

Abstract
Optical modulation of proteins provides superior spatiotemporal resolution for understanding biological processes, and photoswitches built on light-sensitive proteins have been significantly advancing neuronal and cellular studies. Small molecule photoswitches could complement protein-based switches by mitigating potential interference and affording high specificity for modulation sites. However, genetic encodability and responsiveness to non-ultraviolet light, two desired properties possessed by protein photoswitches, are challenging to be engineered into small molecule photoswitches. Here we developed a small molecule photoswitch that can be genetically installed onto proteins in situ and controlled by visible light. A pentafluoro azobenzene based photoswitchable click amino acid (F-PSCaa) was designed to isomerize in response to visible light. After genetic incorporation into proteins via the expansion of the genetic code, F-PSCaa reacts with a nearby cysteine within the protein generating an azo bridge in situ. The resultant bridge built by F-PSCaa is switchable by visible light and allows conformation and binding of CaM to be regulated by such light. This photoswitch should prove valuable in optobiology for its minimal interference, site flexibility, genetic encodability, and response to the more biocompatible visible light.
Graphical Abstract
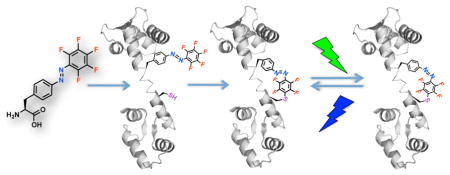
The ability to control protein function with light provides excellent temporal and spatial resolution for precise investigation in situ, and thus is having significant impact on biological studies. Light-sensitive proteins and domains have been exploited for optical modulation of neuronal and cellular processes.1 The bulk of such domains and proteins may interfere with the native function, location, and trafficking of the target protein, limiting aspects of protein functions to be controlled. Installation of photo responsiveness onto proteins using small molecules can mitigate such interference. One elegant approach is to chemically tether a photoswitchable ligand to receptors and channels.2 Unfortunately, ligand tethering has been limited to the extracellular side of membrane proteins. Another approach links two appropriately distanced cysteine residues using symmetric photoswitches bearing two reactive groups,3 but such bivalent reactive units cannot be used in vivo. In addition, while light-sensitive proteins and domains can be genetically encoded providing high selectivity in targeting proteins, cells and tissues, genetically encoding photoswitchable small molecules remains challenging.4–5
In order to selectively install small molecule photoswitches into proteins in vivo for optical modulation, we recently genetically encoded a new class of unnatural amino acids (Uaas), the PhotoSwitchable Click amino acids (PSCaa), into proteins in E. coli and mammalian cells.5 The PSCaa consists of an azobenzene side chain extended by a reactive group, which reacts with a nearby cysteine to form a photoswitchable bridge on the protein in situ.5–6 A drawback of the initial generation of PSCaas is that they isomerize in response to UV light (λ = 365 nm) only, which cannot penetrate deep into tissues and may negatively impact biological processes under study. Here we present the design and genetic incorporation of a new PSCaa, F-PSCaa 1, which generates a photoswitchable protein bridge in situ through a reaction mechanism unreported for proximity-enabled bioreactivity7 (Scheme 1). In addition, 1 enables photoswitching to be modulated by visible light. This new photoswitch should prove valuable for optical modulation of proteins with high specificity and biocompatibility in vivo.
Scheme 1.

In situ formation of an azo bridge on proteins controllable by visible light. The F-PSCaa 1, (S,E)-2-amino-3-(4-((pentafluorophenyl)diazenyl)phenyl)propanoic acid, is genetically incorporated into proteins in vivo through the expansion of the genetic code. After incorporation, 1 reacts with a nearby cysteine residue via nucleophilic aromatic substitution (SNAr) to form an azo bridge in situ. Both the F-PSCaa and the resultant bridge isomerize in response to visible light.
