

Abstract
With the accumulation of protein and its related data on the Internet, many domain-based computational techniques to predict protein interactions have been developed. However, most techniques still have many limitations when used in real fields. They usually suffer from low accuracy in prediction and do not provide any interaction possibility ranking method for multiple protein pairs. In this paper, we propose a probabilistic framework to predict the interaction probability of proteins and develop an interaction possibility ranking method for multiple protein pairs. Using the ranking method, one can discern the protein pairs that are more likely to interact with each other in multiple protein pairs. The validity of the prediction model was evaluated using an interacting set of protein pairs in yeast and an artificially generated non-interacting set of protein pairs. When 80% of the set of interacting protein pairs in the DIP (Database of Interacting Proteins) was used as a learning set of interacting protein pairs, high sensitivity (77%) and specificity (95%) were achieved for the test groups containing common domains with the learning set of proteins within our framework. The stability of the prediction model was also evident when tested over DIP CORE, HMS-PCI and TAP data. In the validation of the ranking method, we reveal that some correlations exist between the interacting probability and the accuracy of the prediction.
INTRODUCTION
The accumulation of protein data and the associated data on the Internet (1–3), gives us the opportunity to computationally find structures and functions of proteins based on the data. More specifically, the accumulation of experimental protein–protein interaction and domain data on the Internet allow us to computationally predict protein–protein interactions.
The benefits of computational prediction of protein–protein interactions are obvious. First and foremost, mass prediction of protein–protein interactions at low cost is possible. This method can also help in finding critical proteins out of numerous candidate proteins without experimental validation. Based on this information, biologists can assign priorities to the proteins or domains to be tested, thereby allowing the construction of a large-scale protein interaction network, and they can also use the information to predict functions of unknown proteins (4).
There are several approaches to the computational prediction of protein–protein interactions (5–8). Finding and analyzing subsequences affecting protein–protein interactions from raw protein sequences is one approach (9). Another is to predict protein interactions by analyzing the physicochemical properties or tertiary structure of proteins (10). Domain-based protein–protein interaction prediction is another approach, and is recently being studied by several research groups (5,8,11,12).
Most domain-based protein–protein interaction prediction methods share the conjecture that such interactions are the result of domain–domain interaction. Those methods infer domain–domain interacting information from protein–protein interaction and then try to predict protein interactions based on the inferred domain–domain interacting information.
Previous domain-based research usually considered the interactions of single domain pairs and assumed the interactions of single domain pairs to be independent of one another, for the convenience of computations. We suspect that such assumptions may be the reason for the limitations of conventional domain-based prediction methods. Protein–protein interaction could be the result of interactions of multiple domain pairs or of groups of domains. As a result, the prediction accuracy of conventional domain-based predictions is not high enough to be effectively used in research or industrial fields. To overcome these limitations, we introduce the notion of domain combination and domain combinations pair (dc-pair) in this paper. The term domain combination is used to denote a set of domains.
In this paper, we propose a domain combination based protein–protein interaction prediction framework. In this framework, protein–protein interaction is interpreted as the result of interactions of multiple domain pairs or of groups of domains, i.e. the prediction model in the framework considers dc-pair as a basic unit of protein interactions (12). This approach is more inclusive than previous domain-based approaches because domain pair information is included in the dc-pair information. Since the proposed framework provides an interacting probability, it is much more natural in predicting the possibility of interaction than conventional methods.
In this paper, we also develop a domain combination based protein–protein interaction possibility ranking method for multiple protein pairs. An interacting probability equation for a protein pair is developed and the ranks for multiple protein pairs are decided by the interacting probabilities computed through the interacting probability equation formulated in this paper. Using the ranking method, biologists can discern the protein pairs that are more likely to interact with each other among multiple protein pairs.
The validity of the prediction model in the framework is evaluated for the interacting set of protein pairs in yeast and also in artificially generated non-interacting sets of protein pairs. When 80% of the set of interacting protein pairs in the DIP (Database of interacting proteins) (3,13) was used as a learning set, on average, 77% sensitivity and 95% specificity were achieved for the test groups containing common domains with the learning set of proteins within our framework.
The model is also proved to be stable as it achieved non-fluctuating and high-prediction accuracy in the evaluations over DIP CORE (14), HMS-PCI (15) and TAP data (16).
In the validity test of our interaction possibility ranking method, the test groups with higher predicted interaction probabilities showed higher sensitivities and specificities. This indicates that the ranking method developed in this paper is valid for assigning ranks to protein pairs.
RELATED WORK
Several attempts have been made to computationally predict protein–protein interactions without domain information. A technique using a support vector machine (SVM) based on primary sequence and associated physicochemical properties has been developed to predict protein–protein interactions (10). A gene fusion method called ‘Rosetta stone’ (6,7) is another computational approach used to identify the functional relations of proteins rather than to predict physical interactions. In another study (17), interacting pairs of yeast proteins and domains in the SCOP (Structural Classification of Proteins) (18) database were used to construct a protein family interaction map. In this algorithm, interactions are predicted based on structural information by parsing the Protein Data Bank (PDB) (2) coordinates to determine if each domain pair could make close contacts.
