


Abstract
We present herein the first complete genome sequence of a thermophilic Bacillus-related species, Geobacillus kaustophilus HTA426, which is composed of a 3.54 Mb chromosome and a 47.9 kb plasmid, along with a comparative analysis with five other mesophilic bacillar genomes. Upon orthologous grouping of the six bacillar sequenced genomes, it was found that 1257 common orthologous groups composed of 1308 genes (37%) are shared by all the bacilli, whereas 839 genes (24%) in the G.kaustophilus genome were found to be unique to that species. We were able to find the first prokaryotic sperm protamine P1 homolog, polyamine synthase, polyamine ABC transporter and RNA methylase in the 839 unique genes; these may contribute to thermophily by stabilizing the nucleic acids. Contrasting results were obtained from the principal component analysis (PCA) of the amino acid composition and synonymous codon usage for highlighting the thermophilic signature of the G.kaustophilus genome. Only in the PCA of the amino acid composition were the Bacillus-related species located near, but were distinguishable from, the borderline distinguishing thermophiles from mesophiles on the second principal axis. Further analysis revealed some asymmetric amino acid substitutions between the thermophiles and the mesophiles, which are possibly associated with the thermoadaptation of the organism.
INTRODUCTION
Many thermophiles and hyperthermophiles have been isolated from hot springs and other thermal environments (1). The complete genome sequences of 19 thermophilic or hyperthermophilic prokaryotic species have been determined. The genomic information should facilitate the study of thermophily in the prokaryotic cells and thermostability of the proteins. In fact, the features of the genomic sequence, which discriminate between thermophiles and mesophiles, can be simply identified by using principal component analysis (PCA) of the amino acid composition (2–4) or relative synonymous codon usage (3–5).
Comparative genomics is another useful approach for extracting candidate genes associated with thermophily. A previous study has shown that a phylogenetic pattern search against the clusters of orthologous groups (COGs) database (6) retrieved only one hyperthermophile-specific gene: reverse gyrase (7). Reverse gyrase, which is similar to some type I DNA topoisomerases from mesophiles, is thought to help DNA to function at high temperatures by increasing topological links between the two DNA strands. Indeed, the reverse gyrase gene has been identified in the genomes of hyperthermophiles, except the recently determined Thermus thermophilus genome (8). Despite this remarkable result, many other crucial genes responsible for thermophily are probably still hidden in the genome. However, identifying such genes through comparison of a variety of genomes is generally not easy, because phylogenetically related thermophiles share many genes that are not directly associated with thermophily, and phylogenetically distant thermophiles may have different mechanisms for thermoadaptation. One of the effective approaches in revealing thermophily-related genes based on genomic information is to compare genomes between closely related organisms, including both thermophiles and mesophiles. This approach is also effective for understanding thermoadaptation from the viewpoint of evolution, although the genomic sequences from an appropriate set of organisms are needed, which have not yet been obtained.
Aerobic endospore-forming Gram-positive Bacillus-related species have been isolated from various terrestrial soils and deep-sea sediments (9–11). It is known that Bacillus-related species can grow in a wide range of environments, at pH 2–12, in temperatures between 5 and 78°C, in salinity from 0 to 30% NaCl, and in pressures from 0.1 MPa (atmospheric pressure) to at least 30 MPa (corresponding to the pressure at a depth of 3000 m) (12,13). The complete genome sequences of five mesophilic bacilli with different phenotypic properties, Bacillus subtilis (14), Bacillus halodurans, (15), Oceanobacillus iheyensis (16), Bacillus cereus (17) and Bacillus anthracis (18), have already been determined, although the complete genome sequence of a thermophilic Bacillus-related species has not yet been established. These species are positioned as representatives of major diverged clusters in the 16S rRNA tree (Supplementary Figure 1).
Geobacillus kaustophilus HTA426, which was isolated from the deep-sea sediment of the Mariana Trench (19,20), is a thermophilic Bacillus-related species whose upper temperature limit for growth is 74°C (optimally 60°C). It is known that there are at least 12 other thermophilic Geobacillus species, which have been reclassified from the genus Bacillus (21).
Here, we report the complete nucleotide sequence of the genome of G.kaustophilus. We provide the first comparative analysis of the thermophilic genome with those of five other phylogenetically related mesophilic bacilli, B.subtilis, B.halodurans, O.iheyensis, B.anthracis and B.cereus, in order to highlight the thermophilic features of the genome. Special emphasis is placed on the mechanisms of adaptation of the bacilli to high-temperature environments.
MATERIALS AND METHODS
Sequencing, gene prediction and annotation
The genome of G.kaustophilus HTA426 was primarily sequenced using the whole-genome random-sequencing method used in our previous studies (15,16). The predicted protein-coding regions were initially defined by searching for open reading frames longer than 100 codons, in a manner similar to previous investigations. Searches of protein databases for amino acid similarities and annotation were performed using the same method as described in the previous study (15,16). The functional assignment for annotated CDSs identified in the G.kaustophilus genome followed the protocol used for B.subtilis (14).
Principal component analysis
The coding sequences of 149 prokaryotic genomes were obtained from the NCBI ftp site (ftp://ftp.ncbi.nih.gov/genbank/genomes/Bacteria). The amino acid composition of the translated sequence and the relative synonymous codon usage (RSCU) of genes in these genomes and the G.kaustophilus genome were subjected to PCA after the elimination of genes smaller than 150 codons. For calculating the amino acid composition, proteins containing at least two transmembrane segments, as predicted by the PSORT program (22), were also eliminated. RSCU for each gene is a 59 dimensional vector, whose elements were defined as
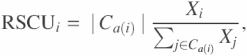
where Xi is the number of occurrences of the i-th codon, which codes for the amino acid a(i), Ca is a set of codons that code for the amino acid a and |C| is the cardinality of the set C (23); we considered 59 codons from which stop codons and Met and Trp codons that have no synonymous codons were excluded. PCA and other statistical analyses were performed using the R statistical package (http://www.r-project.org/).
To examine the effect of overrepresentation of some closely related organisms, we also prepared two additional sets of organisms: one set includes only one strain from each species and the other set includes only one species from each genus except for the genus Bacillus. Since the results of the PCAs with these sets of organisms showed very similar patterns to that of the complete set of organisms, here we show only the result of the complete set of organisms.
Identification of asymmetric amino acid substitutions
The counts of amino acid substitutions between G.kaustophilus and other Bacillus-related species were tabulated using multiple alignments of amino acid sequences in 1056 common orthologous groups (see below) showing one-to-one correspondences across all genomes; the CLUSTALW program (24) was used for creating the alignments. In this statistical analysis, the gaps appearing in the alignment columns are also treated as a single character. Let nAB be the number of sites at which amino acid A in a mesophilic bacillar genome changed to amino acid B in the G.kaustophilus genome, and assume that nAB > nBA. The statistical significance of the observed asymmetry between amino acids A and B was evaluated by the probability P(X ≥ nAB), assuming that X follows the binomial distribution Bi(nAB + nBA, 0.5). Here, we considered the asymmetry significant when P ≤ 10−5. This implies that type I error rate of multiple testing with 210 substitution patterns between 21 characters (including the gap character) is at most 0.21%.
