

Abstract
The function of a protein that has no sequence homolog of known function is difficult to assign on the basis of sequence similarity. The same problem may arise for homologous proteins of different functions if one is newly discovered and the other is the only known protein of similar sequence. It is desirable to explore methods that are not based on sequence similarity. One approach is to assign functional family of a protein to provide useful hint about its function. Several groups have employed a statistical learning method, support vector machines (SVMs), for predicting protein functional family directly from sequence irrespective of sequence similarity. These studies showed that SVM prediction accuracy is at a level useful for functional family assignment. But its capability for assignment of distantly related proteins and homologous proteins of different functions has not been critically and adequately assessed. Here SVM is tested for functional family assignment of two groups of enzymes. One consists of 50 enzymes that have no homolog of known function from PSI-BLAST search of protein databases. The other contains eight pairs of homologous enzymes of different families. SVM correctly assigns 72% of the enzymes in the first group and 62% of the enzyme pairs in the second group, suggesting that it is potentially useful for facilitating functional study of novel proteins. A web version of our software, SVMProt, is accessible at http://jing.cz3.nus.edu.sg/cgi-bin/svmprot.cgi.
INTRODUCTION
Protein functional assignment has been conducted primarily by sequence similarity, clustering and pattern identification methods (1–7). These methods tend to become less effective for novel proteins that have no homolog or whose homolog is of different function (4,5,7,8). Genomes are known to contain a substantial portion of such novel proteins. For instance, 20–100% of the unknown putative protein-coding open reading frames in a number of recently sequenced viral genomes (9–12) are without a single homolog in Swiss-Prot database (13) based on PSI-BLAST search of that database as of September 2004. Hence, there is a need for exploring other functional prediction methods (14,15). Alternative approaches have been developed that explore structural features (16,17), interaction profiles (18,19), protein/gene fusion data (20,21) and functional family assignment by using statistical learning methods including discretized naïve Bayes, C4.5 decision trees, and instance-based leaning (22), neural networks (23) and support vector machines (SVMs) (22,24–29).
In particular, the possibility of using SVM for functional family assignment of distantly related proteins and homologous proteins of different functions has been raised based on testing results of a relatively small number of such proteins (25,27). However, the proteins used in these studies were selected based on BLAST instead of PSI-BLAST results. PSI-BLAST (30) is known to be significantly more sensitive to proteins of weak similarities than BLAST (1). Therefore, proteins selected based on PSI-BLAST results can, in a more critical manner, better test the capability of SVM functional classification of distantly related proteins, particularly those whose function cannot be assigned by sequence alignment and clustering methods. Moreover, the number of proteins used in earlier studies is relatively small, which may not be sufficient for testing the performance of SVM assignment of functional family of novel proteins.
In this work, two groups of enzymes, obtained from unbiased search of protein databases and literatures and subsequently verified by PSI-BLAST, are used to assess the capability of SVM for predicting the functional family of novel proteins. One group includes enzymes that are without a homolog in the protein databases based on PSI-BLAST search of these databases. A similarity E-value threshold of 0.05 is used for homolog searching to ensure maximum exclusion of enzymes that have a homolog. The second group contains pairs of homologous enzymes of different families. A stricter similarity E-value threshold of 10−6 is used for selecting these enzyme pairs to ensure minimum inclusion of non-homologous pairs. In the hypothetical situation that one enzyme in a pair of homologous enzymes of different families is newly discovered and the other is the only known protein of similar sequence, the function of the first enzyme can be incorrectly assigned to that of the second enzyme by using sequence similarity methods. Thus, it is of interest to examine to what extent SVM can be used as an alternative approach for facilitating functional assignment for these enzymes. These two groups of enzymes are further checked to remove those that are in the SVM training sets.
SVM is based on the structural risk minimization principle from statistical learning theory (31). For each protein functional family, it constructs a hyperplane either in an input space or a higher-dimensional hyper-space to maximally separate two groups of proteins, one group is composed of members and the other is composed of non-members of that family. Proteins in a training set, represented by their sequence-derived physicochemical properties, are projected onto this hyperspace where members of a family are separated from the non-members by a hyperplane whose parameters are adjusted by using a testing set of proteins. By projecting a new sequence onto the hyperspace, this SVM system can be used to determine whether it is a member of that family based on its location with respect to the hyperplane.
SVM classifies proteins into functional families defined from activities and physicochemical properties rather than sequence similarity (19,22,24,25,27,28,32). These families are composed of multiple homolog groups and some distantly related proteins. The accuracy of SVM depends on the diversity of the protein samples, the quality of the representation of protein properties, and the efficiency of the statistical learning algorithm. To a certain extent, no sequence similarity is required per se. Thus SVM is an attractive approach for facilitating the functional assignment of novel proteins.
METHODS
SVM protein functional family assignment system is developed in the following manner. First, every protein sequence is represented by specific feature vector assembled from encoded representations of tabulated residue properties including amino acid composition, hydrophobicity, normalized Van der Waals volume, polarity, polarizability, charge, surface tension, secondary structure and solvent accessibility for each residue in the sequence (19,22,24,25,32–34). Similar types of features have been successfully used for predicting enzyme functional (22) and structural classes (22,32) by using statistical learning methods.