Recently, predictions of protein interactions have been performed in the context of domain–domain interactions at the primary sequence level rather than from the PDB coordinates (4,11), using experimentally identified interacting protein pairs in Helicobacter pylori or Saccharomyces cerevisiae. Since domain or motif is a structural and/or functional unit, specific signature sequences are conserved to represent the protein's structure or function through evolution. Therefore, it is not surprising that many of the protein interactions can be reduced to the problem of domain–domain interaction. In addition, it is generally accepted that domain is an independent unit within the protein structure and sequence, and this notion is used in various classification systems such as SCOP, CATH and FSSP (18–20).
Mering et al. (21) described the concept of groups of interacting partners. Although the concept of domain combination is similar to that of groups of interacting partners, the latter has protein granularity while the former has domain granularity.
Deng et al. (5) proposed a probabilistic prediction model for inferring domain interactions from protein interaction data. The maximum-likelihood estimation technique is mainly used in their method. The Pfam database is used to extract domain information and the MIPS database is used to test their model, but they also consider single domain pairs as a basic unit of protein interactions. The approach taken by Kim et al. (22) shares the same assumption with Deng et al. (5), but both approaches suffer from low sensitivity and specificity of predictions. Ng et al. (8) collected data from three sources: (i) the experimentally derived protein interaction data from DIP (3,13); (ii) the intermolecular relationship data from protein complexes; and (iii) the computationally predicted data from Rosetta Stone sequences. They then inferred putative domain–domain interactions based on the collected data through the development of InterDom, a database of interacting domains (http://interdom.lit.org.sg) (8). However, the accuracy of the data inferred from domain–domain interaction is not apparent.
Goffard et al. (23) developed IPPRED, a web-based server for the inference of proteins interactions. IPPRED infers the possibility of the interaction of the two proteins A and B by discovering if there is an interacting protein pair C and D which are homologous to A and B (or B and A).
MATERIALS AND METHODS
Prediction framework
Domain combination and domain combination pair
Before we explain our prediction model, we introduce the notion of domain combination and domain combination pair. When a protein p contains multiple domains, then the domain combination of protein p is all the possible groups of domains that can be formed from the set of domains of protein p. Here, the groups must contain at least one domain. As such the set of all possible domain combinations of protein p can be defined more formally by
![]()
where domain (p) represents the set of domains in protein p. The empty set is eliminated from the definition because the power set operation generates the empty set also. Thus, when a protein contains n domains, 2n − 1 different domain combinations are obtained.
In our prediction model, the domain combination is considered as a basic element of protein interactions, and we assume one or more domain combinations to be involved in invoking protein interactions. In other words, when two proteins interact with each other, their interaction is interpreted as the result of the interaction of the mutual domain combinations. In order to represent this relationship, we introduce a notation of domain combination pairs formed by two proteins. The set of all possible domain combination pairs of two proteins p and q is defined by
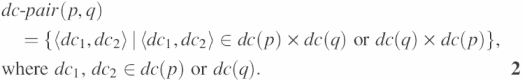
Thus, when two proteins p and q have n and m different domains, respectively, we can construct (2n − 1) × (2m − 1) different dc-pairs from the proteins. Figure 1b illustrates potentially interacting dc-pairs when two proteins with three and two domains interact with each other. Figure 1 also shows the comparison of our domain combination pair based approach with the conventional domain pair based approach. As depicted in Figure 1, the domain combination pair based approach considers not only the interactions of domains but also the interactions of domain combinations. Meanwhile, as there are multiple possible choices for the interaction of domains or domain combinations that can be inferred from a protein interaction, with only the interaction information of two proteins, we cannot figure out which dc-pair plays a decisive role in invoking the interaction. In order to make effective domain or domain combination based models, we need to draw some clues from the role of the domain or domain combination pairs involved in the interaction. This problem is quite difficult to solve with a small amount of interacting protein data. However, the accumulation of the interacting protein pairs on the Internet has made this approach feasible since the extraction of dc-pairs from a number of interacting protein pairs helps to identify and strengthen core dc-pairs in invoking protein interactions. The appropriate weight assignment to strengthen the role of dc-pairs is also important, and we explain this in the following section.
Figure 1.

(a) A conventional domain pair based prediction model versus (b) proposed domain combination pair based new prediction model.
AP matrix
The treatment of the appearance frequencies of domain combinations in a set of protein pairs is simplified by introducing a matrix. When there are n different proteins {p1, p2,…, pn} in a given set of protein pairs and the union of domain combinations of proteins contains m different domain combinations, {dc1, dc2,…,dcm}, i.e. the union of dc(p1), dc(p2),…, and dc(pn) is computed to {dc1, dc2,…, dcm}, and then the m × m AP matrix is constructed. The element APij in the matrix represents the appearance probability of domain combination <dci, dcj> in a given set of protein pairs.
