


Abstract
Chemically modified bases are frequently used to stabilize nucleic acids, to study the driving forces for nucleic acid structure formation and to tune DNA and RNA hybridization conditions. In particular, fluorobenzene and fluorobenzimidazole base analogues can act as universal bases able to pair with any natural base and to stabilize RNA duplex formation. Although these base analogues are compatible with an A-form RNA geometry, little is known about the influence on the fine structure and conformational dynamics of RNA. In the present study, nano-second molecular dynamics (MD) simulations have been performed to characterize the dynamics of RNA duplexes containing a central 1′-deoxy-1′-(2,4-difluorophenyl)-β-d-ribofuranose base pair or opposite to an adenine base. For comparison, RNA with a central uridine:adenine pair and a 1′-deoxy-1′-(phenyl)-β-d-ribofuranose opposite to an adenine was also investigated. The MD simulations indicate a stable overall A-form geometry for the RNAs with base analogues. However, the presence of the base analogues caused a locally enhanced mobility of the central bases inducing mainly base pair shear and opening motions. No stable ‘base-paired’ geometry was found for the base analogue pair or the base analogue:adenine pairs, which explains in part the universal base character of these analogues. Instead, the conformational fluctuations of the base analogues lead to an enhanced accessibility of the bases in the major and minor grooves of the helix compared with a regular base pair.
INTRODUCTION
Chemical modification of the backbone and the nucleobases of RNA can be used to probe the secondary and tertiary structure of RNA and to study the energetic contributions that stabilize or destabilize double-strand formation (1). Recently, fluorobenzene and fluorobenzimidazol derivatives have been suggested as nucleic acid base analogues and universal bases (2–6) and to decompose and analyse the contributions of base stacking and hydrogen bonding to RNA secondary structure stability (5–7). In the case of DNA, such base analogues have been used to study the mechanism of DNA replication [reviewed in (8)]. Self-pairing fluorine-substituted phenyl nucleobases have recently been introduced that allow DNA polymerase-mediated DNA replication and an expansion of the genetic code (9). For RNA, in particular, 1′-deoxy-1′-(2,4-difluorophenyl)-β-d-ribofuranose was found to act as a universal base that pairs with any natural base and stabilizes double-strand RNA (5). Universal bases that stabilize base pair formation can be useful for DNA/RNA hybridization experiments. However, the duplex stabilization with respect to a duplex missing 1 bp was found to be significantly smaller than adding, for example, a natural U:A base pair [∼3 kcal mol−1 (5)]. Similarly, in the case of DNA replacement of a thymine:adenine pair by a difluorotoluene base analogue destabilizes the helix by ∼ 4 kcal mol−1 (2,10).
Spectroscopic studies using circular dichroism indicate that incorporation of a 2,4-difluorophenyl-(F)-base opposite to a natural base at the centre of a duplex RNA resulted in an overall A-form RNA helix (5). The stabilizing contribution of a F-base has been attributed in part to the possible formation of a C–F:H–C bond, which has also been observed in crystal structures of 1′-deoxy-1′-(4-fluorophenyl)-β-d-ribofuranose (4). However, NMR experiments in chloroform failed to identify hydrogen bonding between adenine derivatives and difluorotoluene base analogues (10). Quantum chemical studies also predict a significantly lower stability of pairs formed between fluorine-containing base analogues and adenine compared with a regular T:A pair (11–13). The inability of fluorine to compete with stronger hydrogen bond acceptors such as oxygen and nitrogen is due to its low polarizability and tightly contracted lone pairs (14). In addition, the universal base character of the fluorine-containing analogues indicates that these analogues may only form transiently stable ‘hydrogen bonds’ in the helix.
The thermodynamic and spectroscopic studies give no information on the fine structure and conformational dynamics of difluorophenyl-base containing RNA. Such information would be valuable to estimate the consequence of including such universal bases in larger RNA molecules. Molecular dynamics (MD) simulations are useful for studying the effect of the difluorophenyl-base on the conformational dynamics of duplex RNA at atomic detail. The MD method has already been used successfully to characterize the dynamics of regular RNA as well as RNA molecules that contain non-helical motifs, such as mismatches, bulges and loop structures [reviewed in (15,16)]. The MD method has also been used to investigate the dynamics of difluorotoluene opposite to adenine in B-DNA (17,18). It was found that an adenine:difluorotoluene base pair at the centre of a DNA oligonucleotide preserves overall B-form geometry but does not form a stable base pairing geometry and results in enhanced motions of the adenine:difluorotoluene base pair (17).
In the present study, MD simulations on the nano-second time scale have been used to investigate the dynamics of a difluorophenyl:adenine (F:A) base pair in two different sequence contexts at the centre of an otherwise double-stranded RNA. For comparison, the MD of the corresponding natural uridine:adenine (U:A), a phenyl:adenine (P:A) pair as well as a F:F base pair have also been studied. The study indicates that the F:A, F:F and the P:A containing RNAs stay in an overall A-form structure during the entire simulations. However, the difluorophenyl (and phenyl) containing RNA oligonucleotides show significantly larger conformational fluctuations especially in terms of shear and base pair opening motions than a regular U:A base pair. The study allows drawing important conclusions on the character and magnitude of molecular motions of fluorobenzene analogues in duplex RNA and may give hints on the general most likely motions of nucleotide analogues and mismatches in duplex RNA. It also allows comparison of the effect of such analogues in RNA with previous simulations on DNA containing difluorotoluene bases (17,18).