Amino acid composition can be straightforwardly computed. Methods for computing each of the other properties can be found from the literature (19,24,25,33,34). For each of these properties, amino acids are divided into three groups such that those in a particular group are regarded to have the same property. For instance, amino acids can be divided into hydrophobic (CVLIMFW), neutral (GASTPHY) and polar (RKEDQN) groups. The groupings of amino acids for each of the properties are given in Table 1. Three descriptors, composition (C), transition (T) and distribution (D), are used to describe global composition of each of the properties. C is the number of amino acids of a particular property (such as hydrophobicity) divided by the total number of amino acids in a protein sequence. T characterizes the percent frequency with which amino acids of a particular property is followed by amino acids of a different property. D measures the chain length within which the first, 25, 50, 75 and 100% of the amino acids of a particular property is located respectively.
Table 1. Division of amino acids into three different groups for different physicochemical properties.
| Property | Group 1 | Group 2 | Group 3 |
|---|---|---|---|
| Hydrophobicity | |||
| Type | Polar | Neutral | Hydrophobic |
| Amino acids in the group | RKEDQN | GASTPHY | CVLIMFW |
| Van der Waals volume | |||
| Value | 0–2.78 | 2.95–4.0 | 4.43–8.08 |
| Amino acids in the group | GASCTPD | NVEQIL | MHKFRYW |
| Polarity | |||
| Value | 4.9–6.2 | 8.0–9.2 | 10.4–13.0 |
| Amino acids in the group | LIFWCMVY | PATGS | HQRKNED |
| Polarizability | |||
| Value | 0–0.108 | 0.128–0.186 | 0.219–0.409 |
| Amino acids | GASDT | CPNVEQIL | KMHFRYW |
A hypothetical protein sequence AEAAAEAEEAAAAAEAEEEAAEEAEEEAAE, as shown in Figure 1, has 16 alanines (n1 = 16) and 14 glutamic acids (n2 = 14). The compositions for these two amino acids are n1 × 100.00/(n1 + n2) = 53.33 and n2 × 100.00/(n1 + n2) = 46.67, respectively. There are 15 transitions from A to E or from E to A in this sequence and the percent frequency of these transitions is (15/29) × 100.00 = 51.72. The first, 25, 50, 75 and 100% of As are located within the first 1, 5, 12, 20, and 29 residues, respectively. The D descriptor for As is thus 1/30 × 100.00 = 3.33, 5/30 × 100.00 = 16.67, 12/30 × 100.00 = 40.0, 20/30 × 100.00 = 66.67, 29/30 × 100.00 = 96.67. Likewise, the D descriptor for Es is 6.67, 26.67, 60.0, 76.67, 100.0. Overall, the amino acid composition descriptors for this sequence are C = (53.33, 46.67), T = (51.72), and D = (3.33, 16.67, 40.0, 66.67, 96.67, 6.67, 26.67, 60.0, 76.67, 100.0), respectively. Descriptors for other properties can be computed by a similar procedure.
Figure 1.

The sequence of a hypothetic protein for illustration of derivation of the feature vector of a protein. Sequence index indicates the position of an amino acid in the sequence. The index for each type of amino acids in the sequence (A or E) indicates the position of the first, second, third, … of that type of amino acid (the position of the first, second, third, …, A is at 1, 3, 4, …). A/E transition indicates the position of AE or EA pairs in the sequence.
Overall, there are 21 elements representing these three descriptors: 3 for C, 3 for T and 15 for D (19,25). The feature vector of a protein is constructed by combining the 21 elements of all of these properties and the 20 elements of amino acid composition in sequential order. Table 2 gives the computed descriptors of the human insulin precursor (Swiss-Prot accession no. P01308). The feature vector of a protein is constructed by combining all of the descriptors in sequential order.
Table 2. Characteristic descriptors of human insulin precursor (Swiss-Prot AC P01308).
| Property | Elements of descriptors | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Amino acid composition | 9.09 | 5.45 | 1.82 | 7.27 | 2.73 | 10.91 | 1.82 | 1.82 | 1.82 | 18.18 |
| 1.82 | 2.73 | 5.45 | 6.36 | 4.55 | 4.55 | 2.73 | 5.45 | 1.82 | 3.64 | |
| Hydrophobicity | 24.55 | 38.18 | 37.27 | 15.60 | 16.51 | 30.28 | 5.45 | 40.91 | 54.55 | 80.00 |
| 100.0 | 1.82 | 21.82 | 47.27 | 68.18 | 98.18 | 0.91 | 12.73 | 37.27 | 72.37 | |
| 99.09 | ||||||||||
| Van der waals volume | 40.00 | 41.82 | 18.18 | 29.36 | 11.01 | 13.76 | 1.82 | 21.82 | 52.73 | 71.82 |
| 99.09 | 2.73 | 25.45 | 56.36 | 78.18 | 100.0 | 0.91 | 15.45 | 41.82 | 50.00 | |
| 98.18 | ||||||||||
| Polarity | 40.91 | 32.73 | 26.36 | 24.77 | 20.18 | 13.76 | 0.91 | 14.55 | 38.18 | 74.55 |
| 99.09 | 1.82 | 20.91 | 49.09 | 68.18 | 91.82 | 5.45 | 33.64 | 53.64 | 79.09 | |
| 100.0 | ||||||||||
| Polarizability | 29.09 | 52.73 | 18.18 | 31.19 | 9.17 | 15.60 | 1.82 | 21.82 | 52.73 | 68.18 |
| 91.82 | 2.73 | 25.45 | 56.36 | 79.09 | 100.0 | 0.91 | 15.45 | 41.82 | 50.00 | |
| 98.18 | ||||||||||
The feature vector of this protein is constructed by combining all of the descriptors in sequential order.
