


Abstract
Canine cyclic neutropenia is an autosomal recessive disease in which the number of neutrophils, the primary blood phagocyte, oscillates between almost zero and normal values with two week frequency. We previously found that the causative mutation is an insertion of an extra adenine residue within a tract of nine A's in exon 21 of the 27 exon canine AP3B1 gene. In the course of identifying the mutation, however, we observed an unusual phenomenon: heterozygous carrier dogs, who have one normal allele and one mutant allele, produce a homogeneous population of normal AP3B1 transcripts (containing nine A's), but homozygous affected dogs, who have two mutant alleles, produce a heterogeneous population of AP3B1 mRNA containing mutant transcripts with ten A's and, unexpectedly, wild-type transcripts with nine A's. By RT–PCR subclone analysis and use of an in vitro reporter assay, we show that there is a high frequency of errors made during the transcription of homopolymeric adenine sequences, such that the A tract in the mRNA is frequently shortened or lengthened by an extra residue. Out of frame transcripts are degraded, accounting for this paradox through the preferential accumulation of normal message from mutant alleles.
INTRODUCTION
An implicit conclusion of the central dogma of molecular biology is that the DNA sequence of a gene allows for the prediction of its mRNA and protein sequence. Exceptions are rare, but assume importance in the present era of large-scale sequencing and bioinformatics. One counterexample is the phenomenon of ribosomal frameshifting, where the reading frame of an mRNA is shifted at the time of translation (1).
Another violation of dogma involves changes in genetic information that are introduced post-transcriptionally or during transcription. Some transcripts may undergo RNA editing, in which the mRNA molecule is modified at specific bases through a variety of different mechanisms, such that the final sequence differs from that deduced from genomic DNA (2). There are also several examples in which RNA polymerase makes a high frequency of errors while reading repetitive DNA sequences. Often these errors are frameshifts whose occurrence implies that open reading frames are not simply predictable from genomic sequence. In the Brattleboro rat model of diabetes insipidus, resulting from a +2 frameshift in the vasopressin gene, a tendency toward GA deletion during the transcription of a GAGAG sequence causes a proportion of the transcripts to revert to a normal sequence, leading to amelioration of the disease phenotype (3). Frequent transcriptional frameshift events in similar repetitive sequences in genes encoding beta amyloid precursor protein and ubiquitin are thought to yield proteins unexpectedly corresponding to alternate reading frames, but whose presence, nevertheless, appears detectable by immunocytochemical staining of Alzheimer's disease plaques (4).
Runs of adenine residues also seem to cause RNA polymerase to produce a high rate of errors. Transcription through an A6 tract in the rat p53 gene yields insertion of an extra A in ∼9% of subcloned transcripts (5). A deletion of a T residue that is expected to yield a chain terminating frameshift, and otherwise interrupts a run of ten A's in the factor VIII gene, but that actually produces detectable clotting factor and mild symptoms, is thought potentially attributable, at least in part, to transcriptional infidelity (6). Similarly, in a family with hypobetalipoproteinemia resulting from homozygous mutation of the apolipoprotein B gene, one allele contains a single base deletion creating a continuous tract of eight adenine residues, that results in the appearance of a full-length protein, in addition to the truncated protein anticipated as a result of the frameshift (7). The latter example is reproducible in cultured cells, where expression of a transfected cDNA containing frameshifted A tracts results in ∼10% of corrected transcripts (8). In prokaryotes, transcriptional fidelity through homopolymeric adenine tracts is sufficiently unreliable (or reliable, as the case may be), such that Thermus thermophilus employs a single gene to encode two different subunits of DNA polymerase III (9).
Here, we describe transcriptional slippage occurring in the dog AP3B1 gene in a tract of A's that normally numbers nine bases long, which is extended to ten as a consequence of an autosomal recessive mutation responsible for the disease canine cyclic neutropenia. The mutation, which occurs in exon 21 of the 27 exon gene, is predicted to cause a frameshift and create a premature protein termination signal 11 bp downstream of the A tract. Because of degradation of out of frame transcripts, a paradoxical situation arises in which the population of transcripts expressed from carrier animals that are heterozygotes is homogeneous, while that from affected homozygotes appears heterozygous.