METHODS
The Jumna (Junction Minimization of Nucleic Acids) program (19) in combination with the Cornell et al. (20) force field was used to build double-strand RNA molecules in standard A-form geometry with the sequences: 5′-rCGCUGCG: 5′-rCGCAGCG or a 2,4-difluorophenyl or phenyl base instead of the central uridine or 2,4-difluorophenyl instead of both central uridine and adenine bases. In addition, the F:A pair was also studied in the context of nearest neighbour A:U base pairs. This resulted in six RNA duplexes: 5′-rCGCUGCG: 5′-rCGCAGCG; 5′-rCGCFGCG:5′-rCGCAGCG; 5′-rCGCPGCG:5′-rCGCAGCG; 5′-rCGCFGCG:5′-rCGCFGCG; 5′-rCGAUUGC:5′-rCGUAAGC; 5′-rCGUFAGC:5′-rCGUAACG. Initial energy minimization (EM) was performed using a modified version of the Jumna program (21) employing a generalized Born solvation model (22–24). Force field parameters for the fluorine atom were taken from the Cornell et al. (20) force field. Partial charges were calculated following the RESP protocol based on quantum chemical calculations at the HF/6-31G level using Gaussian98 (25).
All MD simulations were performed with the sander module of the Amber6 (Assisted Model Building with Energy Restraints) package (26) in a periodic box including explicit TIP3 water molecules (27) and using the parm94 force field (20). Initial positions of 16 additional sodium and 4 chloride ions were placed using the xleap module of the Amber package. About ∼2000 water molecules were added to fill the boxes. A 9 Å cutoff for the short range non-bonded interactions was used in combination with the particle mesh Ewald option (28) using a grid spacing of ∼0.9 Å to account for long range electrostatic interactions. The conformations of the solvated RNA molecules were first relaxed via EM. Following minimization, the systems were gradually heated from 50 to 300 K with positional restraints on the RNA atoms over a period of 0.1 ns. During another 0.1 ns simulation time at 300 K, the positional restraining force constant was gradually reduced from 50 kcal mol−1 Å−2 to zero. Each simulation was continued for a total simulation time of ∼5.1 ns. All MD simulations were performed at constant pressure (1 bar) and constant temperature (300 K) with weak coupling to a temperature and pressure bath, respectively (relaxation times: 4 ps). Solute coordinates were stored every 0.25 ps simulation time. The helical parameters and backbone dihedral torsion angles of the generated structures were analysed using the program Curves (29).
RESULTS AND DISCUSSION
EM using Jumna (19) of all RNA duplexes with either a central uridine (U):adenine (A), a 2,4-difluorophenyl (F):A, a F:F or a phenyl (P):A base pair resulted in converged structures with no significant deviation from the A-form starting structure and the base analogues stacked in the helix. This indicates that the nucleobase analogues are sterically compatible with an A-form RNA geometry (Figure 1). The first 2 ns of the MD simulations at 300 K were used to equilibrate the structures. The average inter-helical parameters of the central (5) base pairs of all RNA duplexes during the data gathering period (2–5.1 ns) were close to the values of the corresponding duplexes with natural base pairs (Table 1). The root-mean-square deviation (Rmsd) of the generated structures in the case of the U:A and the F:A containing duplexes during the data gathering period (2–5.1 ns) is illustrated in Figure 2. The RNA duplexes with central 2,4difluorophenyl:A pair showed larger deviations from the start structure and larger shifts of the total Rmsd compared with a regular RNA duplex with a central U:A base pair (Figure 2). Similar observations were made for the other RNA duplexes with central base analogues (data not shown). However, as can be seen from Figure 3, the conformational fluctuations along the RNA strands (with respect to an average structure) are not uniform. The fluctuations observed in the case of the base analogue containing RNAs are quite similar to the fluctuations of the reference RNA with the exception of the region around the central base pair. The region around the central base pair (heavy atoms ∼65–85 in the first strand and ∼215–235 in the second strand, Figure 3) showed strongly enhanced fluctuations not only of the backbone but in particular of the central bases. Interestingly, the enhanced fluctuations were observed for both the base analogue as well as the opposing base in the other strand. Note that the average structures from the data gathering period are for all duplexes with base analogues close to the average A-form structure of the reference duplex (with the U:A pair, illustrated for the F:A and P:A pair in Figure 4) with Rmsds of <1.5 Å and average A-form helical parameters (Table 1). This result indicates that the duplexes with base analogues undergo significant conformational fluctuations but do not result in strand dissociation or an overall conformational drift, e.g. to a non-A-form structure.
Figure 1.
