


Abstract
We have cloned and expressed the ahdIC gene of the AhdI restriction-modification system and have purified the resulting controller (C) protein to homogeneity. The protein sequence shows a HTH motif typical of that found in many transcriptional regulators. C.AhdI is found to form a homodimer of 16.7 kDa; sedimentation equilibrium experiments show that the dimer dissociates into monomers at low concentration, with a dissociation constant of 2.5 μM. DNase I and Exo III footprinting were used to determine the C.AhdI DNA-binding site, which is found ∼30 bp upstream of the ahdIC operon. The intact homodimer binds cooperatively to a 35 bp fragment of DNA containing the C-protein binding site with a dissociation constant of 5–6 nM, as judged both by gel retardation analysis and by surface plasmon resonance, although in practice the affinity for DNA is dominated by protein dimerization as DNA binding by the monomer is negligible. The location of the C-operator upstream of both ahdIC and ahdIR suggests that C.AhdI may act as a positive regulator of the expression of both genes, and could act as a molecular switch that is critically dependent on the Kd for the monomer–dimer equilibrium. Moreover, the structure and location of the C.AhdI binding site with respect to the putative −35 box preceding the C-gene suggests a possible mechanism for autoregulation of C.AhdI expression.
INTRODUCTION
Bacterial restriction-modification (R-M) systems encode enzymes for DNA methylation and endonuclease (ENase) activity. The methyltransferase (MTase) methylates specific bases contained within recognition sequences found throughout their genome, while the restriction ENase cleaves under-methylated DNA after binding to the same recognition site. Only host DNA bears the appropriate methylation pattern, so this system can differentiate ‘self’ DNA from ‘non-self’ and prevent incorporation of foreign DNA into the host and thus acts as a primitive immune system.
However, if the system is not temporally regulated, ENase activity may occur prior to methylation of the host DNA, resulting in auto-restriction and cell death. It is therefore necessary to repress ENase expression until such time as the MTase has methylated all potential host target sites. Many R-M systems contain an additional open reading frame (ORF) coding for a small protein (the controller, or C, protein) that is believed to act as a transcriptional regulator of ENase and/or MTase expression, as well as regulating its own transcription (1–6).
A number of studies have suggested that C-proteins bind to a symmetrical sequence upstream of the C-gene. The binding site has been postulated to range in size from 7 to 26 bases (3,5,7–10). However, to date, the DNA-binding sites of only two C-proteins have been determined experimentally, and the consensus sequence, if any, remains unclear.
The AhdI R-M system from Aeromonas hydrophila is an unusual one; like type I R-M systems, it encodes three genes involved in restriction and/or modification—M, S and R. However, the ENase is coded by a single gene (R) as in type II R-M systems, while the MTase requires both the M and S genes, as for type I R-M systems; moreover, the subunit/domain structure of the MTase also resembles type I MTases (13). An ORF coding for a 74 amino acid (8.4 kDa) protein showing homology to other C-proteins is found just upstream of the R-gene, suggesting that this gene codes for the AhdI controller protein. The M and S genes are transcribed together on a separate operon, with C/R and M/S operons being aligned convergently (see Figure 1a).
Figure 1.

(a) Arrangement of the AhdI R-M genes. The M and S genes, and the C and R genes are arranged convergently, with transcription in the direction shown. The two operons are separated by a central self-complementary region, depicted here as a hairpin. (b) Secondary structure prediction. The amino acid sequence of C.AhdI is given with the putative helix–turn–helix region underlined. Secondary structure predictions are shown from the program PSIPRED with associated reliability indices (Rel) on a scale of 1–10. Predicted helices are denoted as ‘H’.
The amino acid sequence of C.AhdI is shown in Figure 1b, together with the secondary structure prediction. The structure is largely alpha-helical, and the five helices predicted include a helix-turn–helix motif (helix 2–helix 3), as found in many bacterial gene regulatory proteins, as well as in eukaryotic transcription factors such as POU domains. Although there may be secondary structure similarities to such proteins, the degree of sequence identity is low (∼20% when C.AhdI is compared with lambda repressor or the 434 cro protein, or ∼30% when compared with the SinR protein of Bacillus subtilis).
To date, no work has been published on the purification and/or characterization of the C-protein of the AhdI R-M system, C.AhdI (and indeed there has been no biophysical characterization of any members of the C-protein family). Here, we report the expression and purification of C.AhdI, and characterization of the protein in terms of its subunit structure, DNA-binding site and DNA-binding affinity. Structural analysis of the protein by X-ray crystallography is also underway in our laboratory (13).
METHODS
Sub-cloning
The ahdIC gene (GenBank accession no. AY313905) was amplified by PCR using the parental AhdI pUC19 plasmid (New England Biolabs) as the template. The amplified product was gel-purified using the QIAquick Gel Extraction Kit (QIAgen) and digested with BamHI and NdeI restriction ENases prior to ligation using T4 DNA ligase (NEB). The ahdIC gene was initially ligated into both pET-11a and pET-23b expression vectors. However, although the pET-23b vector incorporated an N-terminal hexa-histidine tag to allow simpler purification of the protein, it failed to express sufficient quantities in a soluble form. Therefore, the pET-11a expression vector was used, allowing the production of native C.AhdI.