For the construction of the AP matrix, we first constructed the WF (Weighted Frequency) matrix in which each row and column represent a domain combination and each element of the matrix represents a dc-pair. In the WF matrix, the appearance frequencies of domain combinations in a given set of protein pairs are registered. The element WFab in the matrix holds the weighted appearance frequency of domain combination <a, b> in a given set of protein pairs and its value is computed by
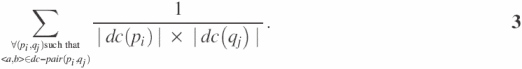
The final result of the equation is computed by adding up the expression 1/(|dc(pi)| × |dc(qj)|) for the entire protein pairs <pi, qj> which contain dc-pair <a, b>. By using Equation 3, the weights of the potential contribution of dc-pair <a, b> in the interactions of a given set of protein pairs holding the dc-pair are computed and then added together.
The weight assignment is based on the conjecture that the dcs or dc-pairs contained in the interaction of proteins with fewer domains are considered more important than those with more domains. Many other strategies could be adopted to give weights in the appearance frequencies of dc-pair and in the computing of WF matrix elements. This is still an open issue and further details are not discussed in this paper.
We use an example to illustrate how Equation 3 is used to compute the elements of the WF matrix. Suppose there are proteins A, B and C with domains domain(A) = {a1, a2}, domain(B) = {b1}, domain(C) = {a1, c1}, and let a set of interaction protein pairs {<A,B>, <A,C>,<B,C>} be given. In order to construct the WF matrix for proteins A, B and C, the matrix elements for all possible dc-pairs of the given set of protein pairs should be computed. As an example, expression 1/(|dc(B)|×|dc(A)|) is used to compute the element WF(b1)(a1) because the domain combination <{b1}, {a2}> appears only in dc-pair(A, B). As dc(A) = {{a1},{a2},{a1,a2}} and dc(B) = {{b1}}, expression 1/(|dc(B)|×|dc(A)|) is computed as 1/3. The other elements of the WF matrix are computed in a similar manner.
Once the WF matrix is constructed, the AP matrix construction is rather straightforward. Each element of the AP matrix is computed by

Then, each element of the AP matrix represents its appearance probability in the whole dc-pair space. Since there are sample spaces on each set of interacting and non-interacting protein pairs, we can generate two AP matrices. Large portions of the two matrices may be shared or can overlap each other, but they need not be similar in shape or share the components of the matrices. We denote the matrices as APi and APr, respectively, and the intersection APi ∩ APr as APc. The definitions are as follows:
APr: AP matrix constructed from a set of non-interacting protein pairs.
APi: AP matrix constructed from a set of interacting protein pairs.
APc: APi ∩ APr.
Once the AP matrices for interacting and non-interacting protein pairs are constructed, we can categorize a dc-pair by discerning to which matrix it belongs; we then name the categories using the APi, APr and APc notations. All the dc-pairs composing the APi matrix constitute APi dc-pair space. In the same way, APr dc-pair and APc dc-pair spaces are constituted.
Primary interaction probability
After the construction of AP matrices, a probability equation to predict the probability of interactions between unknown protein pairs <A, B> based on the two AP matrices, is defined and an undefined constant in the equation is determined. The first thing to be done in this step is to compute all the possible dc-pairs that can be formed from the protein pair <A,B> using Equation 2. Since many dc-pairs can be formed, and there are several categories in the dc-pair space, we classify the dc-pairs by the categories of the dc-pair space, and denote them as follows:
DCc(A,B) = {dc-pair|dc-pair ∈ dc-pair(A,B) and appears in APc space}
DCr−c(A, B) = {dc-pair|dc-pair ∈ dc-pair(A, B) and appears in APr − APc space}
DCi−c(A, B) = {dc-pair|dc-pair ∈ dc-pair(A, B) and appears in APi − APc space}
Figure 2 shows which element belongs to which category when dc-pair(A, B) is formed on the spaces of APi, APr. The elements of dc-pair(A, B) are denoted by special symbols (*, Δ, ×). We now define the basic interaction probability Equation 5 when DCc(A,B) is detected in the APc dc-pair space. The probability implies the likelihood of a protein pair <p, q> to interact when DCc(A,B) appears in the APc dc-pair space. We introduce a random variable X to denote the interacting and non-interacting events. The value 1 is used to represent an interacting event and 0 for a non-interacting event.
Figure 2.

Domain combination categories on AP spaces.
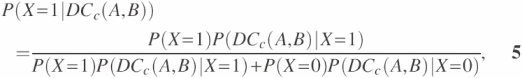
where P(X = 1), P(X = 0), P(DCc(A, B)|X = 1), P(DCc(A,B)|X = 0) are defined by

respectively.