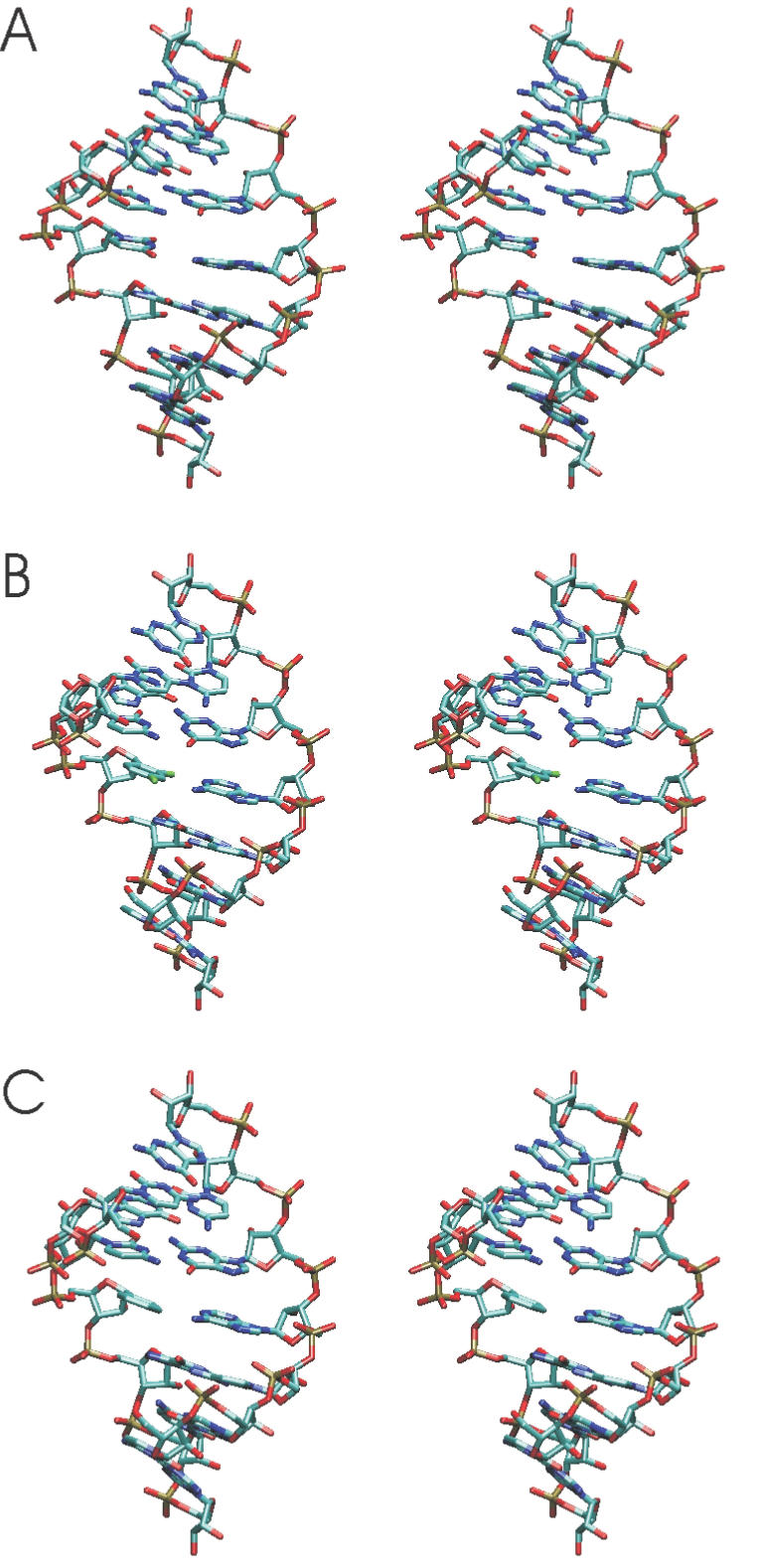
Examples of energy-minimized RNA duplex start structures (in stereo) with the sequence 5′-rCGCXGCG:5′-rCGCAGCG. The central base X was an uridine (A), a 2,4-difluorophenyl (B) base analogue or a phenyl base analogue (C) and standard backbone sugar–phosphate structure. For clarity only heavy atoms are shown.
Table 1. Average helical structure.
| Helical parameter | CGCUGCG | CGCFGCG | CGCPGCG | CGCFGCG | CGUUACG | CGUFACG |
|---|---|---|---|---|---|---|
| GCGACGC | GCGACGC | GCGACGC | GCGFCGC | GCAAUGC | GCAAUGC | |
| <shift> | 0.0 (0.6) | 0.2 (0.9) | −0.2 (1.0) | 0.0 (1.0) | −0.1 (0.7) | 0.0 (0.8) |
| <slide> | −0.4 (0.4) | −0.6 (0.7) | −0.6 (0.8) | −0.6 (0.6) | −0.4 (0.5) | −0.6 (0.7) |
| <rise> | 2.7 (0.5) | 3.0 (0.7) | 2.9 (0.6) | 3.0 (0.7) | 2.8 (0.5) | 3.0 (0.7) |
| <tilt> | 0.2 (5.0) | −1.3 (6.0) | 1.9 (13.0) | 1.0 (7.0) | 0.6 (6.0) | 1.6 (8.0) |
| <roll> | 4.2 (8.0) | 9.2 (7.0) | 8.9 (13.0) | 6.3 (7.0) | 6.8 (9.0) | 6.9 (9.0) |
| <twist> | 32 (8.0) | 29 (5.0) | 30 (11.0) | 29 (9.0) | 29 (5.0) | 29 (8.0) |
Average helical parameters (SDs in parenthesis) were calculated for the central 4 bp steps (bold, second strand in 3′ → 5′ direction) averaged over the entire data gathering period (2–5.1 ns) using Curves (29).