Expression and purification of C.AhdI
E.coli BL21 (DE3) Gold cells containing the pET-11a plasmid plus ahdIC gene were grown at 37°C in 2× YT broth until an OD600 value of 0.6 was obtained. The cells were then induced with 1 mM isopropyl-β-d-thiogalactopyranoside and grown for a further 3 h. The cells were harvested by centrifugation and the pellets were stored at −20°C until required. The cell pellets were resuspended in 40 mM bicine (pH 8.7), 0.1 M NaCl, 3 mM DTT, 5 mM EDTA and lysed by sonication at 4°C. Cell debris was removed by centrifugation at 39 000 g.
The soluble cell lysate was made to 1 M NaCl in a 30% w/v solution of (NH4)2SO4 and the precipitated protein was sedimented by centrifugation at 27 000 g. The protein pellet was solubilized in 5 ml (per pellet) of 40 mM bicine, pH 8.7, 0.1 M NaCl, 1 mM EDTA, 1 mM DTT and dialysed in the same solution to remove (NH4)2SO4. The sample was subsequently bound to a heparin column and upon reaching a baseline, a linear 0.1–1.0 M NaCl gradient in 40 mM bicine, pH 8.7, 1 M NaCl, 1 mM EDTA and 1 mM DTT was used to elute the target protein. Fractions containing the target were pooled and dialysed back down to 0.1 M NaCl overnight, which results in precipitation of the target protein.
The precipitated protein was sedimented by centrifugation at 27 000 g and resuspended in 40 mM tri-sodium citrate, pH 5.6, 0.1 M NaCl, 1 mM EDTA, 1 mM DTT and loaded onto a SP Sepharose column. A linear 0.1–1.0 M NaCl gradient was again used to elute the protein. Following dialysis to reduce the NaCl concentration to 0.1 M and remove DTT, the target protein was further purified on a Sephacryl S100 26/60 HR column and concentrated on a SP Sepharose column using a 0.1–1 M NaCl step gradient. The sample was again dialysed back to 0.1 M NaCl and the purity of the sample was evaluated by UV-fluorescence analysis of a urea-denatured aliquot: the emission maximum was observed at ∼303 nm (tyrosine) rather than at 348 nm (tryptophan) as C.AhdI does not contain tryptophan.
The extinction coefficient was calculated to be 2900 M−1 cm−1 for the monomer at 276 nm (5800 M−1 cm−1 for the dimer) by summing the contributions from the aromatic amino acids. Protein purity at each stage was estimated using 12% tris-tricine PAGE, which allows the resolution of low molecular weight proteins (14), with Coomassie blue staining. Electrospray ionization mass spectrometry was performed at the University of Leeds, UK, using a Platform II mass spectrometer (Micromass UK Ltd) following dissolution in aqueous methanol with 0.5% formic acid. Data were acquired over the m/z range 800–1200 and an expected mass accuracy of 0.01% was achieved by calibrating the system using horse heart myoglobin (Mr = 16 951.49 Da).
Light scattering
Dynamic light scattering (DLS) was performed on a series of C.AhdI concentrations in 40 mM tri-sodium citrate, pH 5.6, 100 mM NaCl, 1 mM EDTA at 20°C using a Protein Solutions DynaPro temperature-controlled microsampler. The technique provides an estimate of particle hydrodynamic radius (Rh) and solution molecular weight, as well as the polydispersity of the sample, by analysis of the autocorrelation function. For globular proteins, the value of Rh can be used to estimate the molecular mass, Mr, using the empirical equation:
![]()
Static (Rayleigh) light scattering measurements were performed on the same instrument to allow an estimate of the Mr independent of shape. This technique requires measurement of light intensity scattered from the sample with respect to a standard scatterer (in this case, toluene). The residual intensity (ILS), the total intensity of scattered light minus the solvent light scattering, is directly proportional to both the molecular mass and concentration of the protein. If protein concentration is varied, molecular mass can be calculated according to the equation:
![]()
where K = instrument constant incorporating the refractive index increment (dn/dc)2 and the wavelength, C = protein concentration in mg/ml, R90 is the ratio of the residual intensity relative to the toluene standard at an angle of 90°, Mr = particle molecular mass and A2 = the second virial coefficient. Plotting the function (K)(C)/Rθ against concentration allows the Mr to be obtained from the intercept.
Analytical ultracentrifugation
A Beckman Optima XL-A analytical ultracentrifuge (Beckman-Coulter, Palo Alto, CA) was used for sedimentation equilibrium experiments. The experiment was performed in six-channel cells of 12 mm optical path length, using 90 μl of solution at protein concentrations ranging from 1 to 60 μM; 100 μl of the sample buffer (40 mM tri-sodium citrate, pH 5.6, 100 mM NaCl, 1 mM EDTA) was loaded into the corresponding channel. The rotor was accelerated to 21 000 and 28 000 r.p.m. and scans of absorbance (276 and 235 nm) versus radial displacement were taken at a resolution of 0.001 cm for times up to 21 h. The temperature was set at 20°C.
