

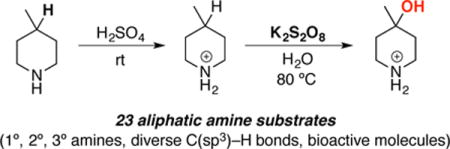
Abstract
This letter describes the development of a method for selective remote C(sp3)–H oxygenation of protonated aliphatic amines using aqueous potassium persulfate. Protonation serves to deactivate the proximal C(sp3)–H bonds of the amine substrates and also renders the amines soluble in the aqueous medium. These reactions proceed under relatively mild conditions (within 2 h at 80 °C with amine as limiting reagent) and do not require a transition metal catalyst. This method is applicable to a variety of types of C(sp3)–H bonds, including 3°, 2° and benzylic C–H sites of in primary, secondary, and tertiary amine substrates.
Graphical Abstract
Aliphatic amines appear in a wide variety of pharmaceuticals and agrochemicals.1 As such, methods for the selective C– H functionalization of aliphatic amines would be valuable for the synthesis and late-stage modification of many biologically active molecules.2 A variety of methods have been developed for the functionalization of the weak C(sp3)–H bonds α-to nitrogen in these substrates.2b,3 In contrast, it remains much more challenging to selectively functionalize less activated C(sp3)–H bonds that are remote from nitrogen.4,5 The incorporation of various nitrogen protecting groups including carbamates,6 amides,7 imides,2a,8 and sulfonamides3f,9 has been used to deactivate the α-C–H bonds and enable C(sp3)–H functionalization at alternate sites. Similarly, directing groups have been appended to nitrogen to enforce remote C(sp3)–H functionalization.2c,10 However, these strategies both require the additional steps of protecting group installation and removal. Furthermore, they are only applicable to 1° and/or 2° amine substrates.
An attractive alternative approach involves the in situ protonation of unprotected aliphatic amines.2d,11 Protonation of the nitrogen reversibly converts it into a strong electron-withdrawing group,12 thereby deactivating the α-C–H bonds.13 We2d,11b and others2a,11a have utilized this protonation strategy to achieve several different types of amine C(sp3)–H functionalization reactions. However, existing methods have significant limitations. For instance, the earliest reported example of this approach exhibited a small substrate scope and required the impractical oxidant methyl(trifluoromethyl)dioxirane (TFDO).11a,14 More recent approaches have utilized transition metal catalysts,2a,2d,11b which can be expensive and also difficult to separate from the products. Finally, in most reported examples, the scope is largely restricted to the oxygenation of remote primary,11b tertiary,2a or benzylic2d C(sp3)–H bonds.
We sought to address these limitations by developing an operationally simple remote C(sp3)–H hydroxylation of protonated amines that is applicable to a broad range of substrates. We report herein a new method that uses aqueous solutions of inexpensive K2S2O8 for this transformation. This system is effective for the oxygenation of tertiary and benzylic C(sp3)–H bonds as well as secondary C(sp3)–H sites. Furthermore, both alcohol and ketone products can be accessed from the latter, depending on the reaction conditions. We further demonstrate that this method can be applied to the C–H oxidation of unprotected amino acids and other amine-containing bioactive molecules.
Our initial studies focused on the hydroxylation of the tertiary C–H bond in 4-methyl piperidine (1) using commercially available and inexpensive K2S2O8 as the oxidant (Table 1).16 Water was selected as the solvent for two reasons. First, unlike most organic solvents, water lacks C–H bonds that could undergo competitive oxidation. Second, both K2S2O8 and the protonated amine substrate are highly soluble in water, while the free amine is not. As such, these conditions were expected to enable selective reaction of the protonated amine.15
Table 1.