Figure 2.

(A) Rmsd time course of RNA heavy atoms (with respect to start structure) during the data gathering phase of the 5′-rCGUUACG/5′-rCGUAACG duplex simulation (black line). (B) Rmsd time course of the duplex simulation with central F:A base pair (black line). For comparison, the Rmsd time course of the 5′-rCGCUGCG/5′-rCGCAGCG duplex simulation is shown as grey dashed line (in A and B).
Figure 3.

Atomic root-mean-square fluctuation (Rmsf) (heavy atoms of RNA) observed during the MD simulations versus heavy atom number. The atom numbers follow the order phosphate, sugar and base of each nucleotide from 5′ to 3′ end. The second strand starts in the middle of each panel. The black curves indicate the atomic fluctuations of the 5′-rCGUUACG/5′-rCGUAACG duplex (A), the 5′-rCGCFGCG/5′-rCGCAGCG duplex (B), the 5′-rCGCPGCG/5′-rCGCAGCG duplex (C), the 5′-rCGCFGCG/5′-rCGCFGCG duplex (D) and the 5′-rCGUFACG/5′-rCGUAACG duplex (E), respectively. For comparison, the atomic fluctuations of the 5′-rCGCUGCG/5′-rCGCAGCG duplex are shown as grey dashed line in each panel.
Figure 4.

Superposition of the average structures (central and neighbouring base pairs) obtained during the data gathering period of the reference RNA simulation (black line) and the simulation on the RNA with central F:A base pair (grey line in A) and the central P:A base pair (grey line in B), respectively. The view is into the major groove.
Typical base pair geometries observed during the MD simulations are illustrated as snapshots for the F:A case in Figure 5. Similar fluctuations were observed for all other base pairs containing base analogues (data not shown). The snapshots illustrate that both central bases can stay in a nearly ‘base-paired’ geometry similar to the geometry of an U:A pair (Figure 5) but can also undergo significant motions towards the major and/or minor groove. Here, both symmetric (e.g. both bases move towards the major grove at the same time) as well as anti-symmetric motions (e.g. one moves toward the minor groove and the other base towards the major groove) were observed during the simulations. This result indicates that the presence of a central base pair with either a F:F, a P:A or a F:A base pair although largely preserving an A-form geometry during the entire simulations do not result in a stable base-paired geometry. Instead, the modified bases and the opposing adenine base sample a variety of substates with no ‘hydrogen bonding’ or only weakly stable ‘hydrogen bonds’ between the central bases. Such sampling of substates without stable ‘hydrogen bonding’ was also observed in MD simulations of the difluorotoluene:adenine pair in DNA termed base pair ‘breathing’ motion (17,18). The lack of a stable hydrogen bonding geometry is compatible with the known character of organic fluorine to seldom form stable hydrogen bonds (14). It also agrees with the experimentally observed universal base character of the base analogues (5) and NMR studies on adenine derivatives and difluorotoluene in chloroform that did not result in stable hydrogen bonding (10).
Figure 5.

Conformational snapshots (A–F) observed during the simulation of the RNA with a central F:A base pair. The view is along the helical axis of the RNA. For clarity, only the central base pair (thick lines) and the neighbouring base pairs (thin lines) are shown.
To further analyse the character of the motions caused by the presence of base analogues, the base–base helical parameters and fluctuations at the duplex centre and nearest neighbours was analysed. Most average base–base parameters of the base pairs adjacent to the central base pairs containing a base analogue are similar to the corresponding averages for the RNA duplex with natural base pairs (Table 2, numbers in parenthesis indicate the SDs over the data gathering period). Slight shifts and enhanced fluctuations were observed in the case of the helical parameters stretch and stagger of the central base pairs in the presence of base analogues (Table 2). However, the most significant increase in fluctuations due to the base analogues compared with the reference duplexes was observed for the base–base helical parameters shear and opening (Table 2) fully compatible with the snapshots illustrated in Figure 5. Shear motion indicates an overall translational motion of the bases in the direction perpendicular to the helical axis and to an axis that connects the two bases at the centre. Opening motion indicates an angular motion in the same direction. Symmetric and anti-symmetric shear and opening motions, respectively, of the bases are distinguished by the sign in the plots. Interestingly, both types of motion with enhanced magnitude compared with standard duplex RNA were seen for the F:F, the F:A as well as the P:A central base pair (Figure 6). The sampling time of a few nanoseconds was sufficient to observe several opening and shear transitions (motions of the bases from a position in the minor to the major groove and vice versa, Figure 6). Interestingly, in both the cases, the same parameter fluctuations for the neighbouring base pairs were much smaller and close to the magnitude observed for the regular RNA duplex (Figure 6). Apparently, the enhanced conformational fluctuations due to the modified nucleotides are localized to the centre of the duplex with only a small influence on the fluctuations of adjacent nucleotides. This was also seen for the F:A pair flanked by A:U base pairs indicating that the context effect is small with little influence of opening and shear motion of the central base on the pairing of the flanking A:U bases (Figure 6). This conclusion is also supported by the results on atomic fluctuations (in Figure 2).