32P-end-labelling of DNA
Approximately 30 μg oligonucleotide was mixed with 50 U T4 polynucleotide kinase and 100 μCi of [γ-32P]ATP. The mixture was incubated at 37°C for 30 min before being supplemented with 1 mM dATP and incubated at 37°C for a further 30 min. The unincorporated [γ-32P]ATP was removed using G-25 spin columns (Amersham Biosciences), ∼30 μg complementary strand was added and the mixture was incubated at 90°C for 10 min. The mixture was then cooled to room temperature over a 2 h period. The procedure was repeated with the other strand to give two duplexes, with one or other of the strands 5′ end-labelled. Each duplex was gel-purified and eluted. The DNA concentration for each duplex was calculated by summation of the nucleotide extinction coefficients and corrected for hypochromicity. The percentage hypochromicity was estimated following phosphodiesterase digestion of the duplexes.
DNA footprinting studies
The Exo III footprinting method was developed from the method described elsewhere (15). A series of protein concentrations were set up in binding buffer (50 mM Tris–HCl, pH 8.0, 10 mM MgCl2 and 1 mM DTT) with 1 μM 35 bp duplex for each labelled strand. The samples were incubated at 4°C for 30 min before the addition of 2 U of Exo III (New England Biolabs) and incubation at 20°C for a further 20 min. The reaction was stopped by the addition of 2 vol of formamide loading buffer and heating to 90°C for 2 min. The samples were then loaded onto a pre-run 20% denaturing polyacrylamide gel.
A 60 bp duplex containing the 35 bp sequence was used for DNase I footprinting studies, and the oligonucleotides were pre-labelled at the 5′ end with a hexachlorofluorescein tag during synthesis. A series of increasing protein concentrations were set up in binding buffer with 4 μM 60 bp duplex for each labelled strand. The samples were incubated at 4°C for 1 h before the addition of 0.2 U of DNase I (Invitrogen) and incubation at 20°C for a further 6 min. The reaction was stopped for the Exo III reactions. The samples were then loaded onto a pre-run 15% denaturing polyacrylamide gel.
The sequences of 35 and 60 bp DNA duplexes used are shown below.
CTGGTACTCATAGTCCGTGGACTTATCGACATCAT
GACCATGAGTATCAGGCACCTGAATAGCTGTAGTA
TCGGTCAAAGTGGCTGGTACTCATAGTCCGTGGACTTATCGACATCATTAAGAGTGTAAT
AGCCAGTTTCACCGACCATGAGTATCAGGCACCTGAATAGCTGTAGTAATTACACTCTTA
Gel retardation analysis
Gel retardation experiments were performed using non-denaturing gel electrophoresis. Aliquots of C.AhdI were incubated at various concentrations with 80, 240 and 800 nM [γ-32P]labelled 35 bp DNA duplex in binding buffer (as described above) at 4°C for a period of 30 min. The samples were then loaded onto a pre-run 8% native polyacrylamide gel. After electrophoresis at 100 V for 90 min, the gels were dried and visualized using a phosphorImager screen. The fraction of bound DNA in each lane was determined using ImageQuant software. Following quantitation of the free and bound fractions, binding constants were estimated by fitting the data to various binding models using ProFit (Cherwell Scientific Publishing).
Surface plasmon resonance (SPR)
All SPR experiments were performed using a BIAcore X system and streptavidin (SA) sensor chip (Biacore AB, Uppsala, Sweden). A synthetic 35 bp oligonucleotide (as described above) was 5′-biotinylated and annealed with a complementary unlabelled strand and gel-purified and precipitated. The biotinylated DNA duplex was bound to the chip until a final immobilization of ∼150 RU of duplex DNA was achieved. The second flow cell was used as a reference cell to subtract the bulk solution contribution from flow cell one. The protein sample was buffer exchanged into Biacore HBS-EP buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% surfactant P20) and a series of protein concentrations was prepared. Sample injections lasted 160 s and a delay of 160 s prior to washing enabled collection of dissociation data. SDS solution (0.05%) was then injected for 1 min to remove any residual protein and an extended period of washing with the buffer followed. All experiments were performed at 20°C. The sensorgrams obtained were fitted using the BiaEvaluation software version 3.0 to give equilibrium and kinetic constants.
Secondary structure analysis
Secondary structure analysis of the C.AhdI ORF sequence (AAP78483) was performed using the program PSIPRED (16).
RESULTS
Protein purification
Following ammonium sulfate precipitation from bacterial cell lysates, C.AhdI was purified to homogeneity (Figure 2) by a combination of heparin, cation-exchange and gel permeation chromatography steps (see Materials and Methods). A single band was observed by Coomassie blue staining, migrating as a protein of ∼8.5 kDa, and was estimated to represent >99% of the protein present. Mass spectrometry indicated a molecular weight of 8369.6 Da, identical to the theoretical molecular weight of C.AhdI calculated from the predicted amino acid sequence. The yield of purified C.AhdI was ∼1–2 mg l−1 of culture.
Figure 2.
