

Abstract
CD48 (SLAMF2) is an adhesion and costimulatory molecule constitutively expressed on hematopoietic cells. Polymorphisms in CD48 have been linked to susceptibility to multiple sclerosis (MS), and altered expression of the structurally related protein CD58 (LFA-3) is associated with disease remission in MS. We examined CD48 expression and function in experimental autoimmune encephalomyelitis (EAE), a mouse model of MS. We found that a subpopulation of CD4+ T cells highly upregulated CD48 expression during EAE and were enriched for pathogenic CD4+ T cells. These CD48++ CD4+ T cells were predominantly CD44+ and Ki67+, included producers of IL-17A, GM-CSF and IFN-γ, and were the majority of CD4+ T cells in the CNS. Administration of anti-CD48 mAb during EAE attenuated clinical disease, limited accumulation of lymphocytes in the CNS, and reduced the number of pathogenic cytokine-secreting CD4+ T cells in the spleen at early time points. These therapeutic effects required CD48 expression on CD4+ T cells but not on antigen presenting cells. In addition, the effects of anti-CD48 were partially dependent on FcγRs, as anti-CD48 did not ameliorate EAE nor reduce the number of cytokine-producing effector CD4+ T cells in Fcεr1γ−/− mice or in wild type mice receiving anti-CD16/CD32 mAb. Our data suggest that anti-CD48 mAb exerts it therapeutic effects by both limiting CD4+ T cell proliferation and preferentially eliminating pathogenic CD48++ CD4+ T cells during EAE. Our findings indicate that high CD48 expression is a feature of pathogenic CD4+ T cells during EAE and point to CD48 as a potential target for immunotherapy.
INTRODUCTION
CD48 (SLAMF2, BLAST-1) and the related gene CD58 have been identified in genome-wide association studies as susceptibility genes in multiple sclerosis (MS)2 (1, 2), a demyelinating disease of the CNS that results in progressive loss of motor and sensory function (3). Functional studies associated a protective allele of CD58 with increased CD58 mRNA expression in PBMCs (1, 4), and CD58 expression in PBMCs was found to increase during remissions in MS patients (4, 5). While this work implicates CD48 and CD58 in MS, little is known about their roles in CNS autoimmunity. However, studies in mice indicate that CD48 can regulate T cell activation and tolerance.
CD48 is a GPI-linked molecule, constitutively expressed on the surface of all hematopoietic cell types and involved in cell adhesion and costimulation through interactions with its ligands CD2 (6) and CD244 (7). On antigen presenting cells (APCs), CD48 promotes immune synapse organization (8) and T cell costimulation (9) through binding to CD2 on T cells. CD48 on T cells enhances TCR signaling through cis interactions with CD2, LAT and Lck (10, 11). CD58 is also a ligand for CD2, but is expressed only in humans (12). Interactions between CD48 and CD244 regulate target cell lysis by NK cells and CTLs, as well as effector and memory T cell responses (13). In addition, binding of bacterial FimH to CD48 on granulocytes and monocytes contributes to innate immune responses to bacteria (14). CD48 expression increases on cells exposed to inflammatory stimuli. CD48 is upregulated on EBV-infected B cells, human PBMCS exposed to interferons, monocytes and lymphocytes from patients with viral and bacterial infections (15), eosinophils from patients with atopic asthma or mice after allergen challenge (14), and mouse T cells during LCMV infection (16) or peptide immunization (17).
CD48 is involved in regulating T cell activation and tolerance in mice. CD48 deficiency exacerbated lupus-like disease in mice on an autoimmune-prone genetic background (18, 19), while CD48 deficiency on T cells and macrophages mitigated disease in a model of inflammatory colitis (20). In addition, treatment with an anti-CD48 blocking mAb attenuated T cell-mediated inflammation in models of colitis (20), delayed-type hypersensitivity (21), and transplantation (22). These immunoregulatory roles, together with human genetic studies implicating CD48 in MS, led us to hypothesize that CD48 may regulate CNS autoimmunity.
We used experimental autoimmune encephalomyelitis (EAE), which replicates many of the features of MS (23), to evaluate the role of CD48 in CNS autoimmunity. We found that CD48 expression increased on antigen-specific CD4+ T cells in mice with EAE. Treatment of mice with an anti-CD48 mAb delayed EAE onset, and reduced incidence and severity. Cellular analyses revealed fewer pathogenic CD4+ T cells both in the periphery and the CNS of anti-CD48 treated mice. Clinical and cellular effects of anti-CD48 were highly dependent on CD48 expression on CD4+ T cells and on FcγRs. Our results indicate that CD48 upregulation is a feature of pathogenic CD4+ T cells during EAE, and point to CD48 as a potential target for immunotherapy.