MATERIALS AND METHODS
Animals
We obtained DNA samples, originally extracted from peripheral blood, from 17 dogs affected with canine cyclic neutropenia, 13 known carriers of canine cyclic neutropenia, and 17 unaffected dogs of unknown carrier status from seven different pedigrees, of which five have been published previously (10), and including one outbred pedigree, as well as 11 controls from different breeds (French bulldog, clumber spaniel, pembroke corgi, poodle, sheltie, springer spaniel, Welsh corgi, Doberman, Irish setter, basenji, Yorkshire terrier and a mongrel). All affected, carrier and control dogs were genotyped for the length of the A tract in exon 21 of AP3B1 by sequencing of genomic DNA, as detailed below, revealing 10A allele homozygosity, 9A/10A allele heterozygosity and 9A homozygosity, respectively. The cDNA sequence and subclone analysis are representative results obtained from an affected, known carrier and non-carrier dog.
Plasmids
Oligonucleotide cassettes containing A-tracts of various lengths were cloned into the NcoI site downstream from the SV40 promoter of the pGL3-Control firefly luciferase reporter and analyzed by luminometry with the Dual-Luciferase Reporter Assay (Promega), internally controlled with Renilla luciferase. A total of 12-well plates of HEK293T cells (ATCC) were transfected in triplicate using 50 ng of reporter and 5 ng of the pRL-TK internal control with LipofectAMINE PLUS reagent (Gibco), using the manufacturer's protocol. Cells were harvested 40 h following transfection. Similar results were obtained with triplicate transfection to 200 ng of reporter (data not shown).
Northern-blot analysis
We loaded 5 μg/lane of total RNA extracted from peripheral blood on a 1% agarose formaldehyde-denaturing gel, with blot-transfer onto a BrightStar-Plus membrane (Ambion) and fixation by ultraviolet photo cross-linking. Hybridization was at 42°C in Ultrahyb (Ambion) with an 800 bp random-primed, 32P-labeled probe representing canine AP3B1 cDNA nucleotides 3129–3297 and internal control probe to human β-actin. Densitometry with Scion Imaging software was performed with the integrated density function of reflective scanned (Epson Perfection 3200 Photo) autoradiograms exposed on XAR-5 film (Kodak).
Subclone analysis
Total RNA was extracted from canine blood using RNAeasy (Qiagen) and reverse-transcribed with Omniscript (Qiagen), using the primer 5′-TGGAGATGACTTCATGATGAGC-3′. For cDNA subcloning, we utilized the Expand High Fidelity Plus DNA polymerase system (Roche) for PCR and subcloning into pGEM-T vector (Promega) with propagation in XL1-Blue Escherichia coli (Stratagene). Similar results were obtained with subcloning of PCR products amplified with Taq DNA polymerase (data not shown). Sequencing was performed with ABI Big Dye terminator chemistry on an ABI 310 capillary device. Primers for PCR subcloning were the following: forward cDNA (exon 20) 5′-ACTTCGGATTCTTCCAGCACTG-3′, forward DNA (intron 20) 5′-ACCCAGGATTGAGTCCCATGTCAGG-3′, and reverse for both cDNA and DNA (exon 21) 5′-CATCCAGGTCTAGAAGTGAAACATC-3′. [We originally reported that the mutation was in exon 20 of 26 of AP3B1; recent revision of the Dog Genome Browser (http://genome.ucsc.edu) indicates that it is actually exon 21 of 27.]