Remote hydroxylation of protonated 4-methylpiperidine with K2S2O8
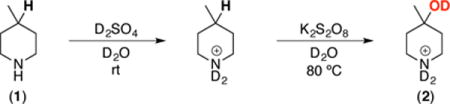
| |||||
|---|---|---|---|---|---|
| entry | K2S2O8 (equiv) | D2SO4 (equiv) | time (h) | conv 1 (%) | yield 2 (%) |
| 1 | 1 | 1.1 | 2 | 75 | 47 |
| 2 | 1 | 1.1 | 4 | 76 | 51 |
| 3 | 2 | 1.1 | 2 | >99 | 53 |
| 4 | 2 | 2.2 | 2 | >99 | 75 |
| 5 | 2 | 3.3 | 2 | >99 | 75 |
| 6 | 2 | none | 2 | 50 | <5 |
Yields and conversions determined by 1H NMR spectroscopy.15
In the event, the combination of 1 equiv of 1 and 1.1 equiv of D2SO4 at room temperature, followed by the addition of 1 equiv of K2S2O8 and heating at 80 °C for 2 h afforded the 3° alcohol product 2 in 47% yield as determined by 1H NMR spectroscopy. A significant quantity of starting material (25%) remained under these conditions, and increasing the reaction time to 4 h did not lead to further conversion (entry 2). This result suggests that the oxidant is consumed within 2 h and thus that additional oxidant is required to increase the conversion/yield. Indeed, the use of 2 equiv of K2S2O8 in conjunction with 2.2 equiv of H2SO4 under otherwise identical conditions resulted in complete consumption of the starting material and the formation of the 2 in 75% yield (entry 4). Importantly, the addition of acid is crucial for accessing product 2. As shown in entry 6, in the absence of acid, <5% of the 3° hydroxylation product 2 was detected.
Overall, this represents an inexpensive and operationally simple method for the selective C–H hydroxylation of 1.17 Furthermore, this method is highly complementary to our recently reported Pt-catalyzed oxidation of protonated amines.11b As shown in eq 1, the Pt-catalyzed reaction of 1 leads to selective functionalization at the least hindered primary C(sp3)–H bond of 1 (to form 3), while the current method affords tertiary C(sp3)–H hydroxylation with high selectivity.
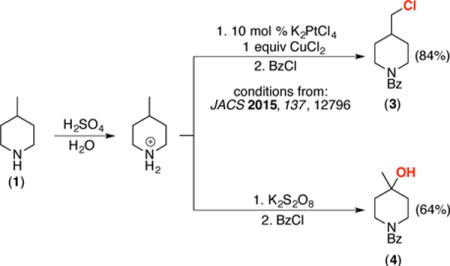 |
(1) |
We next applied this method to the hydroxylation of tertiary C(sp3)–H bonds in a variety of different amine substrates to form products 4–17. Notably, the products derived from primary and secondary amine substrates were subsequently converted to amides to facilitate isolation (Scheme 2a), while the tertiary amine products were isolated directly (Scheme 2b). In all cases, we observed hydroxylation at a remote tertiary C–H site. However, the yield and selectivity vary as a function of substrate. In general, the highest yields and selectivities were obtained when the tertiary C–H bond is two or three carbons from the protonated nitrogen (e.g, forming products 4, 5, 8–10, and 14–16).18 In systems with longer alkyl chains between the protonated nitrogen and the tertiary C–H site (e.g., 11, 12), side products derived from oxidation of secondary C–H bonds along the chain were detected. This resulted in lower isolated yields of the major tertiary C–H oxidation product. These secondary C–H oxidation side reactions could be limited by lowering the reaction temperature to 65 °C. Notably, small quantities of related ring oxidation side products were also observed in systems containing nitrogen heterocycles (13–17).
Scheme 2.