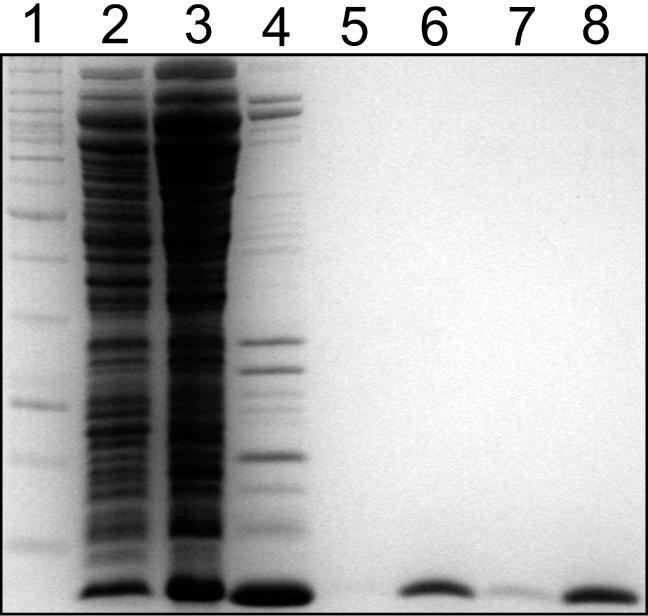
PAGE showing stages in the purification of C.AhdI. Lane 1, molecular weight marker; lane 2, whole cell lysate; lane 3, ammonium sulfate pellet following resuspension and dialysis; lane 4, pooled heparin fractions; lane 5, resuspended protein precipitate; lane 6, pooled cation exchange fractions; lane 7, pooled size exclusion fractions; and lane 8, concentrated C.AhdI.
Oligomeric status
DLS measurements on purified C.AhdI (Figure 3a) indicated a hydrodynamic radius of 1.97 nm. The protein samples all possessed low polydispersity, typically 5–10%. Using the globular proteins model for molecular mass determination, this value corresponds to a Mr of 16.5 kDa, suggesting that C.AhdI forms homodimers in solution. However, DLS does not give an absolute molecular weight as the hydrodynamic radius also depends on shape.
Figure 3.

(a) Dynamic light scattering. The peak corresponds to a hydrodynamic radius of 1.97 nm, equivalent to a molecular mass of 16.5 kDa. Polydispersity is 10%. Protein concentration was 0.2 mg ml−1 (24 μM). (b) Static (Rayleigh) light scattering. A series of C.AhdI protein concentrations (0.099–0.493 mg ml−1) were analysed by static light scattering. The plot of residual intensity is shown in blue, (K)(C)/R90 is shown in red. The intercept on the (K)(C)/R90 axis gives 1/Mr, from which we obtain Mr = 16804 ± 376 Da.
Static (Rayleigh) light scattering analysis allows the measurement of molecular weight independently of shape. Using a range of C.AhdI concentrations (0.1–0.6 mg ml−1), total scattered light intensity was measured. The molecular mass was found to increase slightly with concentration over this range. Plotting (K)(C)/R90 (see Materials and Methods) and extrapolating to zero concentration gives a molecular mass of 16.8 ± 0.4 kDa from the intercept (see Figure 3b), in excellent agreement with the theoretical Mr for a dimer (16.7 kDa).
Sedimentation equilibrium experiments were subsequently carried out, which allowed measurement of the solution molecular weight of C.AhdI over a wider range of protein concentrations and provided an estimation of the Kd for dimerization. At each protein concentration, we initially fitted the analytical ultracentrifugation data to a single-species model. The resulting plot of apparent Mr against protein concentration is shown in Figure 4a, and clearly shows a concentration dependent increase in Mr from that of a monomer (8.4 kDa) to a dimer (16.8 kDa) over this concentration range, with evidence of a small degree of larger aggregate formation at the highest protein concentrations. At the lowest protein concentration, there is a large error due to the low signal. At 15 μM, the error reflects the invalidity of the single species model. From the weight averaged Mr at 15 μM, one can estimate the ratio of monomer and dimer (ca. 1:2) and thus estimate a Kd in the region of 2 μM. The equilibrium distribution curve at 15 μM was subsequently fitted to a monomer–dimer equilibrium model (Figure 4b) which gave a dimer dissociation constant of 2.5 (±0.2) μM and a second order virial coefficient of 10−5.
Figure 4.

Sedimentation equilibrium. (a) Weight-averaged Mr from sedimentation equilibrium runs for C.AhdI at a series of different concentrations, with associated errors from the fit. (b) The sedimentation equilibrium profile at 15 μM protein was fitted to a monomer–dimer equilibrium model, and residuals shown (× 10−3). The centrifuge was run at 21 000 r.p.m. for 13 h (20°C) and absorption monitored at 235 nm.
DNA footprinting studies
Preliminary experiments were carried out using Exonuclease III as the footprinting reagent (Figure 5a). Exonuclease III cuts unidirectionally from the 3′ end of duplex DNA and can be used to locate the 3′ edges of the C.AhdI binding site on each strand. Bands towards the top of the gel are seen (on each strand) that increase in intensity as the protein concentration is increased. These sites are symmetrically disposed, 13 bp either side of the pseudo-dyad axis in the centre of the 35 bp DNA fragment. Additional sites are seen close to the centre of the symmetrical sequence; the intensities of these bands vary in a complex way with protein concentration suggesting the possibility of multiple-binding equilibria (see Discussion).
