer L, Weinreich D, Goia D, Pavel N, McLaughlin RE, Tamarkin L. Colloidal gold: A novel nanoparticle vector for tumor directed drug delivery. Drug Deliv. 2004;11:169–183. doi: 10.1080/10717540490433895. [DOI] [PubMed] [Google Scholar]
- 37.Farma JM, Puhlmann M, Soriano PA, Cox D, Paciotti GF, Tamarkin L, Alexander HR. Direct evidence for rapid and selective induction of tumor neovascular permeability by tumor necrosis factor and a novel derivative, colloidal gold bound tumor necrosis factor. Int J Cancer. 2007;120:2474–2480. doi: 10.1002/ijc.22270. [DOI] [PubMed] [Google Scholar]
- 38.Shenoi MM, Iltis I, Choi J, Koonce NA, Metzger GJ, Griffin RJ, Bischof JC. Nanoparticle delivered vascular disrupting agents (VDAs): Use of TNF-alpha conjugated gold nanoparticles for multimodal cancer therapy. Mol Pharmaceutics. 2013;10:1683–1694. doi: 10.1021/mp300505w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koonce NA, Quick CM, Hardee ME, Jamshidi-Parsian A, Dent JAD, Paciotti GF, Nedosekin D, Dings RPM, Griffin RJ. Combination of Gold Nanoparticle-Conjugated Tumor Necrosis Factor and Radiation Therapy Results in a Synergistic Antitumor Response in Murine Carcinoma Models. Int J Radiat Oncol, Biol, Phys. 2015;93:590–596. doi: 10.1016/j.ijrobp.2015.07.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Visaria R, Loren M, Williams B, Ebbini E, Paciotti GF, Griffin R. Nanotherapeutics for enhancing thermal therapy of cancer. Int J Hyperthermia. 2007;23:501–511. doi: 10.1080/02656730701611241. [DOI] [PubMed] [Google Scholar]
- 41.Myer L, Jones D, Tamarkin L, Paciotti GF. Nanomedicine-based enhancement of chemotherapy. Cancer Res. 2008;68:S5718. [Google Scholar]
- 42.Kingston DGI, Cao S, Zhao J, Paciotti GF, Huhta MS. Thiolated paclitaxels for reaction with gold nanoparticles as drug delivery agents. 8,558,019 B2. US Patent. 2013 Oct 15;
- 43.Mellado W, Magri NF, Kingston DGI, Garcia-Arenas R, Orr GA, Horwitz SB. Preparation and Biological Activity of Taxol Acetates. Biochem Biophys Res Commun. 1984;124:329–335. doi: 10.1016/0006-291x(84)91557-2. [DOI] [PubMed] [Google Scholar]
- 44.Ryu B-Y, Sohn J-S, Hess M, Choi S-K, Choi J-K, Jo B-W. Synthesis and Anti-Cancer Efficacy of Rapid Hydrolyzed Water-Soluble Paclitaxel Pro-Drugs. J Biomater Sci, Polym Ed. 2008;19:311–324. doi: 10.1163/156856208783720994. [DOI] [PubMed] [Google Scholar]
- 45.Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg JM, Sloane BF, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013;73:1524–1535. doi: 10.1158/0008-5472.CAN-12-2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Magri NF, Kingston DGI. Modified taxols, 4. Synthesis and biological activity of taxols modified in the side chain. J Nat Prod. 1988;51:298–306. doi: 10.1021/np50056a017. [DOI] [PubMed] [Google Scholar]
- 47.Deutsch HM, Glinski JA, Hernandez M, Haugwitz RD, Narayanan VL, Suffness M, Zalkow LH. Synthesis of Congeners and Prodrugs. 3. Water-Soluble Prodrugs of Taxol with Potent Antitumor Activity. J Med Chem. 1989;32:788–792. doi: 10.1021/jm00124a011. [DOI] [PubMed] [Google Scholar]