Table 2. Base–base helical parameters.
| Helical parameter | CGCUGCG GCGACGC | CGCFGCG GCGACGC | CGCPGCG GCGACGC | CGCFGCG GCGFCGC | CGUUACG GCAAUGC | CGUFACG GCAAUGC |
|---|---|---|---|---|---|---|
| <shear> | ||||||
| −1 | 0.0 (0.4) | 0.2 (0.5) | 0.1 (0.5) | 0.0 (0.4) | −0.2 (0.4) | 0.2 (0.4) |
| 0 | 0.1 (0.4) | −1.4 (2.7) | −1.2 (2.3) | 0.0 (2.4) | −0.1 (0.5) | 2.7 (2.5) |
| +1 | 0.0 (0.4) | 0.0 (0.4) | 0.1 (0.9) | −0.1 (0.4) | 0.0 (0.5) | 0.2 (0.6) |
| <stretch> | ||||||
| −1 | 0.3 (0.4) | 0.3 (0.4) | 0.2 (0.4) | 0.2 (0.3) | 0.2 (0.5) | 0.2 (0.3) |
| 0 | 0.3 (0.4) | 0.6 (1.0) | −0.9 (1.2) | −0.7 (1.1) | 0.4 (0.6) | −0.1 (1.0) |
| +1 | 0.3 (0.4) | 0.2 (0.3) | 0.5 (0.7) | 0.3 (0.3) | 0.4 (0.7) | 0.1 (0.7) |
| <stagger> | ||||||
| −1 | 0.0 (0.5) | −0.2 (0.4) | −0.3 (0.4) | −0.2 (0.5) | −0.1 (0.5) | −0.3 (0.5) |
| 0 | 0.0 (0.5) | −0.5 (1.0) | −0.6 (1.2) | −0.65 (1.0) | −0.1 (0.5) | 0.0 (1.1) |
| +1 | 0.0 (0.4) | −0.2 (0.4) | −0.4 (0.4) | −0.3 (0.4) | −0.1 (0.5) | −0.2 (0.7) |
| <buckle> | ||||||
| −1 | 2.0 (10) | 4.4 (11) | 13 (13) | 6.9 (11) | −0.7 (14) | −2.7 (11) |
| 0 | −0.6 (9) | −2.0 (11) | 15 (17) | −1.1 (14) | 3.2 (13) | −0.6 (13) |
| +1 | −6.0 (10) | −5.2 (10) | 3 (15) | −7.2 (11) | −1.2 (14) | −1.2 (12) |
| <propeller> | ||||||
| −1 | −16 (11) | −12 (8) | −8 (8) | −10 (9) | −11 (16) | −13 (10) |
| 0 | −16 (10) | −11 (11) | −10 (12) | −10 (11) | −14 (16) | −8 (13) |
| +1 | −17 (11) | −11 (8) | −3 (14) | −11 (8) | −14 (16) | −11 (11) |
| <opening> | ||||||
| −1 | 2.3 (5) | 3.4 (5) | 0.3 (5) | 1.0 (7) | 1.0 (6) | 3.2 (7) |
| 0 | 2.6 (6) | 14.8 (18) | −12 (26) | −7.0 (25) | 4.3 (6) | 2.4 (16) |
| +1 | 2.5 (5) | 1.8 (4) | 4 (9) | −2.1 (0) | 5.5 (6) | 6.7 (9) |
Average inter-base helical parameters (SDs in parenthesis) were calculated for the central base pair (position 0) and the 5′ (−1) and 3′ (+1) neighbouring base pairs (relevant sequence in bold, second strand in 3′ → 5′ direction) averaged over the entire data gathering period (2–5.1 ns) using Curves (29).
Figure 6.

Base pair opening (A) and shear (B) motions of the central base pairs (black lines in right panels, IV–VI) and the base pair before the central base pair (black lines in left panels, I–III) observed during the MD data gathering period of the rCGCFGCG/5′-rCGCAGCG duplex (I and IV), the 5′-rCGCFGCG/5′-rCGCFGCG duplex (II and V) and the 5′-rCGUFACG/5′-rCGUAACG duplex (III and VI), respectively. For comparison, the same motions for the reference duplexes with natural central base pairs are also shown (grey dashed line).