SVM is then trained by using representative proteins of a particular functional family (positive samples) and those that are outside this family (negative samples). The positive samples of a family include all of the known distinct proteins in that family. Because of the enormous number of proteins, the size of negative samples needs to be restricted to a manageable level by using a minimum set of representative proteins. One way for choosing representative proteins is to select one or a few distinct proteins from each protein domain family. The negative samples of a family can be selected from seed proteins of the 7316 curated protein families (domain-based) in the Pfam database (35) excluding those families that have at least one member belonging to the functional class. Pfam families are constructed on the basis of sequence similarity. The purpose of using Pfam proteins is to ensure that the negative samples are evenly distributed in the protein space. Sequence similarity is not required for selecting positive samples. In this sense, SVMProt is to some extent independent of sequence similarity.
The theory of SVM has been described in the literature (19,24,25,33,34). Thus only a brief description is given here. In linearly separable cases, SVM constructs a hyperplane that separates two different groups of feature vectors with a maximum margin. A feature vector is represented by xi, with physicochemical descriptors of a protein as its components. The hyperplane is constructed by finding another vector w and a parameter b that minimizes‖w‖2 and satisfies the following conditions:
![]()
![]()
where yi is the group index, w is a vector normal to the hyperplane, |b|/‖w‖ is the perpendicular distance from the hyperplane to the origin and ‖w‖2 is the Euclidean norm of w. After the determination of w and b, a given vector x can be classified by
![]()
In nonlinearly separable cases, SVM maps feature vectors into a high dimensional feature space using a kernel function K(xi, xj). An example of a kernel function is the Gaussian kernel, which has been extensively used in a number of protein classification studies (19,24,26,31,33,34,36):
![]()
The linear SVM procedure is then applied to the feature vectors in this feature space and the decision function for their classification is given by
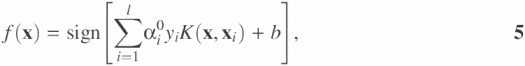
where the coefficients αi0 and b are determined by maximizing the following Langrangian expression:
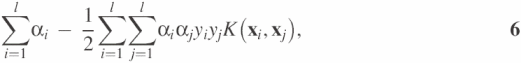
under conditions,
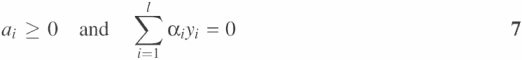
A positive or negative value from Equation 3 or 5 indicates that the vector x belongs to the positive or negative group, respectively. To further reduce the complexity of parameter selection, hard margin SVM with threshold instead of soft margin SVM with threshold is used in SVMProt.
Scoring of SVM classification of proteins has been estimated by a reliability index and its usefulness has been demonstrated by statistical analysis (34). A slightly modified reliability score, R-value, is used in SVMProt:
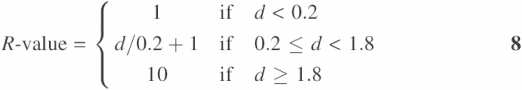
where d is the distance between the position of the vector of a classified protein and the optimal separating hyperplane in the hyperspace. There is a statistical correlation between R-value and expected classification accuracy (probability of correct classification) (34). Thus another quantity, P-value, is introduced to indicate the expected classification accuracy. P-value is derived from the statistical relationship between the R-value and actual classification accuracy based on the analysis of 9932 positive and 45 999 negative samples of proteins (25).
RESULTS AND DISCUSSION
The protein functional family prediction system SVMProt is improved by using training sets of a significantly larger number of proteins than that reported earlier (25,27). The training and testing sets consist of 49 975 representative enzymes from 46 functional families obtained from UniProt version 1.6, and 243 152 non-enzyme representative proteins from 7316 Pfam curated protein families (35). Enzyme functional families are the International Commission (EC) classes (37) up to the second level (from EC1.1 to EC6.5). The procedure for selecting positive samples of a family is as follows. First, all members of this family in UniProt 1.6 are collected and subsequently mapped into the original feature space which is divided into small grid blocks, then one or a few distinct enzymes are selected from those distributed in each of these blocks are selected as the training set of that family. Enzymes in the testing and independent sets are randomly selected from the remaining pool of family members. The negative samples of a family are selected from representative proteins of Pfam families that are non-enzymes or enzymes of other enzyme families.
The statistics of the datasets and the prediction results as well as SVMProt can be accessed at http://jing.cz3.nus.edu.sg/cgi-bin/svmprot.cgi. An independent set of 13 891 enzymes and 122 710 non-enzymes are used to assess the capability of SVM for assignment of enzymes into their respective family (sensitivity) and for assignment of non-member proteins outside that family (specificity). The sensitivity is >85% for 9 families, 70–85% for 21 families, 60–70% for 10 families and 53–60% for 6 families. The specificity is >95% for 38 families and 82–95% for 8 families.
The overall sensitivity for all of the 13 891 enzymes is 86%, which is improved against the accuracy of 68% for the assignment of 14 709 enzymes into their respective EC second level class by using one or more of the three statistical learning methods discretized naïve Bayes, C4.5 decision trees, and instance-based leaning (22). SVM has also been used for classification of enzymes into structural families irrespective of sequence similarity, and the accuracy for assignment of 1178 enzymes is 80% (32). These suggest that statistical learning methods are useful for functional and structural family assignment. The overall sensitivity is however slightly lower than that of 92% for the BLAST assignment of the EC class of 12 900 enzymes (38). Non-the-less, as these are to a certain extent independent of sequence similarity, statistical learning methods such as SVM are useful alternative for studying novel proteins whose function cannot be assigned on the basis of sequence similarity.