MATERIALS AND METHODS
Mice
8-12 week old mice, age and sex matched, were used for all experiments. Wild type (WT) C57BL/6, Thy1.1 (B6.PL-Thy1a/CyJ), Rag1−/− (B6.129S7-Rag1tm1Mom/J), TCRα−/− (B6.129S2-Tcratm1Mom/J) and OTII (B6.Cg-Tg(TcraTcrb)425Cbn/J) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). NK-deficient Rag−/− (B10;B6-Rag2tm1Fwa Il2rgtm1Wjl) and Fcεr1γ−/− (B6.129P2-Fcer1gtm1Rav N12) mice were purchased from Taconic Biosciences (Hudson, NY). 2D2 TCR Tg Foxp3-IRES-GFP were generated by crossing 2D2 TCR Tg mice (24) with Foxp3-IRES-GFP knockin mice (25), and maintained our facility. CD48−/− 2D2 TCR Tg, CD48−/− Rag1−/−, and CD48−/− TCRα−/− were generated by crossing CD48−/− mice generated in our laboratory (B6 background, manuscript in preparation) with 2D2 TCR Tg, Rag1−/−, or TCRα−/− mice, respectively. Mice were housed in a specific pathogen-free animal facility, and used according to the Harvard Medical School Standing Committee on Animals and National Institutes of Health Guidelines.
MOG35-55/CFA immunizations and EAE
Mice were immunized s.c. with 50μg myelin oligodendrocyte glycoprotein 35-55 peptide (MOG35-55; MEVGWYRSPFSRVVHLYRNGK; UCLA Biopolymers Facility) in 100μl PBS emulsified in 100μl CFA with 400μg Mycobacterium tuberculosis H37RA (Difco Laboratories). For EAE, 100ng (or 200ng for Fcεr1γ−/− mice) pertussis toxin (List Biological Laboratories) was given i.p on days 0 and 2 after immunization. Mice were monitored daily and clinical disease scored as: 0, no disease; 1, limp tail; 2, hind limb weakness; 3, hind limb paralysis; 4 hind and fore limb paralysis; and 5, moribund. Average clinical scores for each group include animals that did not develop EAE.
Histopathology
Mice were perfused with PBS, brains and spinal cords fixed in 10% formalin, and paraffin-embedded sections stained with hematoxylin and eosin or Luxol fast blue/periodic acid-Schiff by the Dana Farber/Harvard Cancer Center Rodent Histopathology Core. Inflammatory foci in meninges and parenchyma were counted as described previously (26).
In vivo antibody administration
200μg of antibodies were given i.p. in 200μl PBS, as indicated in figure legends. Anti-NK1.1 (PK136), mouse IgG2a (C1.18), rat IgG2a (2A3), anti-CD48 (HM48-1), and polyclonal Armenian hamster IgG were purchased from BioXCell. Anti-CD16/CD32 (clone 93), anti-CD48 (HM48-1), and hamster isotype control (HTK888) were purchased from BioLegend.
EdU administration and staining
600-800μg of 5-ethynyl-2’-deoxyuridine (EdU, 2mg/mL, Life Technologies) in PBS was injected i.p. 12-16 hours before euthanasia, and EdU incorporation detected with the Click-iT EdU Flow Cytometry Assay Kit (Life Technologies) according to the manufacturer's instructions.
Analysis of mononuclear cells in the blood, lymphoid organs and CNS
Spleen and lymph nodes (LN) were mechanically dissociated through nylon cell strainers (70μm), blood collected by cardiac puncture, and RBC lysed with ACK lysing buffer (Lonza). Mice were perfused with PBS before CNS tissue collection, brains and spinal cords mechanically dissociated through a nylon cell strainer, and mononuclear cells isolated by centrifugation through a Percoll gradient (37% and 70%). Cells were resuspended in culture medium (RPMI 1640 [Gibco] with 10% FBS, 10mM HEPES [Gibco], 100U/mL Penicillin/Streptomycin [Gibco], and 100μM 2-ME [Sigma]).
Flow cytometry
Cells from lymphoid organs, blood, CNS, or in vitro cultures were resuspended in staining buffer (PBS with 1% FBS and 2mM EDTA) and stained with directly-conjugated antibodies for: CD3 (145-2C11), CD4 (RM4-5), CD8α (53-67), CD11b (M1/70), CD11c (N418), CD44 (IM7), CD45 (30-F11), CD48 (HM48-1), CD49b (DX5), CD49d (R1-2), CD62L (MEL-14), CD69 (H1.2F3), CD90.1 (OX-7), CD90.2 (30-H12), B220 (RA3-6B2), I-A/I-E (M5/114.15.2), and NK1.1 (PK136). For intracellular staining, cells were fixed and permeabilized with the Foxp3/Transcription Factor Staining kit (eBioscience) and stained for: Foxp3 (FJK-16s), GM-CSF (MP1-22E9), IFN-γ (XMG1.2), IL-2 (JES6-5H4), IL-10 (JES5-16E3), IL-17A (TC11-18H10.1), and Ki67 (B56). Antibodies were from BioLegend, BD Biosciences, or eBioscience. Flow cytometry data were acquired on a FACSCalibur (BD) or LSRII (BD) using FACSDiva software (BD), and analyzed with FlowJo software (FlowJo LLC).