![]()
In Equation 5, P(X = 1) represents the ratio of the set of interacting dc-pairs to the total dc-pairs in APc, whereas P(X = 0) represents the ratio of the set of non-interacting dc-pairs to the total dc-pairs in APc. Itotal and Rtotal in the above equations represent the total number of interacting and non-interacting protein pairs, respectively. The constant k is inserted into the equation because the exact ratio of Itotal and Rtotal in nature is unknown, and the value of optimal k is estimated by maximum-likelihood estimation. The set of dc-pairs DCc(A, B) appears in APi space, and P(DCc(A, B)|X = 0) denotes the probability of the set of dc-pairs DCc(A, B) appearing in APr space. APIc and APRc denote APc in interacting dc-pair space and non-interacting dc-pair space, respectively.
Equivalently, the interaction probability equation when the domain combinations DCi−c(A,B) are detected in APi−APc space is defined as in Equation 6 where, P(X = 1) is computed to 1 and P(X = 0) is computed to 0. Thus, the final probability is computed to 1. Using the probability Equations 5 and 6, Primary Interaction Probability (PIP) of a protein pair (A, B) with dc-pairs DCc(A,B) is defined by
![]()
PIP distribution and interaction prediction
Once the final equation of PIP is obtained in the second step, we can compute the PIP values by applying Equation 7 to the interacting and non-interacting sets of protein pairs. When all the PIP values of each set are computed, we get PIP distributions, and then we normalize the distributions to compare them. From this we can interpret PIP function as one that maps a protein pair onto a real number in the range of 0–1.
After the distributions are obtained, the interaction prediction of a protein pair is reduced to a two-category classification problem on the distributions. In short, to predict whether or not the two proteins in a given protein pair interact, we have to decide to which distribution the PIP value of the protein pair belongs to.
Interaction possibility ranking method
One may consider using the PIP equation directly for assigning an interaction probability to a protein pair. However, when we observe the PIP value distributions of interacting and non-interacting sets of protein pairs, some interacting protein pairs show low-PIP values, whereas some non-interacting protein pairs show high-PIP values (see Figure 3). This indicates that rather than using a PIP value directly as an interaction probability of a protein pair, it is desirable to devise another probability equation based on interacting and non-interacting PIP value distributions. The method proposed in this section involves using the probability of a PIP value to appear in a specific distribution between interacting and non-interacting PIP distributions. In other words, for a protein pair (A, B), its PIP value is computed and its interacting probability is determined by computing the probability of the PIP value to appear in the interacting PIP distribution.
Figure 3.

PIP distribution (y-axis is log scaled).
In this section, we devise probability equations to compute the interaction probability. Since the PIP value obtained may or may not have a matching PIP value in the PIP distributions, we considered the cases in terms of whether or not there exists a PIP value that matches to the target PIP value of a protein pair (A, B), between interacting or non-interacting PIP distributions.
Interaction probability for a protein pair with a matching PIP value
If there exists a PIP value that matches to PIP(A, B) in the interacting or non-interacting PIP value distributions, the computation of the interacting probability is rather straightforward. The interaction probability of a protein pair (A, B) with a PIP value, PIP(A, B), is computed by Equation 8.
In Equation 8, P(X = 1) is the ratio of interacting protein pairs in the total protein pairs; P(X = 0) is the ratio of non-interacting protein pairs in the total protein pairs; freqix is the number of samples with value PIPix in the set of interacting protein pairs; and freqiy is the number of samples with value PIPiy in the non-interacting set of protein pairs. Also the constant k is the same as the one used in Equation 5. P(p = PIP(A, B)|X = 1) is the probability of the random variable p to be PIP(A, B) in the interacting set of protein pairs. P(p = PIP(A, B)|X = 0) is the probability of the random variable p to be PIP(A, B) in the set of non-interacting protein pairs. When there are enough samples with PIP value in the PIP distributions, the interaction probability computed using the above equation could be quite reliable. However, if the numbers of the samples are not large enough, we should be more conservative in using the computed interaction probability. Further discussion on this matter is not in the scope of this paper.
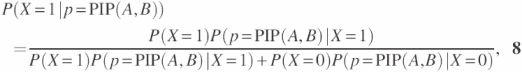
where P(X = 1), P(X = 0), P(p = PIP(A, B)|X = 1), P(p = PIP(A, B)|X = 0) are defined by

respectively.
![]()
![]()
Interaction probability for a protein pair with non-matching PIP value
If there is no PIP value to match PIP(A,B) in interacting or non-interacting PIP value distributions, the PIP values for all possible domain combination pairs that can be formed by proteins A and B are obtained. Among these PIP values, a PIP value that has a matching PIP value in the PIP distributions and is close to PIP(A, B) is selected, and the selected PIP value is used instead in computing protein pair (A, B)'s interaction probability. The PIP value must be within the predefined distance from PIP(A, B). We assume that the distance is decided by users, or the system may use a default value instead. Once the PIP value is decided, the interaction probability is computed using Equation 8.