The origin of the increased shear and opening motions in terms of backbone dihedral torsion angle fluctuations around the central base pair was investigated. The presence of central base analogues did not affect the sugar conformation significantly as indicated by the torsion angle δ which mainly affects the sugar pucker state (Table 3). In all the cases, the δ angle stayed close to values compatible with a C′3-endo sugar pucker state with similar fluctuations in the modified nucleotides and also at the flanking nucleotides. A similar small influence was observed for the torsion angles α, β and γ (data not shown). A number of crank shift motions (also called α–γ flips) were observed (both α and γ change from −gauche and gauche to trans, respectively) but did not show any correlation between the observed opening and shear motions. The average ɛ and ζ backbone dihedral angles were similar to the corresponding values for the natural duplexes (Table 3). However, the ɛ and ζ fluctuations were larger than for the natural duplexes at the central nucleotide and also at the 5′-neighbouring nucleotide (Table 3). Molecular mechanics studies on DNA have indicated that base pair opening motions correlate with changes in the backbone dihedral torsion angle ζ (30). MD simulations on difluorotoluene base analogues opposite to adenine in DNA have found little correlation between the observed base pair ‘breathing’ motions and changes in ɛ and ζ dihedral angles (17). Such breathing motions or in the present study base pair shear and opening motion are, however, affected by the motions of two bases in opposite strands. In Figure 7, the shift motion (motion in the base pair plane perpendicular to the direction that connects two bases on opposite strands) of single bases (base analogue or opposing base) with respect to the previous base and the following base versus simulation time are plotted (for the duplex: 5′-CGUFACG/5′-CGUAACG). In addition, the changes of the ɛ and ζ angles (at the central nucleotide and the 5′ nucleotide) versus simulation time are plotted and show at least a qualitative correlation with respect to the shift motion of the central base. Interestingly, no transitions of the dihedral torsion angles to new substates (e.g. −gauche→trans transistions, separated by torsional barriers) were observed, but only shifts by ∼5–10° that appear to be sufficient to mediate significant translational/rotational shifts of the bases. No such coupling is seen for the α and δ (Figure 7) and other dihedral torsion angles at the central base. The observed coupling between ɛ /ζ and the base shift motions could be due to the fact that the C3′–O3′ bond (ɛ-rotation) and the O3′–P bond (ζ-rotation) have a significant component in the direction of the RNA helical axis. Consequently, any rotation around these bonds affects base motions in the plane perpendicular to the helical axis (like shift and opening motions).
Table 3. Backbone dihedral angle fluctuations.
| Dihedral angle | CGCUGCG | CGCFGCG | CGCPGCG | CGCFGCG | CGUUACG | CGUFACG |
|---|---|---|---|---|---|---|
| δ(−1) | 77 (6) | 79 (7) | 79 (7) | 80 (7) | 81 (7) | 79 (7) |
| δ(0) | 76 (6) | 82 (7) | 78 (7) | 76 (6) | 77 (6) | 79 (7) |
| δ(+1) | 78 (7) | 78 (7) | 79 (7) | 79 (7) | 79 (7) | 80 (7) |
| ε(−1) | −158 (10) | −168 (11) | −168 (13) | −159 (15) | −153 (11) | −154 (16) |
| ε(0) | −157 (9) | −151 (13) | −151 (14) | −157 (14) | −156 (10) | −164 (12) |
| ε(+1) | −155 (10) | −168 (10) | −152 (11) | −155 (11) | −156 (10) | −156 (11) |
| ζ(−1) | −67 (8) | −69 (10) | −79 (12) | −70 (13) | −67 (8) | −66 (14) |
| ζ(0) | −68 (8) | −65 (12) | −64 (13) | −70 (13) | −69 (8) | −75 (13) |
| ζ(+1) | −67 (8) | −69 (9) | −68 (9) | −68 (9) | −69 (9) | −67 (9) |
Average dihedral torsion angles (SDs in parenthesis) were calculated for the central nucleotide of the first strand (position 0) and the 5′ (−1) and 3′ (+1) neighbouring nucleotides (indicated in bold) averaged over the entire data gathering period (2–5.1 ns).
Figure 7.

Shift motion (motion in the base pair plane perpendicular to the direction that connects two bases on opposite strands) of the difluorophenyl-base analogue with respect to the 3′-flanking (black line) and the 5′-flanking base (grey line) in the rCGUFACG/5′-rCGUAACG duplex versus simulation time. For comparison, the changes in the backbone torsion angles ɛ and ζ at the central bases (black line) and at the 5′-flanking bases (grey dashed line) are shown, respectively. In addition, the changes of dihedral angles δ (fourth panel) and α (fifth panel) at the central base versus simulation time are indicated. The panel series (B) shows the same as in (A) but for the adenine nucleotide in the opposite strand. Each data point represents an average over 25 ps simulation time.