RESULTS
Genetic evidence that AP3B1 mutation causes canine cyclic neutropenia
Canine cyclic neutropenia, also known as ‘gray collie syndrome’, is an unusual autosomal recessive disease of dogs in which the peripheral neutrophil count cycles on a calendrical schedule (11). Neutrophils oscillate in number, nearly sinusoidally, between almost zero and normal with an approximate two week frequency. Other blood lineages (monocytes, lymphocytes, reticulocytes and platelets) also cycle, but do so in a phase opposite to that of neutrophils. Affected dogs exhibit coat color dilution and are vulnerable to periodic infections on account of their neutropenia. The disease is similar to (12), although clinically and genetically distinct from, human cyclic neutropenia, where neutrophils and monocytes reciprocally cycle with three week frequency, and which is caused by heterozygous mutations in the ELA2 gene, encoding the neutrophil granule serine protease, neutrophil elastase (13). After excluding ELA2 mutations as the cause of gray collie syndrome, we considered AP3B1 as a candidate because mutations of this gene cause disorders with some similar hematological and/or pigmentary features in humans (Hermansky Pudlak syndrome type 2), mice (pearl strain) and Drosophila (ruby strain).
We employed radiation hybrids to map canine AP3B1 to dog autosome 3. Genetic linkage and linkage disequilibrium analysis with markers from this region suggested AP3B1 as causative with a high degree of statistical certainty (10).
AP3B1 encodes the beta subunit of the ubiquitously expressed heterotetrameric adapter protein complex AP3, involved in post-translational trafficking of cargo proteins between lysosomes and the trans-Golgi network (14). Mutations in the beta subunit of the complex promote instability of the total heterotetramer (15,16). We have shown that the mu subunit of AP3 associates with neutrophil elastase, through a tyrosine based signal near its C-terminus (10), and have proposed that neutrophil elastase is a cargo protein transported by AP3 (10). We have suggested that mutations of neutrophil elastase or of AP3 might therefore have similar consequences because of their participation in a common pathway (12).
Molecular evidence that AP3B1 mutation causes canine cyclic neutropenia
Because the human AP3B1 gene is large (27 exons) and the sequence of canine AP3B1 had not yet been determined, in order to evaluate the contributions of this gene to the disease, we first obtained partial cDNA sequence of canine AP3B1 to generate a probe for northern-blot analysis of total RNA extracted from the blood of dogs [Figure 1d of Ref. (10)]. The ∼4.2 kb AP3B1 transcript appeared absent in an affected dog and approximately half the quantity in a carrier dog, compared with a control dog from a different breed. This result strongly suggested that a mutation of this gene causes the disease. Curiously, though, a small amount of residual transcript does appear in an affected dog, however, upon a longer exposure of the autoradiogram (Figure 1).
Figure 1.
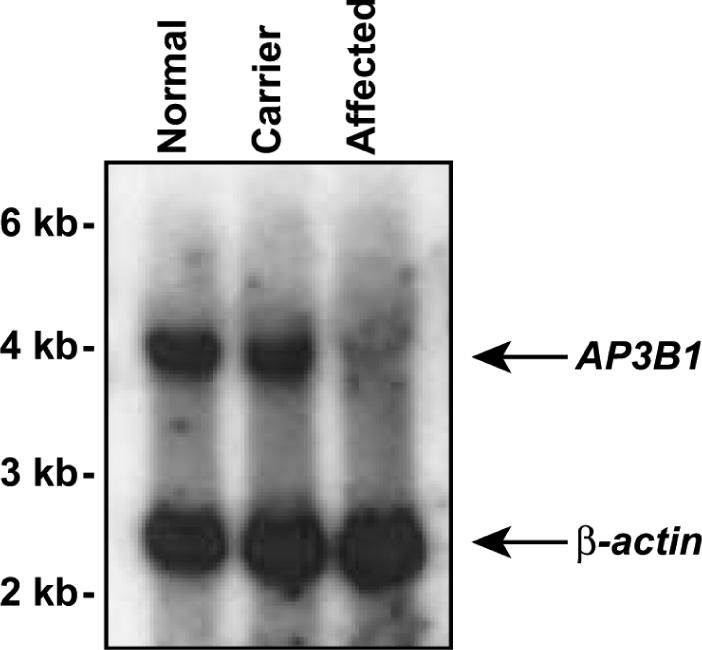
Northern blot of 5 μg total RNA from peripheral blood of normal, carrier or affected dogs, probed with canine AP3B1 and human β-actin cDNAs. Blot was exposed to film at −70°C with intensifying screens for three days.