The rationale behind this approach is based on the fact that the domain or the domain combination pairs formed by protein pair (A, B) will play a decisive role in invoking the interaction of proteins A and B. Therefore, we expect that the PIP value of a domain combination pair may be able to reflect the characteristics of a protein pair containing the domain combination pair. In addition, to reflect the characteristics of a protein pair (A, B) to the fullest extent, we chose the PIP value that was close to the original PIP value, PIP(A, B).
If any of the matching PIP values is not detected by the above processing methods, we considered the two cases in terms of their distance from the closest PIP value, PIPN(A, B), to PIP(A, B) in the PIP distributions. We developed and used different methods for the computation of the interacting probability for each case.
Case 1. |PIP(A, B) − PIPN(A, B)|<δ: In this case, we use a similar technique to the k-nearest-neighbor estimation technique. For the given PIP value of a protein pair (A, B), a resizable window size w′ is set and examined if the number of interacting protein pairs within the range exceedsthe number of k. If the number of interacting protein pairs is under k, the window size w′ is increased to include more interacting protein pairs within the range. This process is repeated until the number of interacting protein pairs exceeds k and the window size w used at that point becomes the final window size. After the window size w is decided, the interaction probability of the protein pair (A, B) is computed by Equation 9. Except for the range notations, the terms in the equation are similarly defined as those of Equation 8. In Equation 9, P(X = 1) is the ratio of interacting protein pairs in the total protein pairs P(X = 0) is the ratio of non-interacting protein pairs in the total protein pairs; freqix is the number of samples with value PIPix in the set of interacting protein pairs; and freqiy is the number of samples with value PIPiy in the non-interacting set of protein pairs. P(|p − PIP(A, B)| ≤ w/2|X = 1) is the probability that the random variable p is to be in the range of PIP(A, B) − w/2 and PIP(A, B) + w/2 in the interacting set of protein pairs. P(|p − PIP(A, B)| ≤ w/2|X = 0) is the probability of the random variable p occurring in the range of PIP(A, B) − w/2 and PIP(A, B) + w/2 in the set of non-interacting protein pairs.
Case 2. |PIP(A, B) − PIPN(A, B)| >δ: In this case, the interaction probability is decided by the value of PIP(A, B) and the PIP value itself becomes the interaction probability. This is represented by Equation 10.
RESULTS
In this section, the validation result of the proposed prediction model and the interaction possibility ranking method is illustrated. For the validation, two sets of protein pairs were prepared. One is the interacting set of protein pairs acquired from DIP database (http://dip.doe-mbi.ucla.edu) (13), where 15 174 interacting protein pairs in yeast were prepared for the validation. Since not all the proteins in the protein pairs have domain information, only 7500 interacting protein pairs could be used in the evaluation. The domain information for the proteins is extracted from the PDB (http://www.ebi.ac.uk/proteome/) (1,2).
On the other hand, the non-interacting set of protein pairs is artificially generated by randomly pairing the reported proteins with domain information in yeast. Note that there is no publicly announced information of the non-interacting set of protein pairs. Approximately 6000 proteins are known from yeast. Among them, 2700 proteins have domain information and they can be used in the creation of non-interacting sets of protein pairs. Altogether, 127 700 protein pairs were generated by randomly pairing the 2700 proteins. Then the negative sets of protein pairs were created by randomly selecting required numbers of protein pairs from the prepared set when necessary. Since interacting protein pairs could be included as well in the prepared set of protein pairs, we eliminated interacting protein pairs when selecting protein pairs for the preparation of non-interacting sets of protein pairs.
Figure 3 shows the distributions of PIP values from interacting (A) and non-interacting (B) sets of protein pairs, respectively. The same size of interacting and non-interacting sets of protein pairs are used for generation of the distributions. The PIP values of each set of protein pairs were mapped onto almost all the ranges from 0 to 1, with some overlapping between the two distributions. However, most PIP values from the interacting set of protein pairs are detected near 1 while most PIP values from the non-interacting set of protein pairs are detected near 0. Note that the scale of the y-axis in the graphs is represented in log scale. This indicates that the PIP equation could be a good classifier in discerning interacting and non-interacting protein pairs.
For testing the prediction accuracy of our method, we divided the interacting and non-interacting sets of protein pairs into learning and testing sets of protein pairs, respectively. Among the data, 80% are used for learning sets and 20% are reserved for test. For the precise evaluation of our protein–protein interaction prediction method, we increased the size of the non-interacting set of protein pairs because it is more natural to assume that there are more non-interacting protein pairs than interacting protein pairs. For the measurement, the protein pairs without overlapping domains in AP matrix are not included in the test data. Note that when there is no common domain between the test protein pairs and the constructed AP matrix, the application of the prediction method is meaningless.