Orthologous gene grouping
Orthologous groups in the Bacillus-related species were established from the all-against-all similarity results of applying the clustering program on the MBGD database (25). The common orthologs conserved in all Bacillus-related genomes were then selected for constructing multiple alignments. Since these orthologs generally display complicated mutual relationships, such as many-to-many correspondences and fusion or fission of domains, we selected only those orthologs with one-to-one correspondences without domain splitting, in order to simplify the analysis.
RESULTS AND DISCUSSION
General features
The genome of G.kaustophilus is composed of a single circular chromosome (3 554 776 bp) and a plasmid (47 890 bp), with a mean G+C content of 52.1 and 44.2%, respectively (Table 1 and Figure 1). We identified 3498 protein-coding sequences (CDSs), with a mean size of 862 nt, and the coding sequences were found to cover 86% of the chromosome. Predicted protein sequences were compared with sequences in a non-redundant protein database, and biological roles were assigned to 1914 CDSs (54.7%). In this database search, 1096 CDSs (31.3%) were identified as conserved proteins of unknown function in comparison with proteins from other organisms (Figure 2D). We found that 75.2% of the genes started with ATG, 13.5% with GTG and 11.3% with TTG. These values are similar to those of other Bacillus-related species, except that the ratio of ATG (83.1%) in the initiation codon of B.anthracis is a little bit higher than that of the others. On the other hand, 42 CDSs were identified in the plasmid designated as pTHA426 (Figure 1), and the mean size of the CDSs (906 nt) was much larger than that identified in the plasmids of B.cereus (645 nt) and B.anthracis (639–645 nt). There is a difference in the pattern of the initiation codon between the chromosome and the plasmid of G.kaustophilus. The ratio of ATG in the chromosome is more than 11 percentage points higher and the ratio of GTG is 8 percentage points lower than the ATG and GTG ratios of the plasmid, respectively (Table 1). The number of predicted proteins with biological roles was 24 (57.1%). The G.kaustophilus genome was found to contain nine copies of the rRNA operon and 87 tRNA species organized into 11 clusters involving 81 genes plus 6 single genes.
Table 1. General features of the G. kaustophilus genome and its comparison with those of Bacillus-related species.
| General features | G.kaustophilus HTA426 | O.iheyensis HTE831 | B.halodurans C-125 | B.cereus ATCC14579 | B.anthracis Ames | B.subtilis 168 | |
|---|---|---|---|---|---|---|---|
| Chromosome | |||||||
| Size (bp) | 3 544 776 | 3 630 528 | 4 202 352 | 5 411 809 | 5 227 293 | 4 214 630 | |
| G+C content (mol%) | |||||||
| Total genome | 52.1 | 35.7 | 43.7 | 35.3 | 35.4 | 43.5 | |
| Coding region | 52.9 | 36.1 | 44.4 | 35.9 | 36.0 | 43.4 | |
| Non-coding region | 47.0 | 31.8 | 39.8 | 32.7 | 32.6 | 43.6 | |
| Predicted CDS number | 3498 | 3496 | 4066 | 5234 | 5311 | 4106 | |
| Average length (bp) | 862 | 883 | 879 | 835 | 794 | 896 | |
| Coding region (%) | 86 | 85 | 85 | 81 | 81 | 87 | |
| Initiation codon (%) | |||||||
| AUG | 75.2 | 79.5 | 78 | 75.3 | 83.1 | 78 | |
| GUG | 13.5 | 7.8 | 12 | 11.9 | 9.1 | 9 | |
| UUG | 11.3 | 12.7 | 10 | 12.8 | 7.8 | 13 | |
| Stable RNA (%) | 1.70 | 1.04 | 1.02 | 1.2 | 1.09 | 1.27 | |
| Number of rrn operon | 9 | 7 | 8 | 13 | 11 | 10 | |
| Mean G+C content | 58.5 | 52.7 | 54.2 | 52.6 | 52.6 | 54.4 | |
| Number of tRNA | 87 | 69 | 78 | 108 | 86 | 86 | |
| Mean G+C content | 59.1 | 58.8 | 59.5 | 59.2 | 59.1 | 58.2 | |
| Plasmid | pHTA426 | — | — | pBClin15 | pX01 | pX02 | — |
| Size (bp) | 47 890 | — | — | 15 100 | 181 677 | 94 829 | — |
| G+C content (mol%) | 44.2 | — | — | 38.1 | 32.5 | 33.0 | — |
| Non-coding region | 44.5 | — | — | 38.4 | 33.7 | 34.2 | — |
| Coding region | 43.8 | — | — | 35.4 | 29.7 | 30.6 | — |
| Predicted CDS number | 42 | — | — | 21 | 217 | 113 | — |
| Average length (bp) | 906 | — | — | 645 | 645 | 639 | — |
| Coding region (%) | 79.5 | — | — | 89.8 | 77.1 | 76.2 | — |
| Initiation codon (%) | |||||||
| AUG | 64.3 | — | — | 75.3 | 75.3 | 75.3 | — |
| GUG | 21.4 | — | — | 11.9 | 11.9 | 11.9 | — |
| UUG | 14.3 | — | — | 12.8 | 12.8 | 12.8 | — |
Figure 1.
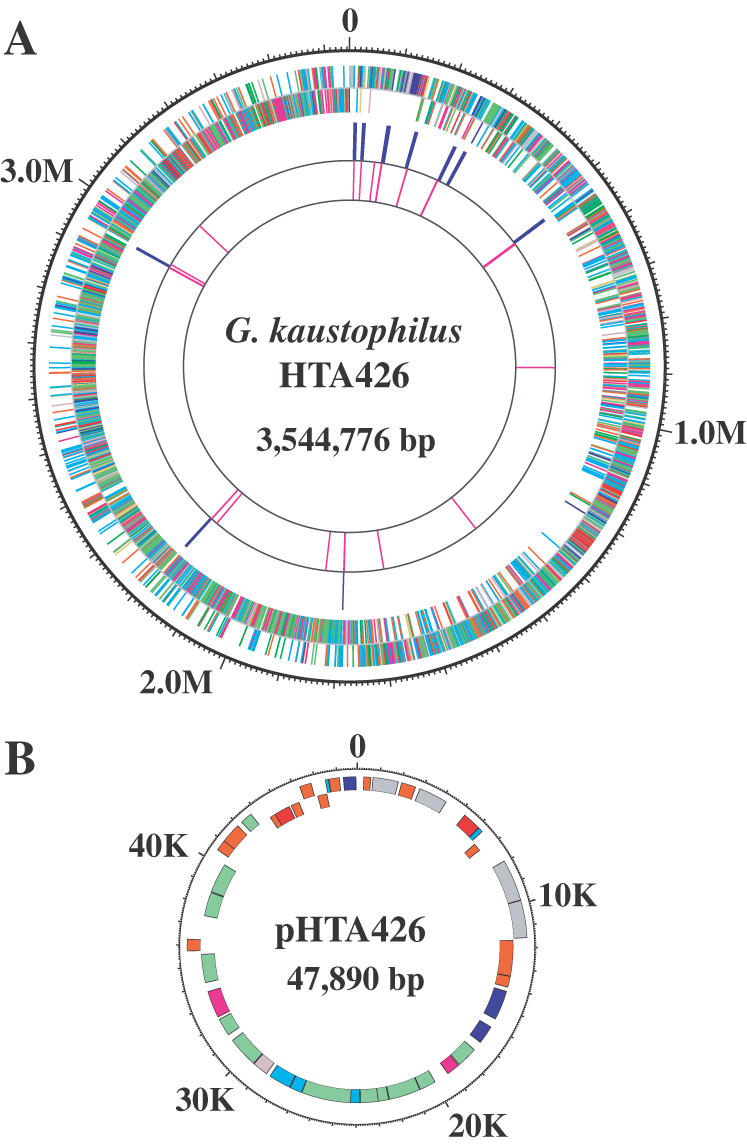
Circular representation of the thermophilic G.kaustophilus HTA426 genome. (A) chromosome; (B) plasmid pHTA426. The distribution of CDSs is depicted by colored boxes according to the functional category and the direction of transcription (the outer circle is the plus strand; the inner circle is the minus strand; red represents the cell wall, sensors, motility and chemotaxis, protein secretion, cell division and transformation/competence; magenta represents transport/binding proteins and lipoproteins and membrane bioenergetics; gold represents sporulation and germination; yellow-green represents intermediary metabolism; gray represents DNA replication, DNA restriction/modification and repair, DNA recombination, and DNA packaging and segregation; pink represents RNA synthesis; blue represents protein synthesis; forest green represents miscellaneous functions; sky blue represents conserved CDSs with unknown function; and coral represents non-conserved proteins). The third and fourth circles indicate the distribution of rRNA and tRNA in the genome, respectively.