Figure 5.

DNA footprinting of C.AhdI. (a) Exonuclease III footprinting. Cleavage sites are shown for the 5′-labelled top and bottom strands of the 35 bp DNA duplex (1 μM). The G + A ladder is shown to the left of each series, and concentration dependent cutting sites are indicated. The protein concentration range was 0–8 μM. (b) DNase I footprinting of C.AhdI. Cleavage sites are shown for the 5′-labelled top and bottom strands of the 60 bp DNA duplex (4 μM). The G + A ladder is shown to the left of each series, and concentration dependent cutting sites are indicated. The protein concentration range was 0–16 μM. (c) Summary of footprinting data. The sites of enhanced cutting by DNase I are shown as black arrows. The horizontal black bar shows the major regions of DNase I protection, along with secondary regions of protection (dotted line), thicker lines/arrows representing more prominent protection/enhancement. The principal ExoIII stop sites are indicated by open arrows, but there is a major additional stop site near the centre of the footprint which is prominent at intermediate protein concentrations (smaller arrow). The central 12 bp sequence which includes the symmetrical sequence is boxed. The putative −35 box sequence, TTGACT, is underlined and the −10 site, TGTAAT, italicized.
Further experiments were performed using DNase I as the footprinting reagent. Typically in DNase I footprinting, regions of protection from nuclease activity are seen where the protein binds (and a few bases beyond, due to the finite size of the probe), together with sites of hyper-cutting—most frequently at the edges of the footprint (where the nuclease pauses), or at accessible sites within the binding site where changes in the structure of the DNA (e.g. groove dimensions) might allow enhanced cutting.
DNase I footprinting of C.AhdI was carried out using the 60 bp DNA fragment (Figure 5b). On the top strand, there are two sites that are clearly enhanced when the protein is bound, either side of the symmetrical sequence; between these two hypersites, there are regions showing significant protection from DNase I. There is a third site of enhanced cutting a further 7 bases on the 5′ side, and a number of sites up to this point also show some degree of protection. On the bottom strand, a similar situation pertains. Although cutting of the free DNA is less intense on this strand, clear protection is observed between the two main hypersites when the protein is bound. Again, there is an additional site of enhanced cutting by a further 8 bp to the 5′ end of this site, but the enhancement is relatively weak and there is rather less protection observed generally at the 5′ end of the footprint on this strand.
The general features of the footprinting data are summarized in Figure 5c. The major region of protection is centred on the symmetrical 12 bp sequence AGTCCNNGGACT. At the higher protein concentrations used, the 3′ edge of the footprint is well defined (both by Exo III and by DNase I) on both strands and extends some 7–8 bases from the edge of the central symmetrical sequence. The 5′ edge of the binding site is less well defined by DNase I (and is not accessible to Exo III footprinting), since there are two sites of enhanced cutting symmetrically placed on each strand extending ∼8 additional bases on each strand. The binding site extends from approximately −21 to −53 with respect to the C.AhdI start codon (∼−25 to −49 when allowing for the size of the enzymatic probe) and incorporates a potential −35 box in the C.AhdI promoter (see Discussion). This protected region is larger than would be expected from binding of a small dimeric protein, and could suggest that two or more dimers can bind at this site, at least at higher protein concentrations. At lower protein concentrations, a smaller footprint is indicated, though it is less well defined.
DNA-binding studies
Titration of C.AhdI with the 35 bp oligonucleotide (80 nM) containing the recognition sequence produced a strong band that increases with protein concentration, which we attribute to binding of the protein dimer; in addition, there is a very faint band seen only at the lower protein concentrations, which we attribute to weak binding of protein monomers (Figure 6a). Parallel experiments performed in the presence of a 10-fold excess of unlabelled competitor DNA duplex did not appreciably affect the complex formation, indicating that binding is sequence-specific (data not shown). If the experiment is repeated with a 10-fold higher DNA concentration (800 nM), then larger complexes become visible at the highest protein:DNA ratios (data not shown), perhaps corresponding to trimers and tetramers.
Figure 6.

Electrophoretic mobility shift assay. (a) Protein samples of increasing concentration (0–1280 nM) were incubated with 80 nM DNA. The monomer (M) and dimer complexes (D) are indicated, together with the free DNA band (F). (b) The fraction bound was determined using ImageQuant software and the value plotted was the average of values obtained from two gels. The solid line shows the fitted curve for a monomer–dimer equilibrium model (see text) with K1 = 2.5 μM and K2 = 6.4 nM. For comparison, the dotted line shows the theoretical binding curve in the absence of dimer dissociation (K1 = 0 and K2 = 6.4 nM).