Table 1 shows the sensitivities and specificities of each test group depending on the ratios of interacting and non-interacting set of protein pairs. The data in each test group are divided further into two subgroups; one group is the test set of protein pairs which has a matching PIP value in PIP distributions and the other group is the test set of protein pairs without matching PIP value in PIP distribution. As shown in Table 1, very high sensitivities and specificities were achieved for the test groups with matching PIP values, whereas moderate sensitivities and specificities were achieved for the test groups without matching PIP values. In the test, it was revealed that protein pairs with common domains in AP matrix are amenable to having matching PIP values in the PIP distributions. Only less than 5% of the protein pairs with common domains in AP matrix had no matching PIP value in the PIP distributions.
Table 1. The change of sensitivities and specificities by the ratios ofinteracting to non-interacting sets of protein pairs in training sets.
| Ratio | 1.0 | 2.0 | 5.0 | 10.0 | |
|---|---|---|---|---|---|
| I | Sensitivity | 96.77 | 92.96 | 85.98 | 78.73 |
| Specificity | 73.20 | 83.62 | 91.03 | 95.00 | |
| II | Sensitivity | 69.70 | 76.74 | 61.19 | 31.15 |
| Specificity | 62.16 | 64.58 | 76.36 | 81.67 | |
| Total | Sensitivity | 95.93 | 92.27 | 85.08 | 76.95 |
| Specificity | 73.07 | 83.32 | 90.73 | 94.65 |
I, protein pairs with matching PIP values.
II, protein pairs without matching PIP values.
The overall prediction accuracy improved as the relative size of non-interacting set of protein pairs in the training sets increased. When the size of the non-interacting set of protein pairs was 10 times bigger than that of the interacting set of protein pairs, 77% sensitivity and 95% specificity were achieved for the test protein pairs with common domains in AP matrix within our framework.
In order to ascertain if the method has stable prediction accuracies for other datasets, its prediction accuracy was also measured using DIP CORE (14), HMS-PCI (15) and TAP (16) data. Table 2 shows the sensitivities and specificities of each test group. Only the case when there is matching PIP values in the PIP distributions is considered. As shown in Table 2, quite stable and high-prediction accuracies are obtained irrespective of datasets. When the ratio is 10, the accuracy of using DIP data is under those of other cases. This indirectly indicates that DIP data contain more error data than the other data sources. On the other hand, the prediction accuracy of using TAP data was almost perfect. However, when we consider that the size of TAP data was relatively small, this result should be interpreted with caution.
Table 2. The sensitivities and specificities from the experiments using DIP, DIP CORE, MHS-PCI and TAP data.
| Ratio | 1.0 | 2.0 | 5.0 | 10.0 | |
|---|---|---|---|---|---|
| DIP | Sensitivity | 96.77 | 92.96 | 85.98 | 78.73 |
| Specificity | 73.20 | 83.62 | 91.02 | 95.00 | |
| DIP CORE | Sensitivity | 97.89 | 97.19 | 95.40 | 90.50 |
| Specificity | 70.23 | 90.77 | 89.76 | 95.21 | |
| HMS-PCI | Sensitivity | 94.64 | 96.98 | 95.71 | 93.08 |
| Specificity | 62.50 | 72.92 | 91.96 | 93.91 | |
| TAP | Sensitivity | 92.70 | 97.23 | 98.30 | 97.66 |
| Specificity | 86.67 | 97.43 | 97.70 | 98.60 |
Besides, the accuracy should not be directly interpreted as the prediction accuracy of protein–protein interactions. When we take into account that the prediction accuracy of the method is severely influenced by the error rate of input learning data (current protein–protein interaction data sources contain substantial amount of error data), the accuracy result of Tables 1 and 2 must be interpreted carefully. Specially, DIP data are known to contain quite a number of such erroneous data. Thus what the results showed, is the classification capability of our prediction method for two groups of protein pairs, in terms of domain combination pairs. It is expected that as the error rate of input learning data decreases, the prediction accuracy of the method for real protein–protein interaction will be improved gradually.
We also measured the proportion of protein pairs with matching PIP values in PIP distributions. In this measurement, protein pairs without common domains in AP matrix were also included. Table 3 shows the results of Hit ratio, which represent the proportion of protein pairs with matching PIP values to the total number of testing protein pairs.
Table 3. Hit ratios of testing protein pairs' PIP values in PIP distributions.
| 1.0 | 3.0 | 5.0 | 10.0 | 15.0 | 20.0 | |
|---|---|---|---|---|---|---|
| I | 845 | 721 | 733 | 727 | 851 | 780 |
| Hit ratio (%) | 53.14 | 45.35 | 46.10 | 45.72 | 53.52 | 49.06 |
| II | 452 | 362 | 467 | 529 | 540 | 643 |
| Hit ratio (%) | 28.43 | 22.77 | 29.37 | 33.27 | 33.96 | 40.44 |
| Total | 1297 | 1083 | 1200 | 1256 | 1391 | 1423 |
| Hit ratio (%) | 44.53 | 37.79 | 39.59 | 40.48 | 45.93 | 45.16 |
I, number of interacting protein pairs with matching PIP values.
II, number of non-interacting protein pairs with matching PIP values.