Substrate scope containing amines with 2°-C(sp3)–H bonds
We next probed the feasibility of secondary C(sp3)–H oxidation in the absence of competing tertiary C–H sites. Initial studies utilized aliphatic amines containing secondary benzylic C–H bonds. Under our standard reaction conditions these substrates afforded mixtures of alcohol and ketone products. For example, 4-ethyl benzylamine reacted to form a 0.94:1 mixture of alcohol 18-OH to ketone 18-O (61% overall yield of C–H oxygenated products). This ratio could be shifted towards the alcohol by simply adding less oxidant. For example, the use of 1 equiv of K2S2O8 under otherwise identical conditions afforded a 1:0.48 ratio of 18-OH:18-O (55% yield). The alcohol product was also favored when the benzylic C–H bonds were closer to the protonated amine. For example, phenethylamine afforded benzylic alcohol 19-OH as the major product (19-OH:19-O = 1:0.28) under our standard conditions. Oxidation of the alcohol is likely slowed due to the proximity of the electron withdrawing ammonium center. This transformation also afforded secondary alcohol products with the non-benzylic substrate butylamine. Under the standard conditions, butylamine reacted to form a 1:0.29 ratio of the γ:β-hydroxylated products (23-OH-a:23-OH-b) in moderate 32% isolated yield.
The results in Scheme 2 show several marked contrasts with other recently reported systems for the remote C(sp3)–H oxygenation of protonated amines. For instance, our Pt/Cu system selectively afforded primary C(sp3)–H hydroxylation products with substrates such as butylamine.11b White’s Fe(PDP)/H2O2 system provided ketone products selectively, and was only effective for oxygenation of C(sp3)–H bonds that were ≥3 carbons from nitrogen.2a Meanwhile, our FeCl3/TBHP system only oxidized benzylic C–H bonds and selectively provided ketone products.2d Furthermore, no reactivity was observed when the benzylic C–H sites were <3 carbons from the amine. Overall, these comparisons highlight the complementarity of the current method to those reported in the literature.
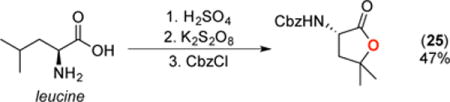
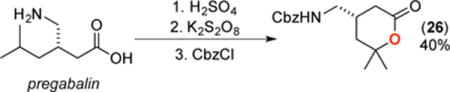
A final set of studies focused on applying this transformation to several amine-containing biologically active molecules. As shown in eq 2, under our standard conditions the Alzheimer’s drug memantine was converted to the corresponding 3° alcohol 24 in moderate yield. Similarly, the amino acid leucine underwent C(sp3)–H oxygenation to afford lactone 25 in 47% isolated yield and 90% ee, after protection of the product and isolation (eq 3). Finally, the epilepsy drug pregabalin underwent selective oxygenation at the tertiary C(sp3)–H site that is remote from nitrogen to afford lactone 26 in 40% yield after protection and isolation (eq 4).19
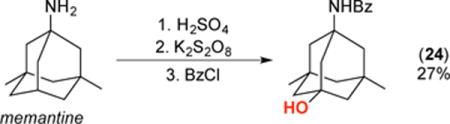 |
(2) |
 |
(3) |
 |
(4) |
In summary, this Letter demonstrates an operationally straightforward method for the remote C(sp3)–H oxygenation of protonated amines. This method uses an inexpensive and safe oxidant (potassium persulfate) and proceeds in water under mild conditions. We demonstrate that this transformation is complementary to several recently reported C–H oxidation reactions of protonated amines, and that it is applicable to bioactive substrates. As such, we anticipate that this method will prove useful in medicinal chemistry, for the late-stage derivatization of amine-containing drug targets.
Supplementary Material
Scheme 1.
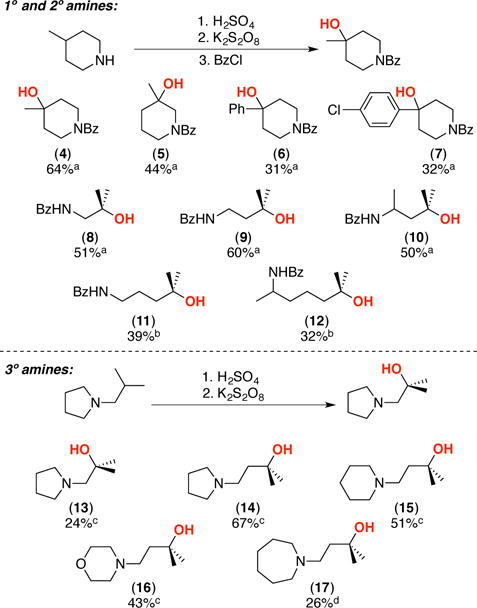
Hydroxylation of remote tertiary C(sp3)–H bonds in a variety of amine substrates.