Enzymes without a homolog of known function are searched from the Swiss-Prot database (13) by using the key word ‘novel’, ‘distinct’, or ‘unrelated’ combined with ‘enzyme’. The next step is to eliminate those with at least one homolog of known function (except for hypothetical proteins) by conducting a PSI-BLAST (1) search against the NR databases that include all non-redundant GenBank, CDS translations, PDB, Swiss-Prot, PIR and PRF databases. This ensures that only those truly having no homolog in protein databases are selected. While the selected enzymes from this process are without a homolog, their function has been determined experimentally and these have been reported in the literature and subsequently described in the Swiss-Prot database. The last step is to remove those present in the SVMProt training sets.
Table 3 gives the 12 enzymes without a homolog in the NR databases (group NR) and additional 38 enzymes without a homolog in the Swiss-Prot database (group SP) selected from this process, none of which are in the SVM training sets; 8 out of 12 (67%) enzymes in group NR and 28 out of 38 (73.7%) enzymes in group SP are correctly assigned to the respective family by SVMProt. The overall accuracy is 72% which is comparable to the average sensitivity for the enzyme families and it is consistent with the sequence-similarity-independent nature of SVM functional assignment. To further facilitate the testing of SVMProt for functional family assignment of novel proteins, a number of proteins of unknown function are selected. These proteins are either without a homolog or without functional indication in Swiss-Prot or NR database as of September 2004 based on PSI-BLAST search. The predicted functional classes of these proteins are given in the Supplementary Material.
Table 3. List of enzymes without a homolog in the NR and Swiss-Prot databases and the results of SVM functional family assignment.
| Enzyme (EC number) [Swiss-Prot accession number] | Database containing no homolog | SVM assigned functional family (probability of correct prediction) | Assignment status |
|---|---|---|---|
| Thiocyanate hydrolase beta subunit (EC 3.5.5.8) [O66186]. | NR | EC 3.5 Hydrolase of non-peptide carbon–nitrogen bonds (98.9%) | + |
| EC 2.6 Transferases of nitrogenous groups (62.2%) | |||
| Potential cysteine protease avirulence protein avrPpiC2 (EC 3.4.22.-) [Q9F3T4]. | NR | EC 4.2 Carbon–oxygen lyase (93.6%) | − |
| EC 2.3 Acyltransferase (83.9%) | |||
| EC 4.1 Carbon–carbon lyase (71.3%) | |||
| Outer membrane (58.6%) | |||
| Extracellular phospholipase (EC 3.1.1.5) [P82476] | NR | EC 3.1 Hydrolase of ester bonds (98.7%) | + |
| Cytochrome c oxidase polypeptide IV, mitochondrial precursor (EC 1.9.3.1) [P30815]. | NR | EC 1.9 Oxidoreductase of a heme group of donors (99.0%) | + |
| Cytochrome c oxidase polypeptide VI (EC 1.9.3.1) [P26310]. | NR | EC 1.9 Oxidoreductase of a heme group of donors (98.4%) | + |
| Transmembrane (98.3%) | |||
| EC 3.1 Hydrolase of ester bonds (62.2%) | |||
| Alginate lyase precursor (EC4.2.2.3) [P39049]. | NR | Transmembrane (65.4%) | − |
| Outer membrane (58.6%) | |||
| EC 2.1 Transferase of one-carbon groups (58.6%) | |||
| DNA α-glucosyltransferase (EC 2.4.1.26) [P04519] | NR | EC 2.4 Glycosyltransferase (80.4%); | + |
| EC 2.7 Transferase of phosphorus-containing groups (68.5%) | |||
| Endonuclease CviAII (EC 3.1.21.4 [P31117] | NR | EC 3.1 Hydrolase of ester bonds (99.0%) | + |
| Type II restriction enzyme CviJI (EC 3.1.21.4) [P52283] | NR | EC 3.1 Hydrolase of ester bonds (99.0%); | + |
| rRNA-binding proteins (98.8%); | |||
| EC 3.4 Peptidase (68.5%) | |||
| DNA-directed RNA polymerase, subunit 10 homolog (EC 2.7.7.6) [P42488] | NR | EC 2.7 Transferase of phosphorus-containing groups (99.0%) | + |
| 7 transmembrane receptor metabotropic glutamate family (58.6%) | |||