Figure 2.

Summary of the orthologous relationships between the six bacillar genomes and the functional assignment of the genes belonging to each group. A, B.anthracis; C, B.cereus; I, O.iheyensis; S, B.subtilis; H, B.halodurans; and K, G.kaustophilus. The figure in the box shows the number of orthologous groups for each combination of species. (A) Breakdown of the genes (221 genes) categorized into 189 common orthologous groups in all bacilli except for G.kaustophilus. (B) Breakdown of genes unique to G.kaustophilus (839 genes) categorized into 757 groups, which have no orthologous relationships to the other five bacilli. (C) Breakdown of the genes (1308 genes) categorized into 1257 common orthologous groups in all six bacilli. (D) Breakdown of all genes identified in the G.kaustophilus genome. Note that the number of orthologous groups does not coincide with the number of genes, because the paralogous genes of G.kaustophilus are included in each orthologous group.
A new database specifically established for the G.kaustophilus sequences, ExtremoBase, will be accessible at http://www.jamstec.go.jp/jamstec-e/bio/exbase.html. The data will be also available through the MBGD server (http://mbgd.genome.ad.jp/) with additional functions for ortholog grouping and comparative genome analysis.
Transposable elements
The G.kaustophilus genome possesses 91 genes encoding putative transposases (Tpases) categorized into 31 groups, which seem to be carried by various insertion sequences (ISs). Although the number of groups was slightly greater than the case of B.halodurans (27 groups), the number of those genes was less than the 112 found in the B.halodurans genome. However, the total number of Tpase genes in G.kaustophilis genome is much greater than that of four other sequenced bacilli, B.subtilis (none), O.iheyensis (21 genes), B.cereus (28 genes) and B.anthracis (12 genes) (Figure 3). Forty-three of the Tpase genes in the G.kaustophilus genome are similar to the sequences present in the genomes of thermophilic bacteria such as Geobacillus stearothermophilus (26), Thermoanaerobacter tengcongensis (27) and Thermotoga maritima (28). The remaining 48 genes are similar to those of mesophilic bacteria such as B.halodurans, O.iheyensis (29) and B.cereus, and also to those of mesophilic archaea such as Methanosarcina mazeri (30) and Methanosarcina acetivorans (31). Of the 48 genes, 9 genes showed significant homology to Tpases carried by IS642, IS653 and IS654 identified in the B.halodurans genome, which are categorized into the IS630 family, the IS650/IS653 family and the IS256 family, respectively (32). The first family has been reported only in B.halodurans and G.stearothermophilus, and the latter two families were known before this study only in B.halodurans among the bacilli (29). Recently, we showed that B.halodurans genome contains a new transposon, which is very similar to the one identified in thermophilic G.stearothermophilus genome (33). Thus, it is clear that insertion sequences, such as the IS and the transposon, are shared not only between thermophiles but also between thermophiles and mesophiles. On the other hand, no evidence of horizontal gene transfer between hyperthermophilic archaea and G.kaustophilus was obtained through the analysis of Tpases in this study.
Figure 3.

Distribution of major Tpase genes in the six bacillar genomes. The Tpase genes categorized into 31 kinds are represented by triangles. The direction of each triangle matches the transcriptional direction. Ori represents the region of the replication origin of the chromosome.
The genome also contains at least 21 putative phage-associated genes, which are similar to those of Streptococcus pyrogenes, Lactococcus lactis, B.subtilis and Clostridium perfringens. These genes are distributed within a 40 kb-region (approximately) corresponding to the region, 535–575 kb from the oriC of the G.kaustophilus genome. The sequence homology and the organization of the phage-related genes in G.kaustophilus genome is comparatively similar to those of prophage 315.1, identified in S.pyrogenes M3, which is classified into the Siphoviridae family (34).
Orthologous relationships among the six bacillar genomes
Out of 3498 genes predicted in the G.kaustophilus chromosome, 839 (24%) categorized into 757 groups were unique genes, which possess no orthologous relationships to the other five sequenced bacillar genomes, and 488 (14%) genes categorized into 419 groups were orphans, showing no significant similarity to any other gene products (Figure 2B and D). The 1257 common orthologous groups composed of 1308 genes are shared among all six sequenced bacillar genomes (Figure 2C and Supplementary Table S1). Recently, it was shown that 271 genes are indispensable for the growth of B.subtilis in nutritious conditions (35). Out of 1308 common genes, 233 show correspondences to the B.subtilis essential genes. Through a series of orthologous analyses of the six bacilli used in this study, four essential genes, ymaA, ydiO, tagB and tagF, associated with purine/pyrimidine biosynthesis, DNA methylation and teichoic acid biosynthesis, were found to be unique to the B.subtilis genome. Teichoic acids are composed of cell-wall teichoic acid and lipoteichoic acid (36). It is known that six genes (tagA, tagB, tagD, tagE, tagF and tagO) are associated with teichoic acid biosynthesis in B.subtilis, with all genes except tagE being essential for growth. The five other Bacillus-related species, however, lack some of these genes; remarkably, G.kaustophilus lacks all genes except tagE, suggesting that these bacilli may have a different pathway for teichoic acid biosynthesis. G.kaustophilus genome lacks eight more B.subtilis essential genes (totally 16 genes), mrpB, mrpC, mrpF, glyQ, glyS, menA, ppaC and ydiP associated with Na+/H+ antiporter for pH homeostasis, glycyl-tRNA synthetase, inorganic pyrophosphatase and DNA methyltransferase.