Substitution on azobenzene has been shown to shift the wavelengths of light driving the cis-trans isomerization. Bulky electron-rich substituents, when installed on the four ortho positions or the para position to the azo group, result in azobenzene derivatives responding to green or red light;8 fluorine atoms substituted at the four ortho positions also shifts azobenzene absorbance to visible light.9 However, bulky substituents are not appropriate for PSCaa because resultant large side chain may make it infeasible to evolve an aminoacyl-tRNA synthetase for genetic incorporation of the PSCaa. We decided to use pentafluorination in designing F-PSCaa 1 based on the following considerations: 1) fluorine has similar size to proton and does not significantly change the bulk of the azobenzene unit; 2) perfluorination of a photoswitch unit can modulate its photochromic performance and shift its absorbance, but it has never been used before to generate a visible light-switchable azobenzene;10 3) perfluoro-benzene allows nucleophilic aromatic substitution (SNAr),11 which may be harnessed for proximity-enabled bioreactivity7 to generate a protein bridge in situ.
We synthesized 1, (S,E)-2-amino-3-(4-((pentafluoro phenyl)diazenyl) phenyl) propanoic acid, by Mills coupling of the Boc-protected 4-amino phenylalanine with the 1,2,3,4,5-pentafluoro-6-nitrosobenzene, which was prepared from 1,2,3,4,5-pentafluoro-6-aminobenzene using Oxone (Figure S1). Subsequent treatment with trifluoroacetic acid gave the final pentafluoro azobenzene amino acid F-PSCaa in acceptable yield and high purity over two steps (44%).
To investigate whether 1 is able to isomerize in response to visible light, we illuminated the amino acid with green or blue light, and determined the E/Z ratios of the corresponding photostationary state (pss) by HPLC. As expected, illumination of 1 with green light (λ = 540 nm) results in E-to-Z isomerization yielding 78% of the Z isomer (Figure 1a, top LC profile). A similar ratio was achieved using common UV activation (λ = 365 nm) (Figure 1a, middle LC profile). Illumination using blue light (λ = 405 nm) results in Z-to-E isomerization restoring 73% of the E photoisomer (Figure 1a, bottom LC profile). The UV-vis spectra of each pure photoisomer show typical features of azobenzene: two absorbance peaks were observed representing π-π* and n-π* transitions, respectively; upon E to Z isomerization the π-π* absorption decreased while the n-π* absorption increased (Figure 1b). Notably, pentafluoro substitution resulted in distinct separation in wavelength of both transition bands between the Z and E isomer, and the separation at the n-π* transition band (Figure 1b insert) enabled the use of green light for selective formation of the Z isomer as observed.
Figure 1.

(a) LC profiles of the photo-equilibrium after illumination of F-PSCaa 1 with green light (λ = 540 nm, top), UV light (λ = 365 nm, middle) and blue light (λ = 405 nm, bottom), respectively. (b) UV-vis spectra of the pure E photoisomer (black) and Z photoisomer (red) of 1. Insert zooms in on the n-π* transition band.
To determine whether F-PSCaa (1) can undergo SNAr with thiols,11 we incubated 1 with Boc-protected cysteine (2) at pH 7.5 and monitored the reaction over time by HPLC-MS. After 2 h of incubation the product 3 was formed (Figure S2). The reaction was also detectable by UV-vis spectroscopy (Figure S3). The absorbance spectrum of the product in E form shows a bathochromic shift as a result of the electron-donating sulfur in para position to the azo group (Figure S3). The π-π* transition band red-shifted from 326 nm (for E form of 1) to 340 nm (for E form of the product 3). Electron donating groups in para to the azo unit can significantly decrease the thermal stability of the Z state,10b however, we didn’t observe a decrease in thermal stability of the Z form of 3. E-to-Z photoisomerization of the product 3 could also be induced by green light illumination at λ = 540 nm, generating an E/Z ratio of 23:77 (Figure S5).