a2.2 equiv H2SO4, 2 equiv K2S2O8, 80 °C, followed by protection with BzCl. b1.1 equiv H2SO4, 2 equiv K2S2O8, 65 °C, followed by protection with BzCl. c2.2 equiv H2SO4, 2 equiv K2S2O8, 80 °C. d1.1 equiv H2SO4, 2 equiv K2S2O8, 65 °C.
Acknowledgments
We acknowledge financial support from NIH NIGMS (GM073836). ML thanks NSF and Rackham Graduate School for graduate fellowships. We also thank Jeff Kampf (University of Michigan) for an X-ray structure determination and Pablo Cabrera (University of Michigan) for assistance with obtaining the data shown in Table 1 as well as the TEMPO and 1,4-dinitrobenzene experiments discussed in reference 17.
Footnotes
Supporting Information
Optimization data, experimental data, complete characterization data for all new compounds, and CIF data for 25 This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Lawrence SA. Amines: Synthesis, Properties and Applications. Cambridge University Press; Cambridge, U.K.: 2004. [Google Scholar]; b Dorwald FZ. Lead Optimization for Medicinal Chemists: Pharmacokinetic Properties of Functional Groups and Organic Compounds. first. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2012. [Google Scholar]; c Vitaku E, Smith DT, Njardarson JT. J Med Chem. 2014;57:10257. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- 2.For examples, see:; (a) Howell JM, Feng K, Clark JR, Trzepkowski LJ, White MC. J Am Chem Soc. 2015;137:14590. doi: 10.1021/jacs.5b10299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) He J, Hamann LG, Davies HML, Beckwith REJ. Nat Commun. 2015;6:5943. doi: 10.1038/ncomms6943. [DOI] [PubMed] [Google Scholar]; (c) Topczewski JJ, Cabrera PJ, Saper NI, Sanford MS. Nature. 2016;531:220. doi: 10.1038/nature16957. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mbofana CT, Chong E, Lawniczak J, Sanford MS. Org Lett. 2016;18:4258. doi: 10.1021/acs.orglett.6b02003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For examples, see:; (a) Mitchell EA, Peschiulli A, Lefevre N, Meerpoel L, Maes BUW. Chem Eur J. 2012;18:10092. doi: 10.1002/chem.201201539. [DOI] [PubMed] [Google Scholar]; (b) Genovino J, Lütz S, Sames D, Touré BB. J Am Chem Soc. 2013;135:12346. doi: 10.1021/ja405471h. [DOI] [PubMed] [Google Scholar]; (c) Legacy CJ, Wang A, O’Day BJ, Emmert MH. Angew Chem Int Ed. 2015;54:14907. doi: 10.1002/anie.201507738. [DOI] [PubMed] [Google Scholar]; (d) Spangler JE, Kobayashi Y, Verma P, Wang DH, Yu JQ. J Am Chem Soc. 2015;137:11876. doi: 10.1021/jacs.5b06740. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Shaw MH, Shurtleff VW, Terrett JA, Cuthbertson JD, MacMillan DWC. Science. 2016;352:1304. doi: 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Osberger TJ, Rogness DC, Kohrt JT, Stepman AF, White MC. Nature. 2016;537:214. doi: 10.1038/nature18941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For representative examples of nitrogen-directed C-H functionalization using unprotected amines as directing groups, see:; (a) Dangel BD, Johnson JA, Sames D. J Am Chem Soc. 2001;123:8149. doi: 10.1021/ja016280f. [DOI] [PubMed] [Google Scholar]; (b) McNally A, Haffemayer B, Collins BSL, Gaunt MJ. Nature. 2014;510:129. doi: 10.1038/nature13389. [DOI] [PubMed] [Google Scholar]; (c) Calleja J, Pla D, Gorman TW, Domingo V, Haffemayer B, Gaunt M. J Nat Chem. 2015;71009 doi: 10.1038/nchem.2367. [DOI] [PubMed] [Google Scholar]