- 48.Senter PD, Pearce WE, Greenfield RS. Development of a Drug-release Strategy Based on the Reductive Fragmentation of Benzyl Carbamate Disulfides. J Org Chem. 1990;55:2975–2978. [Google Scholar]
- 49.Anderson ME. Glutathione: an overview of biosynthesis and modulation. Chem-Biol Interact. 1998;111–112:1–14. doi: 10.1016/s0009-2797(97)00146-4. [DOI] [PubMed] [Google Scholar]
- 50.Kuppusamy P, Li HL, Ilangovan G, Cardounel AJ, Zweier JL, Yamada K, Krishna MC, Mitchell JB. Noninvasive imaging of tumor redox status and its modification by tissue glutathione levels. Cancer Res. 2002;62:307–312. [PubMed] [Google Scholar]
- 51.Jones DP, Carlson JL, Samiec PS, Sternberg P, Jr, Mody VC, Jr, Reed RL, Brown LAS. Glutathione measurement in human plasma: Evaluation of sample collection, storage and derivatization conditions for analysis of dansyl derivatives by HPLC. Clin Chim Acta. 1998;275:175–184. doi: 10.1016/s0009-8981(98)00089-8. [DOI] [PubMed] [Google Scholar]
- 52.Lee KH, Chung YJ, Kim Y-C, Song SJ. Anti-tumor Activity of Paclitaxel Prodrug Conjugated with Polyethylene Glycol. Bull Korean Chem Soc. 2005;26:1079–1082. [Google Scholar]
- 53.Sharifi N, Qi J, Bane S, Sharma S, Li R, Robey R, Figg WD, Farrar WL, Kingston DGI. Survivin Is Not Induced by Novel Taxanes. Mol Pharmaceutics. 2010;7:2216–2223. doi: 10.1021/mp100211k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao J, Bane S, Snyder JP, Huc H, Mukherjee K, Slebodnick C, Kingston DGI. Design and synthesis of simplified taxol analogs based on the T-Taxol bioactive conformation. Bioorg Med Chem. 2011 doi: 10.1016/j.bmc.2011.10.010. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Farma JM, Puhlmann M, Soriano PA, Cox D, Paciotti GF, Tamarkin L, Alexander HR. Direct evidence for rapid and selective induction of tumor neovascular permeability by tumor necrosis factor and a novel derivative, colloidal gold bound tumor necrosis factor. Int J Cancer. 2007;120:2474–2480. doi: 10.1002/ijc.22270. [DOI] [PubMed] [Google Scholar]
- 56.Ebright YW, Chen Y, Kim Y, Ebright RH. S-[2-(4-Azidosalicylamido)ethylthio]-2-thiopyridine: Radioiodinatable, Cleavable, Photoactivatible Cross-Linking Agent. Bioconjugate Chem. 1996;7:380–384. doi: 10.1021/bc9600168. [DOI] [PubMed] [Google Scholar]
- 57.Liu C, Strobl JS, Bane S, Schilling JK, McCracken M, Chatterjee SK, Rahim-Bata R, Kingston DGI. Design, synthesis, and bioactivities of steroid-linked Taxol analogues as potential targeted drugs for prostate and breast Cancer. J Nat Prod. 2003;67:152–159. doi: 10.1021/np030296x. [DOI] [PubMed] [Google Scholar]
- 58.Cao S, Brodie PJ, Miller JS, Randrianaivo R, Ratovoson F, Birkinshaw C, Andriantsiferana R, Rasamison VE, Kingston DGI. Antiproliferative Xanthones of Terminalia calcicola from the Madagascar Rain Forest. J Nat Prod. 2007;70:679–681. doi: 10.1021/np060627g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Louie KG, Behrens BC, Kinsella TJ, Hamilton TC, Grotzinger KR, McKoy WM, Winker MA, Ozols RF. Radiation Survival Parameters of Antineoplastic Drug-sensitive and -resistant Human Ovarian Cancer Cell Lines and Their Modification by Buthionine Sulfoximine. Cancer Res. 1985;45:2110–2115. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.