Quantitation of free and bound DNA bands in the gel retardation assay allowed the binding curve for C.AhdI to be analysed (Figure 6b). The curve is sigmoidal, indicative of cooperative binding, and could not be fitted by the standard single-site binding model. This would be expected if the active binding species were dimeric, as the dimer dissociates at low concentrations. The band that we suggest arises from monomer-DNA binding is extremely weak and the interaction can, to a good approximation, be ignored, the fraction bound by monomeric C.AhdI being almost negligible (representing typically 2–3% of total bound DNA). Including or excluding the small amount of the monomer complex from the calculations made little difference to the fit. An excellent fit to the data could be obtained with a cooperative binding model that allows for the formation of dimers from C.AhdI monomers (with dissociation constant K1) with only the dimer capable of binding to DNA (with dissociation constant K2); the best fit was obtained with K1 = 2.5 (±0.2) μM and K2 = 6.4 (±1.4) nM. In effect, at intermediate protein concentrations, DNA binding is governed by the Kd for dimerization, rather than the Kd for DNA binding (see Figure 6b). The consequences of this are that much higher protein concentrations are required to saturate the DNA-binding site than if the dimer persisted at low protein concentrations, and the binding curve is therefore sigmoidal.
Further analysis of DNA binding was performed by SPR on a BIAcore X. A 35 bp biotinylated DNA duplex was immobilized on a streptavidin surface; the purified C.AhdI protein was injected over the DNA at different protein concentrations, and both the binding and the dissociation phases of the interaction were monitored. The experimental curves were fitted to a number of possible models but using a 1:1 Langmuir binding model with mass transport effects best fitted the data (Figure 7), with a χ2 value of 5.4 for the global fit. The fitting procedure gave values for the on- and off-rates, ka = 4.8 × 106 M−1 s−1 and kd = 0.023 s−1 from which a value for the equilibrium dissociation constant, Kd, of 5 nM is obtained, a very similar value to that obtained from gel retardation analysis.
Figure 7.

Surface plasmon resonance. C.AhdI samples of varying concentration (100, 200 and 300 nM) were injected at 30 μl min−1 and the binding curves were fitted to a 1:1 Langmuir binding model with mass transfer (dotted lines). A protein–DNA dissociation constant of 4.8 nM was calculated from the on-rate (4.8 × 106 M−1 s−1) and off-rate (0.023 s−1).
DISCUSSION
The DNA footprinting experiments show that the binding site for C.AhdI is a sequence of up to 30 bp centred on a 12 bp sequence, AGTCCGTGGACT, in which all but the central 2 bp are self-complementary. It is of interest to compare the C.AhdI binding site with the consensus C-box sequence for the PvuII family (Figure 8a). In the idealized symmetrical sequence proposed by Vijesurier et al. (5), there are two tandem repeats of the ‘dyad consensus sequence’ GACTNNNAGTC, separated by 4 bases including a central and highly conserved GT. This leads to essentially four tetranucleotide sequences of either AGTC or its reverse complement GACT (Figure 8, referred to as boxes 1–4). The central region of the C.AhdI binding site matches box 2 and box 3 completely; however, there is lower homology with box 1 (75%) and box 4 (50%). When all four boxes are compared, the best consensus sequence for C.AhdI is the pentanucleotide sequence GGACT (and its dyad equivalent AGTCC), one base larger than the tetranucleotide repeat (Figure 8d). There are additional nucleotides that are fully conserved with other C-binding sites in the PvuII family that are outside of the symmetrical sequence, including the central GT (Figure 8b). The function of these additional conserved sequences is currently unknown, but presumably these highly conserved sequences must have some functional significance.
Figure 8.

Analysis of C-protein DNA-binding sites. (a) Idealized symmetrical sequence and (b) consensus sequence (with fully conserved bases shown in bold) as proposed by Vijesurier et al. (5). (c) Binding region of C.AhdI: the location of the idealized (pentameric) symmetrical sequence for C.AhdI is shown in red; sequence identities with the consensus sequence of Vijesurier et al. are shown in bold, both within and outside of the symmetrical sequence. The proposed OL and OR sequences for AhdI are compared below, colour coding as above. (d) Comparison of boxes 1–4 in the C.AhdI binding site, with the reverse complementary sequence of boxes 2 and 4 shown to compare the dyad symmetry. Bases conserved in 3 out of 4 of the boxes are indicated in bold. Numbers shown on the right of each box refer to the number of base identities to the C.AhdI consensus sequence. (e) C.AhdI operator site, indicating the −35 sequence (green), the −10 sequence (purple) and the start codon for the C-protein (blue).
At first sight, one might expect that the strongest binding site for the C.AhdI dimer would be the most symmetrical site (i.e. boxes 2 and 3), where there is 100% identity with the C.AhdI consensus sequence GGACT/AGTCC; however, the spacing here (N2 compared with N3) may not be optimal, in which case binding of the dimer to boxes 1 and 2 would be favoured. If protein is in excess, then binding energy will be maximized with two dimers bound, at boxes 1 and 2 (which we will refer to as OL) and boxes 3 and 4 (OR), even if both sites are sub-optimal. OL would be expected to be the preferred site, since it has 9 out of 10 matches to the consensus, compared with 7 out of 10 matches in OR. Thus, as protein concentration increases, the OL site will tend to be filled first, followed by OR. Such a scenario may explain the aspects of the concentration dependent changes in the footprints, most notably the Exo III stop sites near the centre of the footprint observed at intermediate protein:DNA ratios. These sites are principally located in and around box 3, and define the 3′ end (on the top strand) of a smaller binding site at these concentrations. It is also consistent with the generally weaker DNase I footprint found at the 5′ end of the bottom strand (and to a lesser extent, also at the 3′ end of the top strand), since this region corresponds to OR. We cannot rule out the possibility that when a dimer is bound to OL, a C.AhdI monomer may then bind cooperatively to box 3 as the protein concentration increases. Although binding of an isolated monomer to DNA is very weak, it could be stabilized by protein contacts with an existing dimer bound to OL, and as protein concentration increases further, another monomer may be recruited to the weakest binding site (box 4) forming a dimer at OR. Either way, the consequences are much the same: the OR site eventually will be filled at sufficiently high protein concentration.