| Endonuclease IV (EC 3.1.21.-) [P39250] | NR | No function predicted | − |
| Beta-agarase precursor (EC3.2.1.81) [P13734]. | NR | EC 4.1 Carbon–carbon lyase (96.7%) | − |
| EC 2.4 Glycosyltransferase (71.3%) | |||
| Phenylacetaldoxime dehydratase (EC 4.2.1.-) [P82604]. | Swiss-Prot | Transmembrane (98.2%) | − |
| EC 3.4 Peptidase (96.4%) | |||
| EC 3.3 Hydrolase of ether bonds (80.4%) | |||
| EC 2.7 Transferase of phosphorus-containing groups (73.8%) | |||
| ATP synthase H chain, mitochondrial precursor (EC3.6.3.14) [Q12349]. | Swiss-Prot | EC 3.6 Hydrolase of acid anhydrides (99.0%) | + |
| RNA-binding protein (58.6%) | |||
| Peptide-N(4)-(N-acetyl-β-d-glucosaminyl)asparagine amidase F precursor (EC 3.5.1.52) [P21163] | Swiss-Prot | EC 3.5 Hydrolase of non-peptide carbon–nitrogen bonds (99.0%) | + |
| Beta-Barrel porin (58.6%) | |||
| S-Adenosyl-l-methionine hydrolase (EC 3.3.1.2) [P07693] | Swiss-Prot | EC 3.3 Hydrolase of ether bonds (99.0%) | + |
| EC 2.7 Transferase of phosphorus-containing groups (71.3%) | |||
| DNA-binding protein (65.4%) | |||
| Hypothetical 52.8 kDa protein in VPS15-YMC2 intergenic region. (EC 3.1.22.-) [P38257] | Swiss-Prot | DNA-binding protein (89.3%) | − |
| Outer membrane (58.6%) | |||
| Hypothetical protein BBB03 (EC3.1.22.-) [O50979]. | Swiss-Prot | EC 2.7 Transferase of phosphorus-containing groups (88.1%) | − |
| EC 3.4 Peptidase (86.8%) | |||
| EC 2.3 Acyltransferase (71.3%) | |||
| EC 4.1 Carbon–carbon lyase (65.4%) | |||
| Telomere elongation protein (EC2.7.7.-) [P17214]. | Swiss-Prot | EC 2.7 Transferase of phosphorus-containing groups (99.1%) | + |
| DNA-binding protein (78.4%) | |||
| Fucose-1-phosphate guanylyltransferase (EC 2.7.7.30) [O14772] | Swiss-Prot | EC 2.7 Transferase of phosphorus-containing groups (99.1%) | + |
| 7 transmembrane receptor metabotropic glutamate family (58.6%) | |||
| DNA-directed RNA polymerase I 14 kDa polypeptide (EC 2.7.7.6) [P50106]. | Swiss-Prot | EC 2.7 Transferase of phosphorus-containing groups (99%) | + |
| DNA-binding protein (62.2%) | |||
| Beta-Barrel porin (58.6%) | |||
| EC 3.4 Peptidase (58.6%) | |||
| DNA polymerase III, theta subunit (EC 2.7.7.7) [P28689]. | Swiss-Prot | EC 2.7 Transferase of phosphorus-containing groups (99.0%) | + |
| EC 4.2 Carbon–oxygen lyase (58.6%) | |||
| Cytochrome c oxidase polypeptide IV (EC 1.9.3.1) [P77921] | Swiss-Prot | EC 1.9 Oxidoreductase of a heme group of donors (97.0%) | + |
| Envelope protein (58.6%) | |||
| Transmembrane (58.6%) | |||
| Cytochrome c oxidase polypeptide VII (EC 1.9.3.1) [P10174]. | Swiss-Prot | EC 1.9 Oxidoreductase of a heme group of donors (98.3%) | + |
| Transmembrane (58.6%) | |||
| Cytochrome c oxidase polypeptide VIII, mitochondrial precursor (EC 1.9.3.1) [P04039]. | Swiss-Prot | EC 1.9 Oxidoreductase of a heme group of donors (99.0%) | + |
| Transmembrane (58.6%) | |||
| RNA-binding protein (58.6%) | |||
| Cytochrome c oxidase polypeptide VIIA precursor (EC1.9.3.1) [P07255]. | Swiss-Prot | EC 1.9 Oxidoreductase of a heme group of donors (97.8%) | + |
| Transmembrane (93.8%) | |||
| EC 1.10 Oxidoreductase of diphenols and related substances as donors (58.6%) | |||
| Alpha-type channel (58.6%) | |||
| Heme-copper oxidase subunit IV (EC 1.9.3.-) [Q9YDX4]. | Swiss-Prot | EC 1.9 Oxidoreductase of a heme group of donors (99.0%) | + |
| Transmembrane (99.0%) | |||
| Aminoglycoside 2′-N-acetyltransferase (EC 2.3.1.-) [P95219] | Swiss-Prot | EC 2.7 Transferase of phosphorus-containing groups (78.4%) | − |
| EC 4.2 Carbon–oxygen lyase (58.6%) | |||
| Glycosyl transferase alg8 (EC2.4.1.-) [Q887P9]. | Swiss-Prot | Transmembrane (99.0%) | * |
| EC 2.4 Glycosyltransferase (98.6%) | |||
| Beta-agarase B (EC 3.2.1.81) [P48840]. | Swiss-Prot | Outer membrane (58.6%) | − |
| Beta-Barrel porin (58.6%) | |||
| CM (EC 5.4.99.5) [P19080] | Swiss-Prot | EC 5.4. Intramolecular transferase (99.0%) | + |