The presence of fluorine on 1 enabled us to monitor the SNAr reaction of 1 with 2 in DMSO-d6 using 19F-NMR 12 (Figure 2). The 19F NMR spectrum of 1 showed three signals (Figure S11b): a triplet at −155.25 ppm (1F, para), a quartet at −153.95 ppm (2F, ortho), and a doublet of triplets at −164.99 ppm (2F, meta, not shown in the Figure 2). After adding 2, as a result of the nucleophilic attack of cysteine’s thiol at the fluorine in para position to the azo unit, the intensity of the triplet at −155.25 pm for the substituted para 1F decreased with time. Meanwhile, signals for the meta and ortho 2F atoms also decreased with a concomitant arising of two new quartet signals at −136.21 ppm (2F, meta, not shown in Figure 2) and −154.45 (2F, ortho), indicating complete conversion after ~6 h at 25 °C (Figure 2). The two quartet 19F signals correspond to symmetrical location of fluorines in ortho and meta positions on the phenyl ring, confirming that the cysteine thiol was substituted at the para position. Photoisomerization of the SNAr reaction product 3 was characterized with UV-vis, 19F NMR, and 1H NMR spectroscopies (Figure S5–S7).
Figure 2.

SNAr reaction of F-PSCaa 1 (1 mM) with Boc-protected cysteine 2 (3 mM) monitored by 19F-NMR spectroscopy in DMSO-d6.
We next explored whether 1 could be genetically incorporated into proteins through the expansion of the genetic code with an orthogonal tRNA/aminoacyl-tRNA synthetase pair to suppress the amber stop codon TAG.13 We co-expressed in E. coli the orthogonal amber suppressor pair with a calmodulin gene, which encoded an amber TAG codon at position 76 for F-PSCaa incorporation (CaM_76TAG). The MmPSCaaRS is specific for Uaas containing azobenzene and tolerant with different substituents at the meta position of the azo group.5 Because fluorine atoms do not significantly change the bulk of the azobenzene, we thus expect the MmPSCaaRS should also be able to incorporate 1. Indeed, F-PSCaa 1 was successfully incorporated into CaM in E. coli to yield 1.0 mg L−1 of CaM after nickel-nitrilotriacetic acid (Ni-NTA) purification. In the absence of 1 in the culture, no full-length CaM was detected. Electrospray ionization mass spectrometric (ESI-MS) analysis of the purified CaM protein confirmed the incorporation of the intact 1 (Figure S8). An additional peak corresponding to this CaM protein with glutathione (GSH) added to 1 was also detected (Figure S8), indicating that 1 partly reacted intermolecularly with endogenous GSH.
We then investigated whether the genetically encoded F-PSCaa 1 could react with a cysteine at an appropriate distance via proximity-enabled bioreactivity to build an azo bridge on proteins in situ. We co-expressed in E. coli the pair with a calmodulin gene containing the TAG codon at position 76 and a cysteine at position 83 ([Cys83]-CaM_76TAG). As expected, full-length CaM was produced in the presence of 1 (Figure 3a). Analysis of the Ni-NTA purified CaM using high resolution electrospray ionization Fourier transform ion trap mass spectrometry (ESI-FTMS) confirmed the incorporation of 1 and, more importantly, the formation of a protein bridge through 1 reacting with Cys83 (Figure 3b). The mass peak detected at 18116.31 Da corresponds to CaM bearing the covalent azobenzene bridge; a second peak measured at 17985.28 Da corresponds to CaM containing the covalent bridge and lacking the initiating Met. No peak corresponding to non-crosslinked F-PSCaa/cysteine or any other potential modification of 1 was detected, thus indicating formation of the protein bridge through intramolecular SNAr crosslinking in high efficiency. We note that the azobenzene bridge was formed without any further treatment of the CaM protein following expression and purification. The reaction described here between the F-PSCaa and cysteine represents the first SNAr successfully employed for proximity-enabled bioreactivity on proteins in live cells.7,14
Figure 3.