A paradox: homozygous expression from heterozygotes and heterozygous expression from homozygotes
We cloned the full-length canine AP3B1 cDNA by isolating total RNA from dog blood and performing 5′- and 3′-RACE with primers to regions most highly conserved between the human and murine AP3B1 sequences (10). The disease is transmitted with autosomal recessive genetics, suggesting that sequencing of heterozygous carrier dogs would most rapidly pinpoint the location of the mutation by revealing two populations of transcripts. Conversely, a homogeneous population of transcripts would be expected in homozygous, affected dogs. Surprisingly, there were no apparent differences in cDNA sequence obtained on a bulk population of transcripts (i.e. not subcloned) between normal and carrier dogs; however, affected dogs did display two transcript sequence populations that differed by the insertion of an extra A residue in a tract of nine A's (i.e. in Figure 2, beginning at the arrow in the lower panel, it appears that the lower panel represents a superimposition of the traces obtained from the upper and middle panels). We next determined the genomic sequence of AP3B1 by ‘walking’ into introns from the identified exons in the cDNA (14). This allowed us to design primers for the amplification of the AP3B1 A tract from genomic DNA. Here, the results were unambiguously consistent with the expected Mendelian distribution: Affected dogs are homozygous, and carrier dogs are heterozygous, for an insertion of an A residue within the nine A tract, now localized to exon 21 of AP3B1 [see Figure 1c of Ref. (10)]. It is thus clear that mutation of AP3B1 causes canine cyclic neutropenia and that it is the equivalent of the human disease Hermansky Pudlak syndrome type 2. However, we were left with explaining the paradox of how heterozygous carrier dogs produced a single population of transcripts corresponding to only the normal allele, while homozygous affected dogs produced two populations of transcripts, one of which appeared to represent a normal transcript—even though both alleles are mutant.
Figure 2.

Paradoxical heterozygous expression of AP3B1 from homozygous dogs and homozygous expression of AP3B1 from heterozygous dogs. cDNA sequence of the AP3B1 exon 21 A tract is shown. An arrow denotes the start of sequence heterogeneity caused by the presence of both 9A and 10A-containing transcripts in the cDNA population of the affected.
An explanation: transcriptional slippage through homopolymeric adenine tracts
Because of the prior work of others showing an apparently high rate of transcriptional infidelity through repetitive DNA sequences, particularly homopolymeric stretches of adenine, we considered the following hypothesis in order to resolve the paradox. First, the apparent absence of transcript from the mutant allele in heterozygous animals is the result of selective degradation of mutant transcripts. This could be mediated by nonsense mediated decay, a process whereby transcripts encoding a premature chain terminating event, as expected with this frameshift, are subject to degradation prior to translation (17). Second, apparently normal transcripts in affected animals lacking a normal copy of the gene represent transcriptional errors that correct the frameshift and selectively accumulate, because the mutant transcript is again degraded. To test this hypothesis, we sequenced individual cDNAs subcloned in E.coli (Figure 3A). Heterozygous carriers are also heterozygous for a C/T single nucleotide polymorphism in exon 13 not affecting coding, with the C allele segregating on the normal allele, allowing for discrimination of mutant and normal transcripts. In heterozygous carriers, 4 out of 38 (10.5%) transcripts produced from the normal allele containing nine A's (and a C in exon 13) have a mistake (three being shortened by an A residue and one with an extra A inserted). Further, 2 out of 34 (5.9%) normal transcripts in the carrier actually derive from the mutant allele containing 10 A's (and a T in exon 13). In homozygous affected dogs, 10 out of 43 (23%) transcripts (all necessarily derived from a 10A allele) contain errors: 4 (9.3%) are shortened to 9 A residues; 4 (9.3%) are extended to 11 A residues; and 2 of the transcripts of the expected length of 10 A's actually contain base substitutions (an A to C transversion and an A to G transition at the eighth and fourth positions, respectively, within the A tract). Thus, the normal 9A allele manufactures some amount of mutant transcript, and the mutant 10A allele produces measurable quantities of normal transcript.
Figure 3.