CONCLUSION
The present study aimed at characterizing the influence of a 2,4-difluorophenyl nucleobase and a phenyl base analogue opposite to an adenine base on the flexibility of RNA. Both base analogues have been found to act as universal bases able to pair with any natural base (5). However, only the 2,4-difluorophenyl nucleobase was found to stabilize the RNA duplex. In agreement with experimental results, the RNAs with base analogues stayed in an overall A-form geometry throughout the MD simulations. This also agrees with structural studies of difluorotoluene opposite to adenine in DNA that indicated lower duplex stability but overall B-form DNA geometry (31). However, significantly larger conformational fluctuations of the central base pairs in particular opening and shear motions compared with a reference RNA with a central U:A base pair were observed. The motions resulted in many conformations without a ‘base-paired’ geometry of the central base analogue and the opposing adenine. Similar results were obtained in previous MD simulation studies on a difluorotoluene base analogue opposite to adenine in B-DNA duplexes (17,18). This result indicates that the influence of such base analogues on the dynamics of DNA is similar to the effect in RNA. On the present nano-second time scale, several ‘flips’ between completely intra-helical and partially extra-helical base pair geometries were observed. The perturbation of neighbouring base pairs appeared to be small. The simulations indicate that the 2,4-difluorophenyl-nucleobase can only form weak transiently stable ‘hydrogen bonds’ to the opposing base in the RNA duplex. A similar character of enhanced mobility was observed in the case of F:A and F:F base pairs. This result offers an explanation for the experimental observation that the 2,4-difluorophenyl-nucleobase is a universal base and that the stabilization effect of the helix is primarily due to stacking effects (3,5). Any stable ‘hydrogen bonding’ is likely to interfere with the characteristics of a universal base. The simulation studies also give an impression on what types of motions and conformational transitions are expected in the case of incorporation of base analogues or chemically modified bases in duplex RNA. These motions are mainly shear and opening motions of the base analogue and the opposing base. It is likely that similar types of conformational motions and transitions occur in other types of mismatches that weaken intra-strand base pairs. The present simulation studies indicate that the fluorinated base analogues cause an increase in the motion of the backbone torsion angles ɛ and ζ around the base pair with the base analogue and that changes in ɛ and ζ show some correlation with base motions of the same strand. Since the shear and opening motion (or breathing motion) of the complete base pair are affected not only by one base but also by the motion of the opposing base, the correlation with single ɛ and ζ dihedral angles is much weaker. Nevertheless, these dihedral angles seem to play a decisive role to drive the helical base motions. The conformational transitions towards both major and minor grooves lead to a greater chemical accessibility of the base analogues. The result offers an explanation as to why base mismatches although on average in a helical and stacked conformation are often much more sensitive to chemical probing reagents than regular base pairs (32). As demonstrated in the present study, MD simulations can be useful to systematically probe the dynamics of modified bases in RNA at atomic detail. This can be very helpful for the design of base modifications for a specific purpose.
Acknowledgments
ACKNOWLEDGEMENTS
We thank A. Barthel, A. Kloepffer, J. Parsch, D. Roccatano and A. Zivkovicz for helpful discussions. The simulations were performed in part at the Pacific Northwest National Laboratory supercomputer centre supported by DOE grant gc11-2002.
REFERENCES
- 1.Sanger W. (1984) Principles of Nucleic Acid Structure. Springer-Verlag, NY. [Google Scholar]
- 2.Moran S., Ren,R.X.-F., Runny,S. and Kool,E.T. (1997) Difluorotoluene, a nonpolar isostere for thymine, codes specifically and efficiently for adenine in DNA replication. J. Am. Chem. Soc., 119, 2056–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parsch J. and Engels,J.W. (2001) Stacking and stability of RNA duplexes containing fluorobenzene and fluorobenzimidazole nucleosides. Nucleosides Nucleotides Nucleic Acids, 20, 815–818. [DOI] [PubMed] [Google Scholar]
- 4.Bats J.W., Parsch,J. and Engels,J.W. (2000) 1-deoxy-1-(4-fluorophenyl)-β-d-ribofuranose, its hemihydrate, and 1′-deoxy-1′-(2,4-difluorophenyl)-β-d-ribofuranose: structural evidence for intermolecular C-H–F-C interactions. Acta Crystallogr. C., 56, 201–205. [DOI] [PubMed] [Google Scholar]
- 5.Parsch J. and Engels J.W. (2002) C-F–H-C hydrogen bonds in ribonucleic acids. J. Am. Chem. Soc., 124, 5664–5672. [DOI] [PubMed] [Google Scholar]
- 6.Kloepffer A.E. and Engels,J.W. (2004) Synthesis of 2′-aminoalkyl-substitued fluorinated nucleobases and their influence on the kinetic properties of hammerhead ribozymes. Chembiochem, 5, 707–716. [DOI] [PubMed] [Google Scholar]