As shown in Figure 2A, it was found that 189 orthologous groups composed of 221 genes are commonly shared among the five mesophilic bacilli, but not by G.kaustophilus. In these mesophile-specific orthologs, there are 20 genes encoding ABC transporter (10 ATP-binding proteins, 6 permeases and 4 substrate-binding proteins) filed into function category 1.2 (transport/binding proteins and lipoproteins), 20 genes encoding transcriptional regulator (category 3.5.2, regulation), and 11 genes encoding proteins for immunity to bacteriotoxin-related proteins, toxic anion resistance-related proteins and catalase (category 4.2, detoxification) (Figure 2 and Supplementary Table S2). This is one of the significant differences between thermophilic G.kaustophilus and mesophilic bacillar genomes. The set of 839 unique genes of G.kaustophilus contains 22 individual ABC transporter genes (10 ATP-binding proteins, 7 permeases and 5 substrate-binding proteins), whereas there are only six and three genes categorized into 3.5.2 and 4.2, respectively (Figure 2B and Supplementary Table S3).
G.kaustophilus shares 1773–2014 orthologous genes with the other five bacilli, corresponding to 53.4–62.0% of all genes in the HTA426 genome. If the relative physical distribution of orthologous genes in the genomes between G.kaustophilus and other Bacillus-related species is the same, a diagonal line should appear from the lower left to the upper right in Figure 4. However, there are many orthologous genes deviated from the line, although the physical distributions of the orthologous genes between G.kaustophilus and B.subtilis, and between G.kaustophilus and O.iheyensis are largely collinear (Figure 4B and D). The difference in the physical distributions of orthologous genes in the genomes presumably occurred due to various minor inversion and horizontal gene transfer. On the other hand, 1056 are common orthologous genes possessing one-to-one correspondences among the six bacilli and these common orthologs represent 23.7–36% of each genome. Most of the common genes were found to be distributed in the collinear regions, but the direction of collinearity of the orthologs between G.kaustophilus and B.cereus changes at ∼28–33° from the ter region in both directions (Figure 4C). The physical distribution of orthologous genes between G.kaustophilus and B.halodurans is very similar to the case of B.cereus (Figure 4A), and a similar result has been previously documented in a comparison between B.subtilis and B.halodurans. It has been reported that the B.halodurans genome has an inversion between the regions around 112–153° and 212–240°, due to the action of IS elements (14,32).
Figure 4.

Comparison of ortholog organization between G.kaustophilus and four other bacillar genomes. The y- and x-axes show G.kaustophilus and the other genomes, respectively. (A) Orthologs between G.kaustophilus and B.halodurans. (B) Orthologs between G.kaustophilus and B.subtilis. (C) Orthologs between G.kaustophilus and B.cereus. (D) Orthologs between G.kaustophilus and O.iheyensis. Light-colored dots represent the orthologs possessing one-to-one correspondences between the two genomes. Dark-colored dots, which represent the common orthologs across the six bacillar genomes, are overlaid on the light-colored dots.
Principal component analyses for characterizing the genomic features related to thermophily
The features of the genomic sequence determining thermophiles and mesophiles can be easily identified through PCA (or correspondence analysis, a similar technique) of the amino acid composition and the relative synonymous codon usage, as mentioned previously. In both analyses, whereas the first principal component (PC1) correlated with the G+C content of the chromosome, the second PC (PC2) clearly correlated with the optimal growth temperature, so that thermophiles and mesophiles can be distinguished from each other along the second axis. We attempted PCA in order to confirm whether the G.kaustophilus genome has a signature similar to that of other thermophiles (Figure 5 and see also Supplementary Figure 2). In both analyses, all thermophiles whose genomes have already been reported were located above the borderline, distinguishing thermophiles from mesophiles, except Thermosynochococcus elongatus (37), whose upper growth temperature limit (60°C) is rather low in comparison to that of other thermophiles. G.kaustophilus was located above the borderline in the PCA of amino acid composition, but below the borderline in the PCA of synonymous codon usage. Thus, the G.kaustophilus genome is the first complete thermophilic genome that clearly shows different tendencies between the PCAs of synonymous codon usage and amino acid composition.
Figure 5.

Distribution of G.kaustophilus along the first and second axes of the PCA. (A) PCA of the amino acid composition of 150 prokaryotic genomes. (B) PCA of synonymous codon usage in 150 prokaryotic genomes. Mesophiles are denoted by circles. Red: crenarchaeota; orange: euryarchaeota; gray: nanoarchaeota; green: hyperthermophilic bacteria; purple: firmicutes; blue: actinobacteria; magenta: proteobacteria; white: cyanobacteria; and black: others. In addition, aae: Aquifex aerolicus; afu: Archaeoglobus fulgidus; ape: Aeropyrum pernix; mja: Methanococcus jannaschii; mka: Methanopyrus kandleri; mth: Methanobacterium thermoautotrophicum; neq: Nanoarchaeum equitans; pab: Pyrococcus abyssi; pai: Pyrobaculum aerophilum; pfu: Pyrococcus furiosus; pho: Pyrococcus horikoshii; sso: Sulfolobus solfataricus; sto: Sulfolobus tokodaii; tac: Thermoplasma acidophilum; tel: Thermosynechococcus elongatus; tma: Thermotoga maritima; tte: Thermoanaerobacter tengcongensis; tth: Thermus thermophilus; tvo: Thermoplasma volcanicum; BA: B.anthracis; BC: B.cereus; BH: B.halodurans; BS: B.subtilis; GK: G.kaustophilus; and OI: O.iheyensis. The following mesophilic microorganisms above the borderline are shown in red, etc.: Clostridium tetani; cpe: Clostridium perfringens; fau: Fusobacterium nucleatum; mac: Methanosarcina acetivorans; and mma: Methanosarcina mazei.
In the case of the PCA of amino acid composition, we were able to find the borderline distinguishing thermophiles from mesophiles at 0.0164 on the second principal axis. G.kaustophilus is positioned on this borderline, along with the thermophilic Thermoplasma acidophilum (38) and Thermoplasma volcanicum, which can grow at temperatures of up to 62–67°C (39). On the other hand, the PC2 positions of the five mesophilic Bacillus-related species are below the borderline (Figure 5A). Therefore, the genomes of the six Bacillus-related species can serve as good material for studying the mechanisms of thermostabilization of proteins and thermophily of microbes, since a limited number of amino acid changes yielding differences in PC2 positions, as shown in Figure 5, seem to reflect differences in thermophily or thermostability among these six bacilli.
In contrast to the results of the PCA of the amino acid composition, the synonymous codon usage in the G.kaustophilus genome does not show any distinguishable thermophilic pattern (Figure 5B). The thermophilic pattern in the synonymous codon usage is probably due to natural selection related to thermophily acting on the nucleotide sequence; it may be related to mRNA thermostability or the stability of codon–anticodon interactions (4,5). This pattern also seems to reflect the dinucleotide composition of genomic sequences, which may be related to the flexibility of the DNA molecule; purine–purine (RR) or pyrimidine–pyrimidine (YY) dinucleotides are predominant in thermophiles (40) (Supplementary Figure 2B). The G.kaustophilus genome did not appear to have been subjected to such selective pressure [e.g. the RR + YY value of the G.kaustophilus genome (50.7%) was less than that of the B.subtilis genome (53.2%)]. Some specific factors involved in the stabilization of DNA or RNA, of which some candidates will be listed below, might compensate for the lack of this genomic signature.