Transcriptional slippage in A tracts. (A) Histogram of cDNA clones, sorted by length of A tract for carriers heterozygous for both the AP3B1 A-insertion and polymorphic C/T SNP [9A(C)/10A(T)] or affecteds homozygous for A-insertion and SNP [10A(T)/10A(T)]. Blue represents transcripts from the wild type (WT) 9A(C) allele; red, transcripts from mutant 10A(T) allele. (B) Cell culture assay for transcriptional slippage. Luciferase protein sequence is shown starting from the initiation methionine with variable length A tract adenines indicated in red. Subscripted ‘STOP’ represents different, downstream, out of frame (with respect to luciferase protein) termination codons. STOP1 is 91 bp and STOP2 31 bp downstream from the end of the A-tract. Histogram shows normalized luciferase assay for each construct, performed in triplicate. Negative control is luciferase reporter lacking a promoter.
Verification of transcriptional slippage in a reporter assay
It is possible that the heterogeneous population of transcripts observed here and in other reports arises as an artifact of cloning. However, a study (18) addressing the fidelity of PCR of mononucleotide sequences determined that polyadenine tracts shorter than 11 nt (noting that we are dealing with tracts that stretch for 9 or 10 nt) were stably PCR amplified, regardless of which DNA polymerase was used, and that the errors introduced during amplification of tracts longer than 11 nt were primarily contractions, rather than both contractions and expansions, as we observe. Accordingly, we observed that subcloned sequences containing either 9A or 10A tracts stably propagate with PCR and E.coli transformation (data not shown), excluding mutation by DNA polymerase in vitro or genetic instability in bacteria. It is still possible that reverse transcriptase, a low-fidelity enzyme (19), introduces the A-tract mutations during cDNA synthesis. To exclude cloning artifacts, we measured transcriptional slippage in human HEK293T cells by fusing tracts of 7–11 A's upstream of a luciferase reporter gene (Figure 3B). The 9A tract is in-frame with luciferase, but appropriate translation of the other lengths requires that transcriptional slippage generates a corrective frameshift. We assayed luciferase activity in transiently transfected cells, where no vector DNA replication occurs (20). Tracts of 8A's or 11A's are the most slippage prone, leading to frameshifts of 20%, as quantified by relative luciferase activity. For the 9A tract, we also introduced +1 and −1 frameshifts into the vector [9A(+1) and 9A(−1)], allowing determination of the direction of slippage. In the case of the 9A constructs, backward slippage, causing an insertion, occurs about twice as often as forward slippage.
DISCUSSION
Estimates of the fidelity of eukaryotic transcription are difficult to come by. In vitro, the rate of single base misincorporation by RNA polymerase II from wheat germ ranged from 1 in 250 to 1 in 2 × 105, depending upon the template sequence and the nature of divalent metal cofactors (21). An in vivo estimate based on a yeast luciferase reporter assay measuring read through of a termination codon, indicated a maximal single base error frequency not greater than 1/500 (16). There is evidence that RNA polymerase engages in proofreading (22), after incorporation of an incorrect ribonucloetide into a nascent transcript, and that the transcription elongation factor S-II enhances excision of mis-incorporated bases (23). In contrast, here we find transcriptional errors occurring within homopolymeric tracts of A residues, primarily insertion or deletion of a single adenine and therefore, presumably not corrected by proofreading mechanisms, approaching a remarkably high frequency. The cDNA subclone analysis data suggest transcriptional slippage rates of ∼5% in the carrier. This is based on sequence results from 40 clones in the carrier, 20 of which should be derived from the mutant allele if there were no effect on stability of mutant messages. What is seen instead is that two clones are derived from the mutant allele, indicating that the 9A transcript was synthesized from the mutant allele at ∼5% the rate of synthesis from the wild-type allele. In the luciferase reporter assay, the 10A construct (which mirrors the gray collie mutation) shows a 10% slippage rate. These slippage rate values are similar to those reported by others (8). These values are all generally consistent but differ somewhat from the northern blot, where quantitative analysis by densitometry reveals that the affected accumulates only 2% of wild-type transcript levels. In addition to inaccuracies of the various methods, a possible explanation is that RT–PCR can amplify exon-sized fragments of a degraded message, whereas a northern blot more specifically detects only non-degraded, full-length transcript. The observations here are additionally complicated by the cellular phenomenon of nonsense mediated decay, in which messages containing a premature termination codon, an almost inevitable consequence of a frameshift, are preferentially degraded (17). Thus, in homozygous affected dogs, it is possible that faithfully transcribed messages corresponding to the mutant allele are degraded, thereby leading to selective accumulation of abnormal messages arising from a slippage event.