- 5.Example of remote C–H borylation of an unprotected amine:; Li Q, Liskey CW, Hartwig JF. J Am Chem Soc. 2014;136:8755. doi: 10.1021/ja503676d. [DOI] [PubMed] [Google Scholar]
- 6.For examples using carbamate protected amino acids and peptides, see:; (a) Detomaso A, Curci R. Tetrahedron Lett. 2001;42:755. [Google Scholar]; (b) Rella MR, Williard PG. J Org Chem. 2007;72:525. doi: 10.1021/jo061910n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Annese C, D’Accolti L, De Zotti M, Fusco C, Toniolo C, Williard PG, Curci R. J Org Chem. 2010;75:4812. doi: 10.1021/jo100855h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For examples using amide protecting groups, see:; (a) McNeill E, Du Bois J. J Am Chem Soc. 2010;132:10202. doi: 10.1021/ja1046999. [DOI] [PubMed] [Google Scholar]; (b) Pierce CJ, Hilinski MK. Org Lett. 2014;16:6504. doi: 10.1021/ol503410e. [DOI] [PubMed] [Google Scholar]
- 8.For examples using imide protecting groups, see:; (a) Wu H, Xiao Z, Wu J, Guo Y, Xiao JC, Liu C, Chen QY. Angew Chem Int Ed. 2015;54:4070. doi: 10.1002/anie.201411953. [DOI] [PubMed] [Google Scholar]; (b) Wang D, Shuler WG, Pierce CJ, Hilinski MK. Org Lett. 2016;18:3826. doi: 10.1021/acs.orglett.6b01832. [DOI] [PubMed] [Google Scholar]; (c) Zhang X, Yang H, Tang P. Org Lett. 2016;17:5828. doi: 10.1021/acs.orglett.5b03001. [DOI] [PubMed] [Google Scholar]; (d) Li X, Che X, Chen GH, Zhang J, Yan JL, Zhang YF, Zhang LS, Hsu CP, Gao YQ, Shi ZJ. Org Lett. 2016;18:1234. doi: 10.1021/acs.orglett.5b03690. [DOI] [PubMed] [Google Scholar]
- 9.For an example using sulfonyl protecting groups, see:; Adams AM, DuBois J, Malik HA. Org Lett. 2015;17:6066. doi: 10.1021/acs.orglett.5b03047. [DOI] [PubMed] [Google Scholar]
- 10.For representative examples of nitrogen-directed C–H functionalization using protected amines as directing groups, see:; (a) Zaitsev VG, Shabashov D, Daugulis O. J Am Chem Soc. 2005;127:13154. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]; (b) Daugulis O, Do HQ, Shabashov D. Acc Chem Res. 2009;42:1074. doi: 10.1021/ar9000058. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) He G, Chen G. Angew Chem Int Ed. 2011;50:5192. doi: 10.1002/anie.201100984. [DOI] [PubMed] [Google Scholar]; (d) Rodríguez N, Romero-Revilla JA, Fernández-Ibáñez MÁ, Carretero JC. Chem Sci. 2013;4:175. [Google Scholar]; (e) Fan M, Ma D. Angew Chem Int Ed. 2013;52:12152. doi: 10.1002/anie.201306583. [DOI] [PubMed] [Google Scholar]; (f) Rouquet G, Chatani N. Angew Chem Int Ed. 2013;52:11726. doi: 10.1002/anie.201301451. [DOI] [PubMed] [Google Scholar]; (g) Chan KSL, Wasa M, Chu L, Laforteza BN, Miura M, Yu JQ. Nature Chem. 2014;6:146. doi: 10.1038/nchem.