Generally, it is known that the G+C content in rRNA and tRNA, rather than that of the entire genome, linearly correlates with growth temperature in thermophilic archaea, although discriminating moderate thermophiles from mesophiles through the G+C content in the RNA molecules alone is generally not easy (41). Indeed, the mean G+C content in the tRNA of the G.kaustophilus genome was 59.1%, a value slightly lower than in B.halodurans and B.cereus (Table 1). On the other hand, the mean G+C content in the rRNA operons in the G.kaustophilus genome was found to be 58.5% (Table 1). This value is 4–6 percentage points higher than that of mesophilic Bacillus-related species (52.7–54.4%) with maximum temperatures for growth ranging from 42 to 58°C. However, this difference in the rRNAs was less than the difference in the genomic G+C contents between G.kaustophilus (52.1%) and the mesophilic bacilli (35.3–43.7%), probably due to the stronger constraints imposed on the rRNA operons. We examined the relationship between the G+C content in the 16S rRNA and that in the entire genome and confirmed a clear correlation between these values among various mesophiles (Figure 6). On the other hand, the rRNAs of the hyperthermophiles showed apparently higher G+C contents than those of the mesophiles regardless of their genomic G+C contents. The G+C content in the 16S rRNA of G.kaustophilus genome was moderately, but still, significantly higher than those of the mesophiles, even when the difference in the genomic G+C content was taken into consideration (Figure 6). Therefore, we conclude that the higher G+C content in rRNA is one of the thermophilic signatures in the G.kaustophilus genome.
Figure 6.

Relationship between G+C content of 16S rRNA and that of the entire genome. The solid line is the regression line calculated using only the mesophilic genomes; the regression equation is y = 0.17x + 45.36, where x and y are the genomic and the rRNA G+C content, respectively. The dashed lines are the upper and lower limits of the 95% prediction interval. The same symbols, colors and abbreviated species names are used as in Figure 5.
Asymmetric amino acid substitution patterns
We tried to identify amino acid substitutions showing significant asymmetry between G.kaustophilus and other Bacillus-related species, using multiple alignments of 1056 common orthologous groups that have one-to-one correspondences across all genomes. The resulting asymmetric substitutions were plotted on the plane generated by the first two principal components, as in Figure 5, according to the difference in the PC scores yielded by each substitution (Figure 7).
Figure 7.

Asymmetric amino acid substitution patterns across G.kaustophilus and other mesophilic bacilli, observed in the multiple alignments of 1056 common orthologous groups that have one-to-one correspondences. (A) Substitution pattern between G.kaustophilus and B.halodurans. (B) Substitution pattern between G.kaustophilus and B.subtilis. (C) Substitution pattern between G.kaustophilus and B.cereus. (D) Substitution pattern between G.kaustophilus and O.iheyensis. Only substitutions are shown (say, AB) whose frequencies are significantly larger than those in the opposite direction (BA), where AB denotes a substitution pattern in which amino acid A in a mesophilic bacillar genome is changed to amino acid B in the G.kaustophilus genome (see Materials and Methods). Each substitution is plotted on the same principal component plane as the one shown in Figure 6, according to the differences in PC1 and PC2 scores yielded by that substitution; note that the same substitution is plotted at the same position in all plots. The area of the circle is proportional to the difference between the number of substitutions from the number in the opposite direction (nAB − nBA). Red open circles represent asymmetric substitutions commonly identified in all mesophilic bacillar genomes; green open circles represent species-specific asymmetric substitutions whose difference in number from that in the opposite direction is ≥200; black open circles represent other species-specific asymmetric substitutions. The actual substitution pattern (AB for the change from the A of the mesophilic bacillus to the B of G.kaustophilus) is shown for each red or green open circle. A hyphen (-) in the substitution pattern denotes a gap character.
There were 39 asymmetric substitutions commonly identified between G.kaustophilus and all other Bacillus-related species (red circles in Figure 7). Remarkably, all of these substitutions increase the PC1 scores of the G.kaustophilus proteins, corresponding to an increase in the chromosomal G+C content. On the other hand, 24 out of 39 substitutions increase the PC2 scores of the G.kaustophilus proteins, presumably corresponding to an increase in the thermostability of the proteins. These substitutions generally increase the content of Arg, Ala, Gly, Val and Pro in the G.kaustophilus proteins, while decreasing Gln, Thr, Asn and Ser (Figure 7 and see also Supplementary Figure 2A). However, the overall increase in the PC2 score by these common substitutions is less remarkable than that in the PC1 score.
In contrast, among species-specific asymmetric substitutions (those with larger and smaller differences are shown in Figure 7, represented by green and black circles, respectively), we were able to find a substantial number of substitutions that increase the PC2 scores of the G.kaustophilus proteins, especially, when we compared them with those of the B.subtilis or O.iheyensis proteins (Figure 7). Indeed, 8 out of 10 and 15 out of 18 large asymmetric substitutions (green circles) found in B.subtilis and O.iheyensis proteins, respectively, were found to increase the PC2 scores of G.kaustophilus proteins. In particular, there are four asymmetric substitutions (QE, SE, DE and NK) found in the comparison with the O.iheyensis genome and located in the upper-left quadrant of the principal component plane, which cannot be explained by the difference in GC/AT mutation pressure; instead, they are likely to be explained by the difference in selection pressure related to the thermostability of proteins.
The order of the number of asymmetric substitutions was not equivalent to the order of the similarity of each species with the G.kaustophilus proteins. Indeed, the former order was B.halodurans (50 substitutions) < B.subtilis (82 substitutions) < B.cereus (84 substitutions) < O.iheyensis (92 substitutions), whereas the average identities between G.kaustophilus and other Bacillus-related species in the 1056 orthologous protein sequence alignments were B.subtilis (64.0%) > B.cereus (63.0%) > B.halodurans (61.3%) > O.iheyensis (57.0%). Therefore, the difference in Figure 7 cannot be explained by the difference in evolutionary distance only.
The observation that all common asymmetric substitutions between G.kaustophilus and other Bacillus-related species increase the PC1 scores, indicates that the observed substitution bias is mainly due to the GC/AT directional mutation pressure (40), which itself cannot be considered as a direct cause of the thermostabilization of proteins in general. Indeed, as indicated in Figure 5 as well as in the previous studies (2,3,42), increase in G+C content seems to be a completely independent process from thermoadaptation. One of the plausible explanations is that accelerated amino acid changes caused by the GC/AT mutation pressure, combined with the subsequent natural selection, facilitated the adaptation of G.kaustophilus to an environment with a higher temperature through the increase in the thermostability of all its proteins. Recently, Nishio et al. (42) reported a similar observation, according to which the increase in G+C content in the Corynebacterium efficiens genome beyond that in the Corynebacterium glutamicum genome, could account for the major patterns of asymmetric amino acid substitutions, possibly associated with the increase in the thermostability of C.efficiens (43). Thus, such a scenario might apply more generally for mesophilic bacteria in their adaptation to environments with higher temperatures.