| EC 4.2. Carbon–oxygen lyase (58.6%) | |||
| Outer membrane (58.6%) | |||
| DNA β-glucosyltransferase (EC 2.4.1.27) [P04547] | Swiss-Prot | EC 2.4 Glycosyltransferases (95.7%); | + |
| EC 2.5 Transferase of alkyl or aryl groups, other than methyl groups (80.4%) | |||
| dNMPkinase (EC 2.7.4.13) [P04531] | Swiss-Prot | EC 2.7 Transferase of phosphorus-containing groups (99.0%); | + |
| EC 2.4 Glycosyltransferase (96.4%); | |||
| EC 1.1 Oxidoreductase of the CH-OH group of donors (71.3%) | |||
| Endonuclease II (EC 3.1.21.1) [P07059] | Swiss-Prot | EC 3.1 Hydrolase of ester bonds (99.0%) | + |
| Endonuclease V (EC 3.1.25.1) [P04418] | Swiss-Prot | EC 3.1 Hydrolase of ester bonds (99.0%) | + |
| Exonuclease (EC 3.1.11.3) [P03697] | Swiss-Prot | EC 3.1 Hydrolase of ester bonds (99.0%); | + |
| EC 4.1 Carbon–carbon lyases (88.1%); | |||
| EC 2.7 Transferase of phosphorus-containing groups (68.5%); | |||
| EC 1.1 Oxidoreductase of the CH-OH group of donors (58.6%) | |||
| Ribonuclease (EC 3.1.-.-)[P13312] | Swiss-Prot | EC 3.1 Hydrolase of ester bonds (99.0%) | + |
| Intron-associated endonuclease 1 (EC 3.1.-.-) [P13299] | Swiss-Prot | EC 3.1 Hydrolase of ester bonds (99.0%); | + |
| DNA-binding protein (83.9%) | |||
| Intron-associated endonuclease 2 (EC 3.1.-.-) [P07072] | Swiss-Prot | EC 3.1 Hydrolase of ester bonds (99.0%) | + |
| Putative adenine-specific methylase (EC 2.1.1.72) [P51715] | Swiss-Prot | EC 2.1 Transferase of one-carbon groups (99.0%); | + |
| Outer membrane (58.6%); | |||
| mRNA-binding protein (58.6%) | |||
| Protein kinase (EC 2.7.1.37) [P00513] | Swiss-Prot | EC 2.7 Transferase of phosphorus-containing groups (99.0%) | + |
| Slt35 (EC 3.2.1.-) [P41052] | Swiss-Prot | Outer membrane (99.0%) | − |
| EC 1.1. Oxidoreductase acting on the CH-OH group of donors (89.3%) | |||
| EC 4.1. Carbon–carbon lyase (62.2%) | |||
| Ammonia monooxygenase (EC 1.13.12.-)[Q04508] | Swiss-Prot | EC 1.13. oxygenase (99.0%) | + |
| Transmembrane (99.0%) | |||
| EC 2.4. Glycosyltransferases (83.9%) | |||
| 2-Aminomuconate deaminase (EC 3.5.99.5) [P81593] | Swiss-Prot | EC 3.5. Hydrolase acting on carbon–nitrogen bonds, other than peptide bonds (99.0%) | + |
| EC 3.4. Peptidase (58.6%) | |||
| ADP-ribosyltransferase (EC2.4.2.37) [P14299] | Swiss-Prot | Transmembrane (92.9%) | * |
| EC 2.4. Glycosyltransferase (90.3%) | |||
| Outer membrane (58.6%) | |||
| Alpha-N-AFase II (EC 3.2.1.55) [P82594] | Swiss-Prot | EC 3.4. Peptidase (91.3%) | − |
| Aminopeptidase G (EC 3.4.11.-) [Q54340] | Swiss-Prot | EC 3.4. Peptidase (99.0%) | + |
| TC 1.C. Channels/pores—pore-forming toxins (proteins and peptides) (58.6%) | |||
| Alginate lyase (EC 4.2.2.3) [Q59478] | Swiss-Prot | Transmembrane (96.4%) | − |
| EC 3.1. Hydrolase of ester bonds (78.4%) | |||
| Outer membrane (58.6%) | |||
| ATPE_YEAST (EC 3.6.3.14) [P21306] | Swiss-Prot | RNA-binding proteins (58.6%) | − |
| AhdA2cA1c (EC1.14.-.-) [BAC65427.1] | Swiss-Prot | EC 3.1. Hydrolase of ester bonds (82.2%) | − |
| DNA-binding protein (80.4%) | |||
| Transmembrane (58.6%) |
The symbol +, * and − represent the cases that the top of the predicted family, one of the predicted families, and none of the predicted families matches the enzyme function, respectively.
There are eight pairs of homologous enzymes of different families from previous publications (8,27) that satisfy the stricter criterion, which together with SVMProt predicted top family for each enzyme are given in Table 4. It is found that 5 or 62% of these enzyme pairs are correctly assigned by SVMProt, such an accuracy is comparable to the average sensitivity for the enzyme families and indicative of the sequence-similarity-independent nature of SVM functional assignment.
Table 4. List of pairs of homologous enzymes of different families and the results of SVM functional family assignment.