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Han J, Zheng Y, Wang C, Zhu Y, Shi DQ, Zeng R, Huang ZB, Zhao Y. J Org Chem. 2015;80:9297. doi: 10.1021/acs.joc.5b00968. [DOI] [PubMed] [Google Scholar]; (i) Han J, Zheng Y, Wang C, Zhu Y, Shi DQ, Zeng R, Huang ZB, Zhao Y. J Org Chem. 2015;80:9297. doi: 10.1021/acs.joc.5b00968. [DOI] [PubMed] [Google Scholar]; (j) Pasunooti KK, Banerjee B, Yap T, Jian Y, Liu CF. Org Lett. 2015;17:6094. doi: 10.1021/acs.orglett.5b03118. [DOI] [PubMed] [Google Scholar]; (k) Xu Y, Young MC, Wang C, Magness DM, Dong G. Angew Chem Int Ed. 2016;55:9084. doi: 10.1002/anie.201604268. [DOI] [PubMed] [Google Scholar]; (l) Huang Z, Wang C, Dong G. Angew Chem Int Ed. 2016;55:5299. doi: 10.1002/anie.201600912. [DOI] [PubMed] [Google Scholar]; (m) Han J, Zheng Y, Wang C, Zhu Y, Huang ZB, Shi DQ, Zeng R, Zhao Y. J Org Chem. 2016;81:5681. doi: 10.1021/acs.joc.6b00649. [DOI] [PubMed] [Google Scholar]; (n) He J, Wasa M, Chan KSL, Shao Q, Yu JQ. Chem Rev. 2016 doi: 10.1021/acs.chemrev.6b00622. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Asensio G, González-Núñez ME, Gernadini CB, Mello R, Adam W. J Am Chem Soc. 1993;115:7250. [Google Scholar]; (b) Lee M, Sanford MS. J Am Chem Soc. 2015;137:12796. doi: 10.1021/jacs.5b09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isaacs NS. Physical Organic Chemistry. 2nd. Longman Group Limited; London: 1995. pp. 146–192. [Google Scholar]
- 13.Salamone M, Carboni G, Bietti M. J Org Chem. 2016;81:9269. doi: 10.1021/acs.joc.6b01842. [DOI] [PubMed] [Google Scholar]
- 14.Methyl(trifluoro-methyl)dioxirane. e-EROS Encyclopedia of Reagents for Organic Synthesis [Online]; John Wiley & Sons, Posted March 31, 2016. http://onlinelibrary.wiley.com/doi/10.1002/047084289X.rm267.pub3/abstract (accessed Nov 16, 2016).
- 15.A control reaction (Table 1, entry 6) confirmed that 2 is not formed in appreciable quantities in the absence of acid; instead, mostly starting material was recovered. Notably, 1.1 equiv of H2SO4 was added at the end of this reaction to ensure solubility of starting material and products during the NMR analysis of the crude reaction mixture.
- 16.Cat. no. 216224 ($41.09/mol) from Sigma Aldrich Online Catalogue (accessed Dec 4, 2016).
- 17.Under the optimized conditions for the oxidation of substrate 1, the addition of TEMPO or dinitrobenzene led to significantly diminished conversion of 1 and yield of 2. For example, with 1 equiv of TEMPO, the yield of 2 was just 13%, along with 74% of substrate 1 remaining. With 20 mol % of 1,4-dinitrobenzene, the yield of 2 was 44%, along with 49% of substrate 1 remaining. These preliminary results point to a radical pathway, which is fully consistent with the known chemistry of persulfate oxidations. For example, see:; House DA. Chem Rev. 1962;62:185. [Google Scholar]
- 18.The modest yields of 6 and 7 were due to incomplete conversion. Conducting the oxidation of 6 under more forcing conditions (at 100 °C) resulted in formation of side products. The crude NMR spectrum is provided in the Supporting Information.
- 19.Initial attempts to oxidize higher molecular weight amines under our standard conditions were hampered by the modest solubility of many of these substrates in water (even when protonated). Current efforts are focusing on identifying suitable co-solvents and re-optimizing the reaction for such systems.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.