In spite of these complications, the several different assays performed here all indicate that some quantity of normal message is produced from mutant alleles. Even a little may be better than none; for example, the mutant hemophilia A allele that generates a run of 10 adenines, produces 4% of normal factor VIII levels with attendantly mild symptoms (6). It is interesting in this regard that canine neutropenia is milder and cyclical (11), when compared with the severe and static neutropenia in human Hermansky Pudlak syndrome 2 (16), which results from equivalent homozygous null mutations of AP3B1. It is possible that the residual level of AP3B1 mRNA in the dogs ameliorates the phenotype and therefore affords biological proof to substantiate our molecular observations.
In contrast to transcriptional slippage, insertion and deletion mutation within homopolymeric DNA sequences is a well-documented phenomenon, and homopolymeric stretches of A's (or T's) seem particularly vulnerable (24). The mechanism appears to result from misaligned loops, and physiological surveillance for such errors appears to require mismatch correction enzymes (25). In DNA, deletions are more common than insertions and the error frequency increases with increasing length of the homopolymer. In contrast, the results here utilizing the reporter assay, find no direct correspondence between the length of the sequence and the frequency of errors, at least for stretches of A's between 7 and 11 residues long. It has been proposed that frameshift mutations within A tracts may be less a feature of nucleic acid polymerases, in particular, but rather represent a general response to properties of such DNA sequences that are predicted to form a ‘two-faced’ structure with shifted base pairs in the major groove but an apparent normal geometry in the minor groove (26).
AAA codes for lysine, which can also be encoded by the triplet AAG. An analysis of codon usage found that for the peptide sequence Lys-Lys-Lys, triplets of the codon AAA, leading to tracts of nine consecutive A's, were significantly under-represented in comparison to their expectation based on lysine codon distributions in the human genome (8). Thus, there appears to have been evolutionary selection against homopolymeric A tracts in the sense strand of human genes. A comparison of the exon 21 A tract of the AP3B1 gene in dog, human and mouse (Figure 4) shows that the human sequence, encoding amino acids identical to the canine, has interrupted the adenine tract with a guanine, creating two runs of four A's. The mouse sequence also differs from the dog, and does not contain consecutive adenines greater than four.
Figure 4.

Alignment of homologous segments of canine, human and murine AP3B1 exon 21 (NCBI accession nos NM_001002974, NM_003664 and NM_009680). Codons derived from the A tract nucleotide sequence and the corresponding amino acids are indicated in boldface.
A common, cancer-predisposing allele of the APC gene, I1307K, contains a T to A transversion of the final exon [where frameshift mutations are not subject to nonsense-mediated decay (17)] that generates a stretch of eight consecutive A's and appears to be a hotspot for subsequent somatic mutations of this tumor suppressor gene (27). The contributions of this variant to tumorigenesis, however, have remained controversial, because not all colon cancers arising in individuals with the I1307K variant have detectable somatic mutations in the APC gene (28). Our results emphasize the possibility (29) that another effect of this mutation is to produce a high frequency of frameshifted transcripts that produce prematurely truncated proteins in the absence of frank mutation.
Clearly, when evaluating sequences containing extended tracts of adenines, the possibility of unexpected open reading frames and frameshifted transcripts needs to be considered.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs Clinton Lothrop, Glenn Niemeyer and Gregory Acland for providing dog samples and Dr Stephen G. Young for valuable discussion. Supported by grants (to M.H.) from the NIH (DK55820, DK58161) and Burroughs-Wellcome Fund (SATR-1002189).