However, by this hypothesis alone, it is difficult to explain the substantial differences in the substitution patterns among mesophilic bacilli. An alternative hypothesis is that the common ancestors of these bacilli have some thermophilic features, and that the current mesophilic bacilli have lost these features during the course of evolution. In this case, the difference in substitution patterns among the organisms can be explained as a difference in the extent to which the thermostability of proteins has decreased in each organism. In this connection, the position of the Bacillus-related species on the PC2 axis in the PCA of the amino acid composition is remarkable; it is located near the borderline, distinguishing thermophiles from mesophiles (Figure 5).
Candidate genes involved in the thermophilic phenotype
Although it is not clear what the upper temperature limit for bacterial life is, or what specific factors will set this limit, it is generally assumed that the limit will be dictated by molecular instability. Actually, DNA duplex stability is apparently achieved at high temperatures by elevated salt concentrations, polyamines, cationic proteins and supercoiling, rather than the manipulation of G+C ratios (43). RNA stability is enhanced by covalent modification, and the secondary structure is also probably critical. We found some genes in a set of unique G.kaustophilus genes, which seem to be involved in the stabilization of DNA and RNA, such as protamine, spermidine/spermine synthase, tRNA methyltransferase (MTase) and rRNA MTase (Supplementary Table S3).
DNA topology is affected by the interaction with cationic proteins, numerous examples of which have been identified in hyperthermophiles. The small basic proteins bind DNA in vitro with substantial increases in Tm, and have been variously shown to bend DNA or form nucleosome structures (43). Protamine is a protein that binds DNA in sperm, replacing histones and allowing chromosomes to become more highly condensed than it is possible with histones. Surprisingly, a gene (GK 1739) was found to show significant similarity (51%) to sperm protamine P1 from Phascolarctos cinereus (Koala bear). Since there has been no report of prokaryotic protamine-like gene thus far, this is the first such discovery in prokaryotes. Archaeal histones belonging to the HMf family are homologs of eukaryal nucleosome core histones, and have been shown to bind to and compact archaeal DNA both in vitro and in vivo (44). Histone-like proteins from Sulfolobus (45) have no eukaryal homologs, but, like the HMf proteins, they also compact DNA and increase the Tm of DNA in vitro. Therefore, the protamine P1-like protein identified in G.kaustophilus presumably behaves similarly to archaeal histone-like proteins in supporting life at high temperatures.
Polycationic polyamines, which increase the Tm of DNA and protect ribosomes from thermal inactivation in vitro, have been observed in hyperthermophiles. The polyamines participate in many cellular processes through their binding to DNA, RNA and phospholipids, not only in thermophiles but also in mesophiles. There is an interesting report according to which most spermines and spermidines exist as a polyamine–RNA complex in mesophilic Escherichia coli cells (46). A comprehensive analysis of the polyamines in hundreds of bacterial and archaeal species, from mesophiles to hyperthermophiles, has been carried out previously (47–49). The results showed that some kinds of polyamine, such as norspermine and norspermidine, occurred mainly in hyperthermophilic archaea (43). A hyperthermophilic bacterium, T.thermophilus, produced unusual polyamines (homocaldohexamine) in addition to norspermine and norspermidine, and inactivation of the basic genes related to polyamine synthesis, such as speA, speB and speE, resulted in a loss of the hyperthermophilic phenotype (growth defect at 78°C) (50). Thus, it is thought that these polyamines may play unique roles in supporting hyperthermophilic life. On the other hand, thermophilic Geobacillus species do not produce polyamines specific to hyperthermophiles, but produce spermine as a major polyamine; spermine is not produced by mesophilic Bacillus-related species, such as B.halodurans, B.subtilis and B.cereus, which we used for comparative analysis in this study (49). The genomes of five species, i.e. all except for O.iheyensis, contain a common gene for spermidine synthase, and other unique genes for it were identified in the B.anthracis, B.cereus and O.iheyensis genomes in addition to the common gene. Since it became clear that G.kaustophilus possesses unique genes for spermine/spermidine synthase and polyamine ABC transporter (permease) among the six sequenced bacilli, these genes seem to be strong candidates responsible for thermophily in these bacilli.
All types of cellular RNA contain modified nucleosides, but the largest number and greatest variety are found among tRNAs, and >80 different modifications have been identified to date in the tRNAs of various organisms (51). Modifications consist of simple chemical alterations of the nucleoside (e.g. methylation of the base or ribose, base isomerization, reduction, thiolation or deamination) or more complex hypermodifications. The structural stabilization of ribonucleic acids in hyperthermophiles is particularly important in tRNAs, where there is a requirement for the maintenance of a complex three-dimensional structure. Actually, it has recently been reported that the inactivation of the gene for tRNA MTase in T.thermophilus resulted in a thermosensitive phenotype (growth defect at 80°C), which suggests a role for the N1-methylation of tRNA adenosine-58 in the adaptation of life to extreme temperatures (52). The six bacillar genomes were found to share five orthologous tRNA MTases and four orthologous rRNA MTases, and the G.kaustophilus genome was found to contain three more unique tRNA or tRNA/rRNA MTase genes lacking orthologous relationships to the five other bacilli. Thus, these genes also seem to be strong candidates responsible for thermophily, although their specificity in the modification of tRNA is still not clear.