Incorporation of 1 into CaM forming an azobenzene bridge with cysteine in situ. (a) SDS-PAGE stained with Coomassie shows that specifically incorporated 1 into CaM in E. coli. Samples were normalized for constant cell numbers for each lane. The arrow indicates the position of the full-length CaM protein. (b) High resolution ESI-FTMS analysis of intact CaM expressed in the presence of supplemented with 1 mM of 1. Average and monoisotopic (indicated by MI) masses are labeled. The SNAr crosslinking reaction of 1 with cysteine results in the loss of HF. CaM containing the covalent azobenzene bridge: [M-HF+H]+, expected 18116.33 Da, measured 18116.31 Da; [M-Met-HF+H]+, expected 17985.31 Da, measured 17985.28 Da. CaM with non-crosslinked F-PSCaa and cysteine: [M+H]+, expected 18136.32 Da, not detected; [M-Met+H]+, expected 18005.28 Da, not detected. The peak at 18331.43 did not correspond to CaM with F-PSCaa modified by any known potential intracellular reactants such as Cys, GSH or imidazole. In addition, in control protein CaM(F-PSCaa at 76 and Ala at 83), no mass peak corresponding to this 18331.43 peak was detected (Figure S8), suggesting that the 18331.43 peak was not related to F-PSCaa modification.
To determine whether the azobenzene bridge built onto CaM could be directed by visible light, we detected the isomerization process of the purified bridged CaM using UV-vis spectroscopy. Before light illumination, the bridged CaM showed a strong absorbance at 336 nm and a weak band at 445 nm, representing π-π* and n-π* transitions, respectively (black line, Figure 4a). This pattern is characteristic of E azobenzene. After green light illumination at λ = 540 nm, the absorbance at 336 nm decreased substantially, indicating a clear E-to-Z photoisomerization of the azobenzene bridge (red line, Figure 4a). Subsequent illumination with blue light resulted in Z-to-E photoisomerization (blue line, Figure 4a) yielding the photostationary state (pss) of the E state. Successive illumination with either green or blue light allowed for reversible transformation between the two states without showing fatigue of the azo bridge on the protein (Figure 4b). Moreover, photoisomerization of the azo bridge allowed conformational changes of CaM to be driven reversibly as detected by circular dichroism (CD). The recorded spectra are typical for α-helical CaM proteins with a band at 222 nm and 208 nm, respectively (Figure 4c). Upon E-to-Z photosiomerization the intensity of the n-π* transition band at 208 nm decreased (red line, Figure 4c), with magnitude comparable to that of wild-type CaM before and after Ca2+ binding,15 suggesting a clear conformational change of the CaM protein. Notably, the conformation of the ground E state was restored by Z-to-E photoisomerization using blue light subsequently (blue line, Figure 4c).
Figure 4.

The azo bridge built on CaM controls CaM conformation and binding in response to visible light. (a) UV-vis spectra of CaM bearing the azobenzene bridge formed by F-PSCaa76 reacting with Cys83. Green light illumination (λ = 540 nm, 5 min) resulted in E-to-Z photoisomerization of the built-in azo bridge indicative by the substantial decrease of the band at 336 nm (black line, before illumination; red line, after green light illumination). Subsequent blue light illumination (λ = 470 nm, 5 min) induced Z-to-E photoisomerization (blue line). (b) Successive green and blue light illumination does not show fatigue of the azo bridge on CaM. The first cycle was from the E form to the Z pss state and thus had larger change than subsequent cycles switching between the E pss and Z pss states. (c) Circular dichroism (CD) spectra of the E form (black), the Z pss (red) and the E pss (blue) of CaM bearing the azo bridge in PBS buffer (protein conc. 5 μM). After green light activation to the Z pss, the band at 208 nm changed significantly indicating conformational changes within CaM. The conformation of the ground state can be recovered by blue light. (d) Binding of NOS-I peptide to the E state of CaM/Ca2+ studied by fluorescence. Upon addition of NOS-I to the E form (black line), the fluorescence intensity significantly increased (green line, increased by 215%) indicative of peptide binding. (e) CaM/Ca2+ in the E state (black line) was illuminated with green light generating the Z pss state (red line). Subsequent addition of NOS-I peptide to the Z pss state (green line, increased by 50%) showed less increase in intensity in comparison to the E state (green line, Figure 4d). The data obtained suggest that the remaining E form present in the Z pss state binds to the peptide resulting in the slight increase of the intensity. The Z pss state of F-PSCaa 1 consists of 22% E form (Figure 1a).