In the AhdI system, the genes for M and S and the genes for C and R are arranged convergently, with a predicted stem-loop region between the two operons that may act as a terminator of transcription. In the C/R operon, the R-gene is downstream of the C-gene which itself is preceded by a promotor region consisting of a predicted −10 sequence (TGTAAT) and a −35 sequence (TGGACT), which have 5 bases and 4 bases out of 6 in common with their respective consensus sequences (Figure 8e). The central C-protein binding site AGTCCGTGGACT overlaps this potential −35 sequence by 6 bp. However, if the C-protein bound initially to OL, then it would be located immediately 5′ of the −35 sequence and thus in an appropriate position to interact with RNA polymerase bound at this site and activate transcription.
In a number of bacterial promoters, transcriptional activation is achieved through energetically favourable interactions between a gene regulatory protein and the σ70 subunit of RNA polymerase when bound at the −35 sequence (17). There is a highly conserved glutamic acid residue (Glu34) on a short section of the first helix of the HTH motif that, together with the less conserved residue Asp38, defines the positive control surface of the N-terminal domain of the lambda cI protein; the side chain of Glu34 makes a strong electrostatic interaction with an arginine on the σ70 subunit of RNA polymerase. This residue has previously been shown by functional analysis to be absolutely essential for transcription activation. Interestingly, a superposition of the recently solved high-resolution crystal structure of C.AhdI (J.E. McGeehan, S.D. Streeter, I. Papapanagiotou, G.C. Fox and G.G. Kneale, manuscript in preparation) on the HTH motif of lambda cI shows that there is a short helical segment in which a glutamic acid residue (Glu30) is located in an identical position to Glu 34 in the cI protein, consistent with the putative role of the C.AhdI protein as a transcriptional activator. Moreover, Phe34 in C.AhdI is located in an identical position to Asp38 in cI, and indeed the mutation of Asp to Phe at the equivalent site has been shown to enhance transcription activation by lambda cI (18), surprisingly so as this mutation removes the possibility of electrostatic interactions with the sigma subunit at this site. It is notable that Glu30 is highly conserved between C-protein sequences, and that the site equivalent to Phe34 is also typically occupied by one of the residues (Asp, Phe or Tyr) known to promote transcriptional activation (J.E. McGeehan, S.D. Streeter, I. Papapanagiotou, G.C. Fox and G.G. Kneale, manuscript in preparation).
C.AhdI appears to be a transcriptional regulator of both C and R gene expression, and required for efficient transcription of the C/R operon, thus leading to a timing delay in the expression of the ENase, as originally proposed for PvuII (7). Since C.AhdI only binds effectively to its DNA recognition sequence as a dimer, the relatively high Kd for dimerization (2.5 μM) is critical. C.AhdI is unable to activate transcription until a sufficient concentration of the protein accumulates to promote dimerization, and thus transcription of the associated R gene will be similarly delayed. We envisage that the C-protein then binds as a dimer to the sequence we denote as OL, placing it immediately adjacent to the −35 box and in a position to stimulate transcription by interaction with σ70. As C-protein levels increase further, it is proposed that an additional dimer binds to OR (either directly, or via a monomer first binding to box 3), which in either case occludes the −35 sequence and thus represses transcription, in this way acting to autoregulate the expression of the C-protein and hence finely tune the expression of the ENase. Such a mechanism might also apply to PvuII and other members of that family, since in each of the five sequences compared by Vijesurier et al. (5), we note that there are possible −35 sequences (with 3–5 out of 6 bases conserved) overlapping box 3 of the operator site.
The C gene lacks any clear ribosome-binding site (RBS) upstream of the start codon, and indeed may have no leader sequence at all, since the positions of the −35 and −10 sequences suggest that transcription should start at or very close to the ATG. In contrast, there is a predicted RBS sequence (GGAGG) located 11 bp upstream of the R subunit start codon. This could provide a further mechanism for the regulation of expression at the translational level. Translation of the C gene is likely to be very inefficient, further delaying the expression of R. However, once transcription had been activated, translation of the R gene would proceed efficiently. Again, this would further enhance the on-off nature of the switch.
One further level of regulation should also be considered as a theoretical possibility. Intriguingly, the AhdI restriction sequence GACNNNNNGTC is found within the C.AhdI consensus binding sequence GGACTNNNAGTCC, raising the possibility that methylation of the C.AhdI binding site may also play a role in transcriptional control. However, close inspection of the C.AhdI binding site shows that both OL and OR differ by 1 bp from the canonical AhdI restriction sequence. Nevertheless, the precise specificity of the AhdI MTase has yet to be determined experimentally, so such a mechanism cannot be ruled out.