CONCLUSION
In this paper, we have attempted to highlight the genes involved the thermophilic phenotype and the genomic features related to thermophily by PCA of the amino acid composition and asymmetric amino acid substitution based on comparative analysis of thermophilic G.kaustophilus with five other mesophilic Bacillus-related species. G.kaustophilus presumably shares some similar basic mechanisms for thermophily with other thermophiles or hyperthermophiles, while also having mechanisms unique to thermophilic Bacillus-related species. Although we were able to find strong candidates responsible for thermophily in a unique-gene set of G.kaustophilus (839 genes), half of those genes are orphan, and 24.6% of them are conserved in other organisms but are as yet functionally opaque, as shown in Figure 2 and Supplementary Table S3. Therefore, another candidate responsible for thermophily may be among those genes whose function is not yet known. It will be necessary to do further comparative analysis with other thermophilic Bacillus-related species as a second step in revealing hidden capacity for thermophily.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
DDBJ/EMBL/GenBank accession nos+ AP006508–AP006519
REFERENCES
- 1.Gerday C. (2002) Extremophiles: basic concepts. In Knowledge for Sustainable Development. An Insight into the Encyclopedia of Life Support Systems. UNESCO Publishing/EOLSS Publishers, Oxford, Vol. 1, pp. 573–598. [Google Scholar]
- 2.Kreil D.P. and Ouzounis,C.A. (2001) Identification of thermophilic species by the amino acid compositions deduced from their genomes. Nucleic Acids Res., 29, 1608–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suhre K. and Claverie,J.-M. (2003) Genomic correlates of hyperthermostability, an update. J. Biol. Chem., 19, 17198–17202. [DOI] [PubMed] [Google Scholar]
- 4.Singer G.A.C. and Hickey,D.A. (2003) Thermophilic prokaryotes have characteristic patterns of codon usage, amino acid composition and nucleotide content. Gene, 317, 39–47. [DOI] [PubMed] [Google Scholar]
- 5.Lynn D.J., Singer,G.A.C. and Hickey,D.A. (2002) Synonymous codon usage is subject to selection in thermophilic bacteria. Nucleic Acids Res., 30, 4272–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tatusov R.L., Natale,D.A., Garkavtsev,I.V., Tatusova,T.A., Shankavaram,U.T., Rao,B.S., Kiryutin,B., Galperin,M.Y., Fedorva,N.D. and Koonin,E.V. (2001) The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res., 29, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forterre P. (2002) A hot story from comparative genomics: reverse gyrase is the only hyperthermophile-specific protein. Trends Genet., 18, 236–237. [DOI] [PubMed] [Google Scholar]
- 8.Henne A., Brüggemann,H., Raasch,C., Wiezer,A., Hartsch,T., Liesegang,H., Johann,A., Lienard,T., Gohl,O., Martinez-Arias,R. et al. (2004) The genome sequence of the extreme thermophile Thermus thermophilus. Nat. Biotechnol., 22, 547–553. [DOI] [PubMed] [Google Scholar]
- 9.Priest F.G. (1993) Systematics and ecology of Bacillus. In Sonenshein,A.L., Hoch,J.A. and Losick,R. (eds), Bacillus subtilis and Other Gram-positive Bacteria. ASM Press, Washington, DC, pp. 3–16. [Google Scholar]
- 10.Bartholomew J.W. and Paik,G. (1966) Isolation and identification of obligate thermophilic sporeforming bacilli from ocean basin cores. J. Bacteriol., 92, 635–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takami H., Kobata,K., Nagahama,T., Kobayashi,H., Inoue,A. and Horikoshi,K. (1999) Biodiversity in deep-sea sites located near the south part of Japan. Extremophiles, 3, 97–102. [DOI] [PubMed] [Google Scholar]
- 12.Sneath P.H.A. (1986) Endospore-forming Gram-positive rods and cocci. In Sneath,P.H.A., Mair,N.S., Sharp,M.E. and Holt,J.G. (eds), Bergy's Manual of Systematic Bacteriology. Williams and Wilkins, Baltimore, MD, Vol. 2, pp. 1104–1139. [Google Scholar]
- 13.Lu J., Nogi,Y. and Takami,H. (2001) Oceanobacillus iheyensis gen. nov., sp. nov., a deep-sea extremely halotolerant and alkaliphilic species isolated from a depth of 1050 m on the Iheya Ridge. FEMS Microbiol. Lett., 205, 291–297. [DOI] [PubMed] [Google Scholar]
- 14.Kunst F., Ogasawara,N., Moszer,I., Albertini,A.M., Alloni,G., Azevedo,V., Bertero,M.G., Bessieres,P., Bolotin,A., Borchert,S. et al. (1997) The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature, 390, 249–256. [DOI] [PubMed] [Google Scholar]
- 15.Takami H., Nakasone,K., Takaki,Y., Maeno,G., Sasaki,R., Masui,N., Fuji,F., Hirama,C., Nakamura,Y., Ogasawara,N. et al. (2000) Complete genome sequence of the alkaliphilic bacterium Bacillus halodurans and genomic sequence comparison with Bacillus subtilis. Nucleic Acids Res., 28, 4317–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takami H., Takaki,Y. and Uchiyama,I. (2002) Genome sequence of Oceanobacillus iheyensis isolated from the Iheya Ridge and its unexpected adaptive capabilities to extreme environments. Nucleic Acids Res., 30, 3927–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ivanova N., Sorokin,A., Anderson,I., Galleron,N., Candelon,B., Kapatral,V., Bhattacharyya,A., Reznik,G., Mikhallova,N., Lapidus,A. et al. (2003) Genome sequence of Bacillus cereus and comparative analysis with Bacillus anthracis. Nature, 423, 87–91. [DOI] [PubMed] [Google Scholar]
- 18.Read T.D., Peterson,S.N., Tourasse,N., Bailli,L.W., Paulsen,I.T., Nelson,K.E., Tettelin,H., Fouts,D.E., Eisen,J.A., Gill,S.R. et al. (2003) The genome sequence of Bacillus anthracis Ames and comparison to closely related bacteria. Nature, 423, 81–86. [DOI] [PubMed] [Google Scholar]
- 19.Takami H., Inoue,A., Fuji,F. and Horikoshi,K. (1997) Microbial flora in the deepest sea mud of the Mariana Trench. FEMS Microbiol. Lett., 152, 279–285. [DOI] [PubMed] [Google Scholar]
- 20.Takami H., Nishi,S., Lu,J., Shimamura,S. and Takaki,Y. (2004) Genomic characterization of thermophilic Geobacillus species isolated from the deepest sea mud of Mariana trench. Extremophiles, 8, 351–356. [DOI] [PubMed] [Google Scholar]
- 21.Nazina T.N., Tourova,T.P., Poltaraus,A.B., Novikova,E.V., Grigoryan,A.A., Ivanova,A.E., Lysenko,A.M., Petrunyaka,V.V., Osipov,G.A., Belyaev,S.S. et al. (2001) Taxonomic study of aerobic thermophilic bacilli: description of Geobacillus subteaneus gen. nov., sp. nov. and Geobacillus uzenensis sp. nov. from petroleum reservoirs and transfer of Bacillus stearothermophilus, Bacillus thermocatenulatus, Bacillus thermoleovolans, Bacillus kaustophilus, Bacillus thermoglusidasius and Bacillus thermodenitrificans to Geobacillus as the new combinations G.stearothermophilus, G.thermocatenulatus, G.thermoleovolans, G.kaustophilus, G.thermoglusidasius and G.thermodenitrificans. Int. J. Syst. Evol. Microbiol., 51, 433–446. [DOI] [PubMed] [Google Scholar]
- 22.Nakai K. and Horton,P. (1999) PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci., 24, 34–36. [DOI] [PubMed] [Google Scholar]