REFERENCES
- 1.Farabaugh P.J. (2000) Translational frameshifting: implications for the mechanism of translational frame maintenance. Prog. Nucleic Acid Res. Mol. Biol., 64, 131–170. [DOI] [PubMed] [Google Scholar]
- 2.Wedekind J.E., Dance,G.S., Sowden,M.P. and Smith,H.C. (2003) Messenger RNA editing in mammals: new members of the APOBEC family seeking roles in the family business. Trends Genet., 19, 207–216. [DOI] [PubMed] [Google Scholar]
- 3.van Leeuwen F.W., Fischer,D.F., Kamel,D., Sluijs,J.A., Sonnemans,M.A., Benne,R., Swaab,D.F., Salehi,A. and Hol,E.M. (2000) Molecular misreading: a new type of transcript mutation expressed during aging. Neurobiol. Aging, 21, 879–891. [DOI] [PubMed] [Google Scholar]
- 4.van Leeuwen F.W., de Kleijn,D.P., van den Hurk,H.H., Neubauer,A., Sonnemans,M.A., Sluijs,J.A., Koycu,S., Ramdjielal,R.D., Salehi,A., Martens,G.J. et al. (1998) Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer's and Down patients. Science, 279, 242–247. [DOI] [PubMed] [Google Scholar]
- 5.Ba Y., Tonoki,H., Tada,M., Nakata,D., Hamada,J. and Moriuchi,T. (2000) Transcriptional slippage of p53 gene enhanced by cellular damage in rat liver: monitoring the slippage by a yeast functional assay. Mutat. Res., 447, 209–220. [DOI] [PubMed] [Google Scholar]
- 6.Young M., Inaba,H., Hoyer,L.W., Higuchi,M., Kazazian,H.H.,Jr and Antonarakis,S.E. (1997) Partial correction of a severe molecular defect in hemophilia A, because of errors during expression of the factor VIII gene. Am. J. Hum. Genet., 60, 565–573. [PMC free article] [PubMed] [Google Scholar]
- 7.Linton M.F., Pierotti,V. and Young,S.G. (1992) Reading-frame restoration with an apolipoprotein B gene frameshift mutation. Proc. Natl Acad. Sci. USA, 89, 11431–11435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Linton M.F., Raabe,M., Pierotti,V. and Young,S.G. (1997) Reading-frame restoration by transcriptional slippage at long stretches of adenine residues in mammalian cells. J. Biol. Chem., 272, 14127–14132. [DOI] [PubMed] [Google Scholar]
- 9.Larsen B., Wills,N.M., Nelson,C., Atkins,J.F. and Gesteland,R.F. (2000) Nonlinearity in genetic decoding: homologous DNA replicase genes use alternatives of transcriptional slippage or translational frameshifting. Proc. Natl Acad. Sci. USA, 97, 1683–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benson K.F., Li,F.Q., Person,R.E., Albani,D., Duan,Z., Wechsler,J., Meade-White,K., Williams,K., Acland,G.M., Niemeyer,G. et al. (2003) Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nature Genet., 35, 90–96. [DOI] [PubMed] [Google Scholar]
- 11.Jones J.B., Lange,R.D. and Jones,E.S. (1975) Cyclic hematopoiesis in a colony of dogs. J. Am. Vet. Med. Assoc., 166, 365–367. [PubMed] [Google Scholar]
- 12.Horwitz M., Benson,K.F., Duan,Z., Li,F.-Q. and Person,R. (2004) Hereditary neutropenia: dogs explain human neutrophil elastase mutations. Trends Mol. Med., 10, 163–170. [DOI] [PubMed] [Google Scholar]
- 13.Horwitz M., Benson,K.F., Person,R.E., Aprikyan,A.G. and Dale,D.C. (1999) Mutations in ELA2, encoding neutrophil elastase, define a 21-day biological clock in cyclic haematopoiesis. Nature Genet., 23, 433–436. [DOI] [PubMed] [Google Scholar]