When the size of the non-interacting set of protein pairs was 20 times bigger than that of the interacting set of protein pairs, ∼50% of the interacting test set of protein pairs were revealed to have matching PIP values, and ∼45% of non-interacting test set of protein pairs were revealed to have their matching PIP values in the PIP distributions. The numerical precision for the matching test was 1.0E−12.
In the non-interacting set of protein pairs, the proportion of matching protein pairs increases as the size of the total learning set of protein pairs increases. On the other hand, the proportion of matching protein pairs did not change greatly with the increase in the size of the total learning set of protein pairs. That is, PIP values added from non-interacting set of protein pairs by increasing the size of the total learning set rarely matches the PIP values of interacting set of protein pairs. This is another indication that the interacting and non-interacting sets of protein pairs can be divided by PIP values.
In order to validate the ranking method, we counted the number of protein pairs detected in each range of interaction probabilities determined by the ranking method. The size of interacting and non-interacting test groups was 1590 and 1490, respectively. Table 4 shows the result where we can observe that large portions of the interacting test group were detected in the range of high-interacting probabilities, and large portions of the non-interacting test group were detected in the range of low probabilities. Some protein pairs in the interacting test group, however, have low-interacting probabilities and vice versa. This indicates that the ranking method should be applied carefully, and when the interaction probability is in the middle (0.4–0.6), we should be more conservative in applying the ranking method. Nevertheless, we can conclude that the ranking method proposed in this paper is valid to a certain extent.
Table 4. Number of interacting and non-interacting protein pairs and interacting probabilities.
| Interaction probability | No. of interacting protein pairs | No. of non-interacting protein pairs |
|---|---|---|
| 0.0–0.2 | 48 (3.0%) | 884 (59.3%) |
| 0.2–0.4 | 146 (9.2%) | 35 (2.4%) |
| 0.4–0.6 | 202 (12.7%) | 415 (27.9%) |
| 0.6–0.8 | 55 (3.5%) | 26 (1.7%) |
| 0.8–1.0 | 1139 (71.6%) | 130 (8.7%) |
| Total | 1590 (100.0%) | 1490 (100.0%) |
Note. The size ratio of interacting and non-interacting protein pairs used in the test was 15.
DISCUSSION
In this paper, a domain combination based probabilistic framework to predict protein–protein interaction and an interaction probability ranking method for multiple protein pairs are proposed. Evaluation of the techniques was conducted and the validity of the techniques was evident for various data sources. In the evaluation of the domain combination based protein–protein interaction method, we ascertained that the prediction accuracy is dependent on the accuracy of training data sources and the ratios of the sizes of learning sets of interacting and non-interacting protein pairs. As expected, our results confirmed that DIP data contain more noisy data than the other data sources. Nevertheless, it is still valuable data source because the domain coverage of DIP data is wide.
Although the proposed domain combination based prediction method certainly improves the prediction accuracy of the conventional domain based prediction method, it is not without limitations. This is because domain cannot explain all the details of complex protein–protein interactions, and the accumulated data are insufficient and still erroneous. In addition, there is no information on the sets of non-interacting pairs. Hence, we artificially made random pairing of protein and used it as a set of non-interacting protein pairs. This could limit the accuracy of prediction, since it might contain some interacting protein pairs that have not yet been discovered. These limitations, however, will be mitigated if more interacting protein pairs are discovered experimentally.
The contributions of the proposed technique and system can be summarized as follows. First, using this prediction system, biologists can get reliable preliminary information on unknown protein interactions avoiding time-consuming and high-cost experiments. More specifically, biologists can effectively plan their experiments using the ranking information or the services provided by the system. Second, PIP values and distributions can provide useful information in identifying incorrect protein–protein interaction data announced on the Internet. Third, mass prediction on protein interactions makes it possible to construct a large protein interaction network (6), and thus, biologists may be able to easily identify critical proteins from the network. Finally, the proposed technique can be a base for other computational approaches on protein identifications like predicting unknown protein functions.
A service system, named PreSPI (Prediction System for Protein Interaction) is developed using the techniques developed in this paper. The system is accessible on the Internet (http://silver.icu.ac.kr:8080/torajim/index.html). In the future, we are planning to extend the domain combination based protein–protein interaction technique by combining it with a homology search technique (7). Then the prediction for other protein groups, like mouse and human, would be possible. We are also considering applying the technique to the prediction of interactions between protein groups.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Ms Hang-Yi Kim for her pioneering work on protein interaction study in early stages of this research. This work was funded in part by the Korea Institute of Science and Technology Information (Grant No. K-04-BI-16-01S-4) and LG Life Science. J.K.S. was supported partly by the 21C Frontier Microbial Genomics and Application Center Program, Ministry of Science and Technology (Grant No. MG02-0302-003-1-0-0), Republic of Korea.