Analysis of the X-ray structure of C.AhdI is currently in progress, as are structural studies of complexes with C.AhdI operator sequences, in order to elucidate the molecular details of the interactions that constitute this intricate genetic switch.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Professor Robert Blumenthal (Medical College of Ohio) for pointing out similarities between the C.AhdI and C.PvuII operator sequences, and to Dr Darren Mernagh (University of Portsmouth, UK) for advice on surface plasmon resonance. We thank BBSRC for a research grant (to G.G.K.) and studentship (to S.D.S.) and Dr Alison Ashcroft (University of Leeds) for the use of mass spectrometry facilities provided by a Wellcome Trust grant.
REFERENCES
- 1.Tao T. and Blumenthal,R.M. (1992) Sequence and characterization of pvuIIR, the PvuII endonuclease gene, and of pvuIIC, its regulatory gene. J. Bacteriol., 174, 3395–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ives C.L., Nathan,P.D. and Brooks,J.E. (1992) Regulation of the BamHI restriction-modification system by a small intergenic open reading frame, bamHIC, in both Escherichia coli and Bacillus subtilis. J. Bacteriol., 174, 7194–7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rimseliene R., Vaisvila,R. and Janulaitis,A. (1995) The eco72IC gene specifies a trans-acting factor which influences expression of both DNA methyltransferase and endonuclease from the Eco72I restriction-modification system. Gene, 157, 217–219. [DOI] [PubMed] [Google Scholar]
- 4.Lubys A., Jurenaite,S. and Janulaitis,A. (1999) Structural organization and regulation of the plasmid-borne type II restriction-modification system Kpn2I from Klebsiella pneumoniae RFL2. Nucleic Acids Res., 27, 4228–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vijesurier R.M., Carlock,L., Blumenthal,R.M. and Dunbar,J.C. (2000) Role and mechanism of action of C·PvuII, a regulatory protein conserved among restriction-modification systems. J. Bacteriol., 182, 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cesnaviciene E., Mitkaite,G., Stankevicius,K., Janulaitis,A. and Lubys,A. (2003) Esp1396I restriction-modification system: structural organization and mode of regulation. Nucleic Acids Res., 31, 743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tao T., Bourne,J.C. and Blumenthal,R.M. (1991) A family of regulatory genes associated with type II restriction-modification systems. J. Bacteriol., 173, 1367–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anton B.P., Heiter,D.F., Benner,J.S., Hess,E.J., Greenough,L., Moran,L.S., Slatko,B.E. and Brooks,J.E. (1997) Cloning and characterization of the BglII restriction-modification system reveals a possible evolutionary footprint. Gene, 187, 19–27. [DOI] [PubMed] [Google Scholar]
- 9.Bart A., Dankert,J. and van der Ende,A. (1999) Operator sequences for the regulatory proteins of restriction modification systems. Mol. Microbiol., 31, 1277–1278. [DOI] [PubMed] [Google Scholar]
- 10.Zheleznaya L.A., Kainov,D.E., Yunusova,A.K. and Matvienko,N.I. (2003) Regulatory C protein of the EcoRV modification-restriction system. Biochemistry (Moscow), 68, 125–132. [DOI] [PubMed] [Google Scholar]
- 11.Kita K., Tsuda,J. and Nakai,S.-y. (2002) C.EcoO109I, a regulatory protein for production of EcoO109I restriction endonuclease, specifically binds to and bends DNA upstream of its translational start site. Nucleic Acids Res., 30, 3558–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marks P., McGeehan,J., Wilson,G., Errington,N. and Kneale,G.G. (2003) Purification and characterisation of a novel DNA methyltransferase,M.AhdI. Nucleic Acids Res., 31, 2803–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGeehan J.E., Streeter,S., Cooper,J.B., Mohammed,F., Fox,G.C. and Kneale,G.G. (2004) Crystallization and preliminary X-ray analysis of the controller protein C.AhdI from Aeromonas hydrophilia. Acta Crystallogr. D Biol. Crystallogr., 60, 323–325. [DOI] [PubMed] [Google Scholar]
- 14.Schägger H. and von Jagow,G. (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem., 166, 368–379. [DOI] [PubMed] [Google Scholar]
- 15.Metzger W. and Heumann,H. (1994) Footprinting with Exonuclease III. In Kneale,G.G (ed.), DNA–Protein Interactions: Principles and Protocols. Methods in Molecular Biology. Humana Press Inc., Totowa, NJ, Vol. 30, pp. 11–20. [DOI] [PubMed] [Google Scholar]
- 16.McGuffin L.J., Bryson,K. and Jones,D.T. (2000) The PSIPRED protein structure prediction server. Bioinformatics, 16, 404–405. [DOI] [PubMed] [Google Scholar]
- 17.Jain D., Nickels,B.E., Sun,L., Hochschild, A and Darst,A.A. (2004) Structure of a ternary transcription activator complex. Mol. Cell, 13, 43–53. [DOI] [PubMed] [Google Scholar]
- 18.Kolkhof P. and Muller-Hill,B. (1994) Lambda cI repressor mutants altered in transcriptional activation. J. Mol. Biol., 242, 23–36. [DOI] [PubMed] [Google Scholar]