- 14.Boehm M. and Bonifacino,J.S. (2002) Genetic analyses of adaptin function from yeast to mammals. Gene, 286, 175–186. [DOI] [PubMed] [Google Scholar]
- 15.Feng L., Seymour,A.B., Jiang,S., To,A., Peden,A.A., Novak,E.K., Zhen,L., Rusiniak,M.E., Eicher,E.M., Robinson,M.S. et al. (1999) The beta3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky-Pudlak syndrome and night blindness. Hum. Mol. Genet., 8, 323–330. [DOI] [PubMed] [Google Scholar]
- 16.Dell'Angelica E.C., Shotelersuk,V., Aguilar,R.C., Gahl,W.A. and Bonifacino,J.S. (1999) Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol. Cell, 3, 11–21. [DOI] [PubMed] [Google Scholar]
- 17.Wormington M. (2003) Zero tolerance for nonsense: nonsense-mediated mRNA decay uses multiple degradation pathways. Mol. Cell, 12, 536–538. [DOI] [PubMed] [Google Scholar]
- 18.Clarke L.A., Rebelo,C.S., Goncalves,J., Boavida,M.G. and Jordan,P. (2001) PCR amplification introduces errors into mononucleotide and dinucleotide repeat sequences. Mol. Pathol., 54, 351–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Svarovskaia E.S., Cheslock,S.R., Zhang,W.H., Hu,W.S. and Pathak,V.K. (2003) Retroviral mutation rates and reverse transcriptase fidelity. Front. Biosci., 8, d117–d134. [DOI] [PubMed] [Google Scholar]
- 20.Cox L.S. and Lane,D.P. (1995) Tumour suppressors, kinases and clamps: how p53 regulates the cell cycle in response to DNA damage. Bioessays, 17, 501–508. [DOI] [PubMed] [Google Scholar]
- 21.de Mercoyrol L., Corda,Y., Job,C. and Job,D. (1992) Accuracy of wheat-germ RNA polymerase II. General enzymatic properties and effect of template conformational transition from right-handed B-DNA to left-handed Z-DNA. Eur. J. Biochem., 206, 49–58. [DOI] [PubMed] [Google Scholar]
- 22.Thomas M.J., Platas,A.A. and Hawley,D.K. (1998) Transcriptional fidelity and proofreading by RNA polymerase II. Cell, 93, 627–637. [DOI] [PubMed] [Google Scholar]
- 23.Koyama H., Ito,T., Nakanishi,T., Kawamura,N. and Sekimizu,K. (2003) Transcription elongation factor S-II maintains transcriptional fidelity and confers oxidative stress resistance. Genes Cells, 8, 779–788. [DOI] [PubMed] [Google Scholar]
- 24.Kunkel T.A. (1990) Misalignment-mediated DNA synthesis errors. Biochemistry, 29, 8003–8011. [DOI] [PubMed] [Google Scholar]
- 25.Gragg H., Harfe,B.D. and Jinks-Robertson,S. (2002) Base composition of mononucleotide runs affects DNA polymerase slippage and removal of frameshift intermediates by mismatch repair in Saccharomyces cerevisiae. Mol. Cell. Biol., 22, 8756–8762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Timsit Y. (1999) DNA structure and polymerase fidelity. J. Mol. Biol., 293, 835–853. [DOI] [PubMed] [Google Scholar]
- 27.Laken S.J., Petersen,G.M., Gruber,S.B., Oddoux,C., Ostrer,H., Giardiello,F.M., Hamilton,S.R., Hampel,H., Markowitz,A., Klimstra,D. et al. (1997) Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC. Nature Genet., 17, 79–83. [DOI] [PubMed] [Google Scholar]
- 28.Sieber O., Lipton,L., Heinimann,K. and Tomlinson,I. (2003) Colorectal tumourigenesis in carriers of the APC I1307K variant: lone gunman or conspiracy? J. Pathol., 199, 137–139. [DOI] [PubMed] [Google Scholar]
- 29.Raabe M., Linton,M.F. and Young,S.G. (1998) Long runs of adenines and human mutations. Am. J. Med. Genet., 76, 101–102. [DOI] [PubMed] [Google Scholar]