| Enzyme E1 (Swiss-Prot accession number) | EC class (F1) | Enzyme E2 (Swiss-Prot accession number) | EC class (F2) | Sequence similarity (BLAST E-value) | SVM functional family assignment | Assignment status |
|---|---|---|---|---|---|---|
| Glycolateoxidase (P05414) | EC 1.1 | IPP isomerase (Q8PW37) | EC 5.3 | 3.00E−07 | E1->F1;E2->F2 | + |
| Creatine amidinohydrolase (P38488) | EC 3.5 | Prolinedipeptidase (O58885) | EC 3.4 | 3.00E−15 | E1->F1;E2->F2 | + |
| Cystathionine gamma-synthase (P38675) | EC 2.5 | Methionine gamma-lyase (P13254) | EC 4.4 | 2.00E−15 | E1->W;E2->F2 | − |
| Exocellobiohydrolase 1 (P38676) | EC 3.2 | Cystathionine gamma-lyase (Q8VCN5) | EC 4.4 | 1.00E−12 | E1->W;E2->F2 | − |
| Maleylacetoacetate isomerase (P57109) | EC 5.2 | Glutathione S-transferase zeta class (P57108) | EC 2.5 | 1.00E−51 | E1->F1;E2->F2 | + |
| Tyrosine-protein kinase FRK (P42685) | EC 2.7 | Intestinalguanylate cyclase (P70106) | EC 4.6 | 2.60E−12 | E1->F1;E2->F1 | − |
| Glutamate-1-semialdehyde aminotransferase (Q06774) | EC 5.4 | 4-aminobutyrate aminotransferase (P22256) | EC 2.6 | 5.70E−32 | E1->F1;E2->F2 | + |
| Exodeoxyribonuclease (P37454) | EC 3.1 | DNA- (apurinic or apyrimidinic site) lyase (P43138) | EC 4.2 | 1.60E−96 | E1->F1;E2->F2 | + |
E1-> F1 or E2 -> F2 indicates that enzyme E1 or E2 is assigned into family F1 and F2 respectively. E1-> W or E2 -> W indicates that enzyme E1 or E2 is assigned into a wrong family respectively. The symbol + or − represents the cases that SVM is able or unable to distinguish the two enzymes and exclusively assign them into the respective family.
These results suggest that SVM has some capability for functional family assignment of novel proteins having no homolog, and for distinguishing homologous proteins of different functions. The overall accuracy of SVM is not yet at the same level of that of sequence alignment for homologous proteins. One reason is the imbalance between the number of positive and negative samples. The total number of distinct enzymes in some families is <200, which is significantly smaller than that of a few thousand representative proteins used as the negative samples of the respective family. Such a large data imbalance is known to affect the accuracy of a SVM classification system, and methods for solving these problems are being developed (39). It is likely that not all possible types of proteins, particularly those of distantly related members, are adequately represented in some families. This can be improved along with the availability of more protein data. Not all distantly related proteins of the same function have similar structural and chemical features due to the flexibility at the active site (17). This plasticity needs to be properly formulated. These improvements will enable the development of SVM into a useful tool for facilitating functional study of novel proteins.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
REFERENCES
- 1.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baxevanis A.D. (1998) Practical aspects of multiple sequence alignment. Methods Biochem. Anal., 39, 172–188. [DOI] [PubMed] [Google Scholar]
- 3.Bork P. and Koonin,E.V. (1998) Predicting functions from protein sequences—where are the bottlenecks? Nature Genet., 18, 313–318. [DOI] [PubMed] [Google Scholar]
- 4.Eisen J.A. (1998) Phylogenomics: improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res., 8, 163–167. [DOI] [PubMed] [Google Scholar]
- 5.Enright A.J. and Ouzounis,C.A. (2000) GeneRAGE: a robust algorithm for sequence clustering and domain detection. Bioinformatics, 16, 451–457. [DOI] [PubMed] [Google Scholar]
- 6.Hodges H.C. and Tsai,J.W. (2002) 3D-Motifs: an informatics approach to protein function prediction. FASB J., 16, A543. [Google Scholar]
- 7.Whisstock J.C. and Lesk,A.M. (2003) Prediction of protein function from protein sequence and structure. Q. Rev. Biophys., 36, 307–340. [DOI] [PubMed] [Google Scholar]
- 8.Rost B. (2002) Enzyme function less conserved than anticipated. J. Mol. Biol., 318, 595–608. [DOI] [PubMed] [Google Scholar]
- 9.He R., Dobie,F., Ballantine,M., Leeson,A., Li,Y., Bastien,N., Cutts,T., Andonov,A., Cao,J., Booth,T.F. et al. (2004) Analysis of multimerization of the SARS coronavirus nucleocapsid protein. Biochem. Biophys. Res. Commun., 316, 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makeyev E.V. and Bamford,D.H. (2004) Evolutionary potential of an RNA virus. J. Virol., 78, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rustici G., Milne,R.G. and Accotto,G.P. (2002) Nucleotide sequence, genome organisation and phylogenetic analysis of Indian citrus ringspot virus. Brief report. Arch. Virol., 147, 2215–2224. [DOI] [PubMed] [Google Scholar]
- 12.Sabanadzovic S., Ghanem-Sabanadzovic,N.A., Saldarelli,P. and Martelli,G.P. (2001) Complete nucleotide sequence and genome organization of Grapevine fleck virus. J. Gen. Virol., 82, 2009–2015. [DOI] [PubMed] [Google Scholar]