- 7.Kloepffer A.E. and Engels,J.W. (2003) The effect of universal fluorinated nucleobases on the catalytic activity of ribozymes. Nucleosides Nucleotides Nucleic Acids, 22, 1347–1350. [DOI] [PubMed] [Google Scholar]
- 8.Kool E.T. (2001) Hydrogen bonding, base stacking, and steric effects in DNA replication. Annu. Rev. Biophys. Biomol. Struct., 30, 1–22. [DOI] [PubMed] [Google Scholar]
- 9.Henry A.A., Goldenbech-Olsen,A., Matsuda,S., Yu,C., Geierstanger,B.H. and Romesberg,F.E. (2004) Efforts to expand the genetic alphabet: identification of a replicable unnatural DNA self-pair. J. Am. Chem. Soc., 126, 6923–6931. [DOI] [PubMed] [Google Scholar]
- 10.Schweitzer E. and Kool,E.T. (1995) Hydrophobic, non-hydrogen-bonding bases and base pairs in DNA. J. Am. Chem. Soc., 117, 1863–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer M. and Suehnel,J. (1997) Quantum-chemical ab initio study on the adenine–difluorotoluene complex-A mimic for the adenine-thymine base pair. J. Biomol. Struct. Dyn., 15, 619–624. [DOI] [PubMed] [Google Scholar]
- 12.Sponer J., Leszcynski,J. and Hobza,P. (1996) Hydrogen bonding and stacking of DNA bases: a review of quantum-chemical ab initio studies. J. Biomol. Struct. Dyn., 14, 117–135. [DOI] [PubMed] [Google Scholar]
- 13.Barsky D., Kool,E.T. and Colvin,M.E. (1999) Interaction and solvation energies of nonpolar DNA base analogues and their role in polymerase insertion fidelity. J. Biomol. Struct. Dyn., 16, 1119–1134. [DOI] [PubMed] [Google Scholar]
- 14.Dunitz J.D. (2004) Organic fluorine: odd man out. Chembiochem, 5, 614–621. [DOI] [PubMed] [Google Scholar]
- 15.Auffinger P. and Westhof,E. (1998) Simulations of the molecular dynamics of nucleic acids. Curr. Opin. Struct. Biol., 8, 227–236. [DOI] [PubMed] [Google Scholar]
- 16.Zacharias M. (2000) Simulation of the structure and dynamics of nonhelical RNA motifs. Curr. Opin. Struct. Biol., 10, 307–311. [DOI] [PubMed] [Google Scholar]
- 17.Cubero E., Sherer,E.C., Luque,F.J., Orozcp,M. and Laughton,C.A. (1999) Observation of spontaneous base pair breathing events in the molecular dynamics simulation of a difluorotoluene-containing DNA oligonucleotide. J. Am. Chem. Soc., 121, 8653–8654. [Google Scholar]
- 18.Cubero E., Laughton,C.A., Luque,F.J. and Orozcp,M. (2000) Molecular dynamics study of oligonucleotides containing difluorotoluene. J. Am. Chem. Soc., 122, 6891–6899. [Google Scholar]
- 19.Lavery R., Zakrzewska,K. and Sklenar,H. (1995) JUMNA (junction minimization of nucleic acids). Comput. Phys. Commun., 91, 135–158. [Google Scholar]
- 20.Cornell W.D., Cieplak,P., Bayley,C.I., Gould,I.R., Merz,K.M., Ferguson,D.M., Spellmeyer,D.C., Fox,T., Caldwell,J.W. and Kollman,P.A. (1995) A second generation force field for simulation of proteins, nucleic acids and organic molecules. J. Am. Chem. Soc., 117, 5179–5197. [Google Scholar]
- 21.Zacharias M. (2001) Conformational analysis of DNA-trinucleotide-hairpin-loop structures using a continuum solvent model. Biophys. J., 80, 2350–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Still W.C., Tempczyk,A., Hawley,R.C. and Hendrikson,T. (1990) Semi-analytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc., 112, 6127–6129. [Google Scholar]
- 23.Hawkins G.D., Cramer,C.J. and Truhlar,D.G. (1995) Pairwise solute descreening of solute charges from a dielectric continuum. Chem. Phys. Lett., 246, 122–129. [Google Scholar]
- 24.Hawkins G.D., Cramer,C.J. and Truhlar,D.G. (1996) Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem., 100, 19824–19839. [Google Scholar]
- 25.Frisch M.J., Trucks,G.W., Schlegel,H.B., Scuseria,G.E., Robb,M.A., Cheeseman,J.R., Zakrzewski,V.G., Montgomery,J.A., Stratmann,R.E., Burant,J.C. et al. (1998) Gaussian 98, Revision E.4 Gaussian Inc., Pittsburgh, PA. [Google Scholar]
- 26.Pearlman D.A., Case,D.A., Caldwell,J.W., Ross,W.S., Cheatham,T.E., Debolt,S., Ferguson,D., Seibel,G. and Kollman,P.A. (1995) Comput. Phys. Commun., 91, 1–41. [Google Scholar]
- 27.Jorgensen W.L., Chandrasekhar,J., Madura,J., Impey,R.W. and Klein,M.L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys., 79, 926–935. [Google Scholar]
- 28.Darden T., York,D. and Pedersen,L. (1993) Particle mesh Ewald: an NlogN method for Ewald sums in large systems. J. Chem. Phys., 98, 10089–10092. [Google Scholar]
- 29.Lavery R. and Sklenar,H. (1988) The definition of generalized helicoidal parameters of nucleic acids. J. Biomol. Struct. Dyn., 6, 63–91. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y.Z., Mohan,V. and Griffley,R.H. (1998) The opening of a single base without perturbations of neighboring nucleotides: a study on crystal B-DNA duplex d(CGCGAATTCGCG)2. J. Biomol. Struct. Dyn., 15, 765–777. [DOI] [PubMed] [Google Scholar]
- 31.Guckian K.M., Krugh,K.T.R. and Kool,E.T. (1998) Solution structure of a DNA duplex containing a replicable difluorotoluene-adenine pair. Nature Struct. Biol., 5, 954–959. [DOI] [PubMed] [Google Scholar]
- 32.Brunel C. and Romby,P. (2000) Probing RNA structure and RNA–ligand complexes with chemical probes. Meth. Enzymol., 318, 3–21. [DOI] [PubMed] [Google Scholar]