- 23.Sharp P.M. and Li,W.-H. (1986) An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol., 24, 28–38. [DOI] [PubMed] [Google Scholar]
- 24.Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uchiyama I. (2003) MBGD: microbial genome database for comparative analysis. Nucleic Acids Res., 31, 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu K., He,Z.-Q., Mao,Y.-M., Sheng,R.-Q. and Sheng,Z.-J. (1993) On two transposable elements from Bacillus stearothermophilus. Plasmid, 29, 1–9. [DOI] [PubMed] [Google Scholar]
- 27.Bao Q., Tian,Y., Li,W., Xu,Z., Xuan,Z., Hu,S., Dong,W., Yang,J., Chen,Y., Xue,Y., Xu,Y. et al. (2002) A complete sequence of the Thermoanaerobacter tengcongensis genome. Genome Res., 12, 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson K.E., Clayton,R.A., Gill,S.R., Gwinn,M.L., Dodson,R.J., Haft,D.H., Hickey,E.K., Peterson,J.D., Nelson,W.C., Ketchum,K.A. et al. (1999) Evidence for lateral gene transfer between archaea and bacteria from genome sequence of Thermotoga maritima. Nature, 399, 323–329. [DOI] [PubMed] [Google Scholar]
- 29.Takaki Y., Matsuki,A., Chee,G.-J. and Takami,H. (2004) Identification and distribution of new insertion sequences in the genome of extremely halotolerant and alkaliphilic Oceanobacillus iheyensis HTE831. DNA Res., 11, 233–245. [DOI] [PubMed] [Google Scholar]
- 30.Deppenmeier U., Johann,A., Hartsch,T., Merkl,R., Schmitz,R.A., Martinez-Arias,R., Henne,A., Wiezer,A., Bäumer,S., Jacobi,C. et al. (2002) The genome of Methanosarcina mazei: evidence for lateral gene transfer between bacteria and archaea. J. Mol. Microbiol. Biotechnol., 4, 453–461. [PubMed] [Google Scholar]
- 31.Galagan J., Nusbaum,C., Roy,A., Endrizzi,M.G., Macdonald,P., FitzHugh,W., Calvo,S., Engels,R., Smirnov,S., Atnoor,D., Brown,A. et al. (2002) The genome of M.acetivorans reveals extensive metabolic and physiological diversity. Genome Res., 12, 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takami H., Han,C.G., Takaki,Y. and Ohtsubo,E. (2001) Identification and distribution of new insertion sequences in the genome of alkaliphilic Bacillus halodurans C-125. J. Bacteriol., 183, 4345–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takami H., Matsuki,A. and Takaki,Y. (2004) Wide-range distribution of insertion sequences identified in B.halodurans among bacilli and a new transposon disseminated in alkalipilic and thermophilic bacilli. DNA Res., 11, 153–162. [DOI] [PubMed] [Google Scholar]
- 34.Canchaya C., Proux,C., Fournous,G., Bruttin,A. and Brüssow,H. (2003) Prophage genomics. Microbiol. Mol. Biol. Rev., 67, 238–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kobayashi K., Ehrlich,S.D., Albertini,A., Amati,G., Andersen,K.K., Arnaud,M., Asai,K., Ashikaga,S., Aymerich,S., Bessineres,P. et al. (2003) Essential Bacillus subtilis genes. Proc. Natl Acad. Sci. USA, 100, 4678–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neuhaus F.C. and Baddiley,J. (2003) A continuum of anionic charge: structures and functions of d-alanyl-teichoic acids in Gram-positive bacteria. Microbiol. Mol. Biol. Rev., 67, 686–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamaoka T., Satoh,K. and Katoh,S. (1978) Photosynthetic activities of a thermophilic blue-green alga. Plant Cell Physiol., 19, 943–954. [Google Scholar]
- 38.Ruepp A., Graml,W., Santos-Martinez,M.L., Koretke,K.K., Volker,C., Mewes,H.W., Frishman,D., Stocker,S., Lupas,A. and Baumeister,W. (2000) The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum. Nature, 407, 508–513. [DOI] [PubMed] [Google Scholar]
- 39.Kawashima T., Amano,N., Koike,H., Makino,S., Higuchi,S., Kawashima-Ohya,Y., Watanabe,K., Yamazaki,M., Kanehori,K., Kawamoto,T. et al. (2003) Archaeal adaptation to higher temperatures revealed by genomic sequence of Thermoplasma volcanium. Proc. Natl Acad. Sci. USA, 97, 14257–14262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sueoka N. (1988) Directional mutation pressure and neutral molecular evolution. Proc. Natl Acad. Sci. USA, 85, 2653–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakashima H., Fukuchi,S. and Nishikawa,K. (2003) Compositional changes in RNA, DNA and proteins for bacterial adaptation to higher and lower temperatures. J. Biochem., 133, 507–513. [DOI] [PubMed] [Google Scholar]
- 42.Nishio Y., Nakamura,Y., Kawarabayasi,Y., Usuda,Y., Kimura,E., Sugimoto,S., Matsui,K., Yamagishi,A., Kikuchi,H., Ikeo,K. and Gojiobori,T. (2003) Comparative complete genome sequence analysis of the amino acid replacements responsible for the thermostability of Corynebacterium efficiens. Genome Res., 13, 1572–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daniel R.M. and Cowan,D.A. (2000) Biomolecular stability and life at high temperatures. Cell. Mol. Life Sci., 57, 250–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soares D., Dahlke,I., Li,W.-T., Sandman,K., Hethke,C., Thomm,M. and Reeve,J. (1998) Archaeal histone stability, DNA binding and transcription inhibition above 90°C. Extremophiles, 2, 75–81. [DOI] [PubMed] [Google Scholar]
- 45.Green G.R., Searcy,D.G. and DeLange,R.J. (1983) Histone-like protein in the archaebacterium Sulforlobus acidocaldarius. Biochim. Biophys. Acta, 741, 251–257. [DOI] [PubMed] [Google Scholar]
- 46.Miyamoto S., Kashiwagi,K., Ito,K., Watanabe,S. and Igarashi,K. (1993) Estimation of polyamine distribution and polyamine stimulation of protein synthesis in Escherichia coli. Arch. Biochem. Biophys., 300, 63–68. [DOI] [PubMed] [Google Scholar]
- 47.Kneifel H., Stetter,K.O., Andreesen,J.R., Weigel,H., Köning,H. and Schoberth,S.M. (1986) Distribution of polyamines in representative species of archaebacteria. System Appl. Microbiol., 7, 241–245. [Google Scholar]
- 48.Hamana K., Hamana,H., Niitsu,M., Samejima,K., Sakane,T. and Yokota,A. (1994) Occurrence of tertiary and quaternary branched polyamines in thermophilic archaebacteria. Microbios, 79, 109–119. [PubMed] [Google Scholar]
- 49.Hamana K. (1999) Polyamine distribution catalogues of clostridia, acetogenic anaerobes, actinobacteria, bacilli, helicobacteria and haloanaerobes within Gram-positive eubacteria.—Distribution of spermine and agmatine in thermophiles and halophiles.—Microbiol. Cult. Coll., 15, 9–28. [Google Scholar]
- 50.Oshima T., Hamasaki,N., Uzawa,T. and Friedman,S.M. (1989) Biochemical functions of unusual polyamines found in the cells of extreme thermophiles. In Goldemberg,S.H. and Algranati,I.D. (eds), The Biology and Chemistry of Polyamines. IRL Press, Oxford, pp. 1–10. [Google Scholar]
- 51.McCloskey J.A. and Crain,P.F. (1998) The RNA modification database—1998. Nucleic Acids Res., 26, 196–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Droogmans L., Roovers,M., Bujnicki,J.M., Tricot,C., Hartsch,T., Stalon,V. and Grosjean,H. (2003) Cloning and characterization of tRNA (m1A58) methyl-transferase (Trml) from Thermus thermophilus HB27, a protein required for cell growth at extreme temperatures. Nucleic Acids Res., 31, 2148–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.