- 13.Boeckmann B., Bairoch,A., Apweiler,R., Blatter,M.C., Estreicher,A., Gasteiger,E., Martin,M.J., Michoud,K., O'Donovan,C., Phan,I. et al. (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res., 31, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eisenberg D., Marcotte,E.M., Xenarios,I. and Yeates,T.O. (2000) Protein function in the post-genomic era. Nature, 405, 823–826. [DOI] [PubMed] [Google Scholar]
- 15.Smith T.F. and Zhang,X. (1997) The challenges of genome sequence annotation or ‘the devil is in the details’. Nat. Biotechnol., 15, 1222–1223. [DOI] [PubMed] [Google Scholar]
- 16.Teichmann S.A., Murzin,A.G. and Chothia,C. (2001) Determination of protein function, evolution and interactions by structural genomics. Curr. Opin. Struct. Biol., 11, 354–363. [DOI] [PubMed] [Google Scholar]
- 17.Todd A.E., Orengo,C.A. and Thornton,J.M. (2001) Evolution of function in protein superfamilies, from a structural perspective. J. Mol. Biol., 307, 1113–1143. [DOI] [PubMed] [Google Scholar]
- 18.Aravind L. (2000) Guilt by association: contextual information in genome analysis. Genome Res., 10, 1074–1077. [DOI] [PubMed] [Google Scholar]
- 19.Bock J.R. and Gough,D.A. (2001) Predicting protein–protein interactions from primary structure. Bioinformatics, 17, 455–460. [DOI] [PubMed] [Google Scholar]
- 20.Enright A.J., Iliopoulos,I., Kyrpides,N.C. and Ouzounis,C.A. (1999) Protein interaction maps for complete genomes based on gene fusion events. Nature, 402, 86–90. [DOI] [PubMed] [Google Scholar]
- 21.Marcotte E.M., Pellegrini,M., Ng,H.L., Rice,D.W., Yeates,T.O. and Eisenberg,D. (1999) Detecting protein function and protein–protein interactions from genome sequences. Science, 285, 751–753. [DOI] [PubMed] [Google Scholar]
- 22.des Jardins M., Karp,P.D., Krummenacker,M., Lee,T.J. and Ouzounis,C.A. (1997) Prediction of enzyme classification from protein sequence without the use of sequence similarity. Proc. Int. Conf. Intell. Syst. Mol. Biol., 5, 92–99. [PubMed] [Google Scholar]
- 23.Jensen L.J., Gupta,R., Blom,N., Devos,D., Tamames,J., Kesmir,C., Nielsen,H., Staerfeldt,H.H., Rapacki,K., Workman,C. et al. (2002) Prediction of human protein function from post-translational modifications and localization features. J. Mol. Biol., 319, 1257–1265. [DOI] [PubMed] [Google Scholar]
- 24.Karchin R., Karplus,K. and Haussler,D. (2002) Classifying G-protein coupled receptors with support vector machines. Bioinformatics, 18, 147–159. [DOI] [PubMed] [Google Scholar]
- 25.Cai C.Z., Han,L.Y., Ji,Z.L., Chen,X. and Chen,Y.Z. (2003) SVM-Prot: web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res., 31, 3692–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai Y.D. and Lin,S.L. (2003) Support vector machines for predicting rRNA-, RNA-, and DNA-binding proteins from amino acid sequence. Biochim. Biophys. Acta, 1648, 127–133. [DOI] [PubMed] [Google Scholar]
- 27.Cai C.Z., Han,L.Y., Ji,Z.L. and Chen,Y.Z. (2004) Enzyme family classification by support vector machines. Proteins, 55, 66–76. [DOI] [PubMed] [Google Scholar]
- 28.Han L.Y., Cai,C.Z., Lo,S.L., Chung,M.C. and Chen,Y.Z. (2004) Prediction of RNA-binding proteins from primary sequence by a support vector machine approach. RNA, 10, 355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhasin M. and Raghava,G.P. (2004) Classification of nuclear receptors based on amino acid composition and dipeptide composition. J. Biol. Chem., 279, 23262–23266. [DOI] [PubMed] [Google Scholar]
- 30.Altschul S.F. and Koonin,E.V. (1998) Iterated profile searches with PSI-BLAST—a tool for discovery in protein databases. Trends Biochem. Sci., 23, 444–447. [DOI] [PubMed] [Google Scholar]
- 31.Burges C. (1998) A tutorial on support vector machine for pattern recognition. Data Min. Knowl. Disc., 2, 121–167. [Google Scholar]
- 32.Dobson P.D. and Doig,A.J. (2003) Distinguishing enzyme structures from non-enzymes without alignments. J. Mol. Biol., 330, 771–783. [DOI] [PubMed] [Google Scholar]
- 33.Ding C.H. and Dubchak,I. (2001) Multi-class protein fold recognition using support vector machines and neural networks. Bioinformatics, 17, 349–358. [DOI] [PubMed] [Google Scholar]
- 34.Hua S. and Sun,Z. (2001) A novel method of protein secondary structure prediction with high segment overlap measure: support vector machine approach. J. Mol. Biol., 308, 397–407. [DOI] [PubMed] [Google Scholar]
- 35.Bateman A., Coin,L., Durbin,R., Finn,R.D., Hollich,V., Griffiths-Jones,S., Khanna,A., Marshall,M., Moxon,S., Sonnhammer,E.L. et al. (2004) The Pfam protein families database. Nucleic Acids Res., 32, D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan Z., Burrage,K. and Mattick,J.S. (2002) Prediction of protein solvent accessibility using support vector machines. Proteins, 48, 566–570. [DOI] [PubMed] [Google Scholar]
- 37.Enzyme-Nomenclature. (1992) Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Academic Press, NY. [Google Scholar]
- 38.Shah I. and Hunter,L. (1997) Predicting enzyme function from sequence: a systematic appraisal. Proc. Int. Conf. Intell. Syst. Mol. Biol., 5, 276–283. [PMC free article] [PubMed] [Google Scholar]
- 39.Kim H. and Park,H. (2004) Prediction of protein relative solvent accessibility with support vector machines and long-range interaction 3D local descriptor. Proteins, 54, 557–562. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.