To test whether the binding function of CaM could be photo-modulated by the azo bridge, we measured the binding of CaM to the CaM binding domain of the neuronal nitric oxide synthase (NOS-I). Upon binding CaM changes its conformation and wraps around the NOS-I peptide,16 which has been reliably monitored with fluorescence of dyes labeled onto CaM.17 We labeled the azo bridge-containing CaM with dansyl chloride in the presence of Ca2+ and recorded dansyl fluorescence upon binding of the NOS-I peptide. After addition of NOS-I peptide (10 μM) to the E form of the bridged CaM/Ca2+ (0.1 μM), the fluorescence intensity markedly increased by 215% and the emission peak slightly shifted to shorter wavelength (from 517 to 492 nm), which is indicative of the NOS-I peptide binding to CaM (green line, Figure 4d). In contrast, the change of fluorescence intensity upon addition of NOS-I to the Z pss of CaM/Ca2+ is much less (by 50%, green line, Figure 4e), indicating that photoswitching the azo bridge to Z form decreased CaM binding to the peptide. Illumination of the CaM/Ca2+ in the absence of peptide neither influenced the emission wavelength nor intensity (black and red line, Figure 4e).
In summary, we have developed and genetically encoded the pentafluoro photoswitchable click amino acid F-PSCaa 1, which isomerizes upon visible light and enables in situ formation of a visible light-controllable bridge onto proteins through intramolecular SNAr reaction with a cysteine. The F-PSCaa and its bridge formed in situ represent the first genetically encoded small molecule photoswitch that can be modulated by visible light. In comparison with protein-based photoswitches, 1 can be selectively incorporated into any site of protein in vivo, providing greater flexibility in target site selection and expanding the scope of proteins addressable by light. In addition, a light-controllable bridge generated onto proteins in situ makes it possible to optically modulate protein secondary structures and domains spanning multiple residues in addition to a single residue. Moreover, responsiveness to visible light avoids negative effects induced by UV light and makes 1 and its bridge more compatible with in vivo usage. We thus expect that F-PSCaa and its bridge will find broad applications in the emerging field of optobiology for its minimal interference, site-specificity, genetic encodability, and response to the more biocompatible visible light.
Supplementary Material
Acknowledgments
C.H. gratefully acknowledges a fellowship by the Deutsche For-schungsgemeinschaft DFG (HO 4778/1-1) and the Pioneer Foundation. S.C. acknowledges support from U.S. National Institute of Health (GM098630). L.W. acknowledges support from U.S. National Institutes of Health (1DP2OD004744).
Footnotes
The authors declare no competing financial interests.
ASSOCIATED CONTENT
Synthesis of F-PSCaa 1, experimental details, additional 1H- and 19F-NMR spectra, and Figures S1–S11 are provided as supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Fenno L, Yizhar O, Deisseroth K. Annu Rev Neurosci. 2011;34:389–412. doi: 10.1146/annurev-neuro-061010-113817. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bernstein JG, Boyden ES. Trends Cogn Sci. 2011;15:592–600. doi: 10.1016/j.tics.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yazawa M, Sadaghiani AM, Hsueh B, Dolmetsch RE. Nat Biotechnol. 2009;27:941–945. doi: 10.1038/nbt.1569. [DOI] [PubMed] [Google Scholar]; (d) Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, Hahn KM. Nature. 2009;461:104–108. doi: 10.1038/nature08241. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL. Nat Methods. 2010;7:973–975. doi: 10.1038/nmeth.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Levskaya A, Weiner OD, Lim WA, Voigt CA. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Crefcoeur RP, Yin R, Ulm R, Halazonetis TD. Nat Commun. 2013;4:1779. doi: 10.1038/ncomms2800. [DOI] [PubMed] [Google Scholar]; (h) Zhou XX, Chung HK, Lam AJ, Lin MZ. Science. 2012;338:810–814. doi: 10.1126/science.1226854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Szobota S, Isacoff EY. Annu Rev Biophys. 2010;39:329–348. doi: 10.1146/annurev.biophys.093008.131400. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fehrentz T, Schonberger M, Trauner D. Angew Chem Int Ed Engl. 2011;50:12156–12182. doi: 10.1002/anie.201103236. [DOI] [PubMed] [Google Scholar]
- 3.(a) Schierling B, Noel AJ, Wende W, Hien le T, Volkov E, Kubareva E, Oretskaya T, Kokkinidis M, Rompp A, Spengler B, Pingoud A. Proc Natl Acad Sci U S A. 2010;107:1361–1366. doi: 10.1073/pnas.0909444107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang F, Zarrine-Afsar A, Al-Abdul-Wahid MS, Prosser RS, Davidson AR, Woolley GA. J Am Chem Soc. 2009;131:2283–2289. doi: 10.1021/ja807938v. [DOI] [PubMed] [Google Scholar]
- 4.Bose M, Groff D, Xie J, Brustad E, Schultz PG. J Am Chem Soc. 2006;128:388–389. doi: 10.1021/ja055467u. [DOI] [PubMed] [Google Scholar]
- 5.Hoppmann C, Lacey VK, Louie GV, Wei J, Noel JP, Wang L. Angew Chem Int Ed Engl. 2014;53:3932–3936. doi: 10.1002/anie.201400001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoppmann C, Schmieder P, Heinrich N, Beyermann M. ChemBioChem. 2011;12:2555–2559. doi: 10.1002/cbic.201100578. [DOI] [PubMed] [Google Scholar]
- 7.Xiang Z, Ren H, Hu YS, Coin I, Wei J, Cang H, Wang L. Nat Methods. 2013;10:885–888. doi: 10.1038/nmeth.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Beharry AA, Sadovski O, Woolley GA. J Am Chem Soc. 2011;133:19684–19687. doi: 10.1021/ja209239m. [DOI] [PubMed] [Google Scholar]; (b) Samanta S, Beharry AA, Sadovski O, McCormick TM, Babalhavaeji A, Tropepe V, Woolley GA. J Am Chem Soc. 2013;135:9777–9784. doi: 10.1021/ja402220t. [DOI] [PubMed] [Google Scholar]; (c) Kienzler MA, Reiner A, Trautman E, Yoo S, Trauner D, Isacoff EY. J Am Chem Soc. 2013;135:17683–17686. doi: 10.1021/ja408104w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bleger D, Schwarz J, Brouwer AM, Hecht S. J Am Chem Soc. 2012;134:20597–20600. doi: 10.1021/ja310323y. [DOI] [PubMed] [Google Scholar]
- 10.(a) Irie M. Chem Rev. 2000;100:1685–1716. doi: 10.1021/cr980069d. [DOI] [PubMed] [Google Scholar]; (b) Matsui M, Funabiki K, Shibata K. Bull Chem Soc Jpn. 2002;75:531–536. [Google Scholar]
- 11.Spokoyny AM, Zou Y, Ling JJ, Yu H, Lin YS, Pentelute BL. J Am Chem Soc. 2013;135:5946–5949. doi: 10.1021/ja400119t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marsh EN, Suzuki Y. ACS Chem Biol. 2014;9:1242–1250. doi: 10.1021/cb500111u. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Brock A, Herberich B, Schultz PG. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 14.(a) Xiang Z, Lacey VK, Ren H, Xu J, Burban DJ, Jennings PA, Wang L. Angew Chem Int Ed Engl. 2014;53:2190–2193. doi: 10.1002/anie.201308794. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen XH, Xiang Z, Hu YS, Lacey VK, Cang H, Wang L. ACS Chem Biol. 2014;9:1956–1961. doi: 10.1021/cb500453a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shifman JM, Choi MH, Mihalas S, Mayo SL, Kennedy MB. Proc Natl Acad Sci U S A. 2006;103:13968–13973. doi: 10.1073/pnas.0606433103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valentine KG, Ng HL, Schneeweis L, Kranz JK, Frederick KK, Alber T, Wand AJ. PDB code 2O60 2006 [Google Scholar]
- 17.(a) Censarek P, Beyermann M, Koch KW. Biochemistry. 2002;41:8598–8604. doi: 10.1021/bi025681k. [DOI] [PubMed] [Google Scholar]; (b) Spratt DE, Taiakina V, Guillemette JG. Biochim Biophys Acta. 2007;1774:1351–1358. doi: 10.1016/j.bbapap.2007.07.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.