REFERENCES
- 1.Apweiler R., Attwood,T.K., Bairoch,A., Bateman,A., Birney,E., Biswas,M., Bucher,P., Cerutti,L., Corpet,F., Croning,M.D. et al. (2001) The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res., 29, 37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berman H.M., Westbrook,J., Feng,Z., Gilliland,G., Bhat,T.N., Weissig,H., Shindyabu,I.N. and Bourne,B.E. et al. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xenarios I. and Eisenberg,D. (2001) Protein interaction databases. Curr. Opin. Biotechnol., 12, 334–339. [DOI] [PubMed] [Google Scholar]
- 4.Sprinzak E. and Margalit,H. (2000) Correlated sequence-signatures as markers of protein–protein interaction. J. Mol. Biol., 311, 681–692. [DOI] [PubMed] [Google Scholar]
- 5.Deng M., Mehta,S., Sun,F. and Chen,T. (2002) Inferring domain–domain interactions from protein–protein interactions. Genome Res., 12, 1540–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enright A.J., Iliopoulos,I., Kyrpides,N.C. and Ouzounis,C.A. (1999) Protein interaction maps for complete genomes based on gene fusion events. Nature, 402, 86–90. [DOI] [PubMed] [Google Scholar]
- 7.Marcotte E.M., Pellegrini,M., Ng,H.L., Rice,D.W., Yeates,D.O. and Eisenberg,D. (1999) Detecting protein function and protein–protein interactions from genome sequences. Science, 285, 751–753. [DOI] [PubMed] [Google Scholar]
- 8.Ng S., Zhang,Z., Tan,S. and Lin,K. (2003) InterDom: a database of putative interacting protein domains for validating predicated protein interactions and complexes. Nucleic Acids Res., 31, 251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enright A.J. and Ouzounis,C.A. (2002) Protein-Protein Interactions: A Molecular Cloning Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 10.Bock J.R. and Gough,D.A. (2001) Prediction of protein–protein interaction from primary structure. Bioinformatics, 17, 455–460. [DOI] [PubMed] [Google Scholar]
- 11.Wojcik J. and Schachter,V. (2001) Protein–protein interaction map inference using interacting domain profile pairs. Bioinformatics, 17 (Suppl.), S296–S305. [DOI] [PubMed] [Google Scholar]
- 12.Han D., Kim,H., Seo,J. and Jang,W. (2003) Domain combination based probabilistic framework for protein–protein interaction prediction. Genome Informatics, 14, 250–259. [PubMed] [Google Scholar]
- 13.Salwinski L., Miller,C.S., Smith,J.A., Petit,F.K., Bowie,J.W. and Eisenberg,D. (2004) The Database of Interacting Proteins: 2004 update. Nucleic Acids Res., 32, D449–D451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deane C.M., Salwinski,L., Xenarios,I. and Eisenberg,D. (2002) Protein interactions: two methods for assessment of the reliability of high throughput observations. Mol. Cell. Proteomics, 1, 349–356. [DOI] [PubMed] [Google Scholar]
- 15.Ho Y., Gruhler,A., Heilbut,A., Bader,G.D., Moore,L., Adams,S.L., Millar,A., Taylor,P., Bennet,K., Boutilier,K. et al. (2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature, 415, 180–183. [DOI] [PubMed] [Google Scholar]
- 16.Gavin A.C., Bosche,M., Krause,R., Grandi,P., Marzioch,M., Baver,A., Shultz,J., Rick,J.M., Michon,A.M., Cruciat,C.M. et al. (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature, 415, 141–147. [DOI] [PubMed] [Google Scholar]
- 17.Park J., Lappe,M. and Teichmann,S. (2001) Mapping protein family interactions: intramolecular and intermolecular protein family interaction repertoires in the PDB and yeast. J. Mol. Biol., 307, 929–938. [DOI] [PubMed] [Google Scholar]
- 18.Murzin A.G., Brenner,S.E., Hubbard,T. and Chothia,C. (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol., 247, 536–540. [DOI] [PubMed] [Google Scholar]
- 19.Holm L. and Sander,C. (1996) FSSP database: fold classification based on structure–structure alignment of proteins. Nucleic Acids Res., 24, 206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pearl F.M.G., Lee,D., Bray,J.E., Sillitoe,I., Todd,A.E., Harrison,A.P., Thornton,J.M. and Orengo,C.A. (2000) Assigning genomic sequences to CATH. Nucleic Acids Res., 28, 277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mering V.C., Krause,R., Snel,B., Cornell,M., Oliver,S.G., Fields,S.G. and Bork,P. (2002) Comparative assessment of large-scale data sets of protein–protein interactions. Nature, 417, 399–403. [DOI] [PubMed] [Google Scholar]
- 22.Kim W., Park,J. and Suh,J. (2002) Large scale statistical prediction of protein–protein interaction by potentially interacting domain (PID) pair. Genome Informatics, 13, 42–50. [PubMed] [Google Scholar]
- 23.Goffard N., Iragne,F., Groppi,A. and de Daruvar,A. (2003) IPPRED: server for proteins interactions inference. Bioinformatics, 19, 903–904. [DOI] [PubMed] [Google Scholar]