2D2 CD4+ T cell adoptive transfers to Rag−/− and WT mice
CD4+ T cells were isolated from spleen and LN of donor mice (Thy1.1 2D2 TCR Tg, Foxp3.GFP.2D2 TCR Tg, or Thy1.1/1.2 2D2 TCR Tg CD48−/−) using CD4 Micro-Beads (Miltenyi Biotec), or purified by cell sorting to yield total CD4+ T cells (CD4+ CD8α- B220- CD11c-), and 3-10×106 CD4+ T cells injected i.v. or i.p. Recipients were immunized two weeks (Rag−/− strains) or one day (WT) after cell transfer.
2D2 Th1 CD4+ T cell in vitro and adoptive transfer studies
CD4+ T cells from 2D2 TCR Tg or CD48−/− 2D2 TCR Tg mice were cultured at 2×106/mL with 107/mL irradiated feeder cells (WT splenocytes depleted of CD4+ T cells with CD4+ microbeads), IL-12 (10ng/mL, PeproTech), anti-IL-4 (20μg/mL, 11B11, BioXCell), and anti-CD3 (2.5-3μg/mL, 2C11, BioXCell), and supplemented with 100U/mL rhIL-2 (R&D Systems) every 2 days. On day 6 live cells were collected using Lymphocyte Separation Media (Mediatech, Inc). For in vitro studies, cells were labeled with CellTrace Violet (5μM, Life Technologies), cultured at 2×106/mL with 107/mL TCRα−/− or CD48−/− TCRα−/− splenocytes plus MOG35-55 (0-100μg/mL) and cIgG or anti-CD48 (10μg/mL) for 3 days, then analyzed by flow cytometry for CellTrace Violet dilution. For adoptive transfers, cells were restimulated on anti-CD3/anti-CD28 (37.51, BioXCell) coated plates (3μg/mL) for 16-24 hours, and 2-6×106 CD4+ T cells injected i.v. or i.p. Animals were monitored for EAE or sacrificed before disease onset for cellular analysis.
In vitro CD4+ T cell stimulation to assess CD48 expression
Microbead-purified CD4+ T cells from 2D2 and OTII TCR Tg mice were cultured at 2×106/mL with 107/mL TCRα−/− splenocytes plus MOG35-55 (0-100μg/mL), OVA323-339 (ISQAVHAAHAEINEAGR; New England Peptide; 0-50μg/mL), or anti-CD3 (3μg/mL). Naïve CD4+CD62L+CD44-Foxp3.GFP-T cells were purified by cell sorting (98% purity), and 5x104 CD4+ T cells/well cultured in a 96-well tissue culture plate (Falcon) pre-coated with 0, 1, 2, 4, or 8μg/mL each of anti-CD3 and anti-CD28. Cells were analyzed by flow cytometry for CD48 expression 1 or 2 days later.
Cytokine measurements
Total cells from lymphoid organs or CNS, or CD48++ and CD48+ 2D2 cells (Thy1.2+Thy1.1-CD4+CD44+Foxp3.GFP-) purified by cell sorting from Thy1.1 WT recipients on day 10 after MOG/CFA immunization and mixed at a 1:5 ratio with TCRα−/− splenocytes, were restimulated in vitro with 0-100μg/mL MOG35-55 for 3 days. Supernatants were analyzed by Cytokine Bead Array (BD). For intracellular cytokine staining, cells were restimulated in vitro with 50ng/mL PMA and 500ng/mL ionomycin (Sigma-Aldrich) plus GolgiStop (1:1500 dilution, BD) at 37°C for 3-5 hours.
Statistical analysis and software
Analysis was performed using Prism (GraphPad), statistical significance calculated using Student's t test, and data plotted as mean ±SEM unless otherwise noted. EAE clinical scores were analyzed by Mann-Whitney test. P values less than 0.05 were considered statistically significant. *=p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001.
RESULTS
CD48 expression increases on activated CD4+ T cells during EAE
We examined CD48 expression on CD4+ T cells during MOG35-55/CFA-induced EAE, and found that a subpopulation of CD4+ T cells expressed high levels of CD48 (CD48++), and were increased during EAE (Figure 1A-B). At the peak of disease, ~60% of CD4+ T cells in the CNS were CD48++ compared to 13% in the spleen, and 4% in spleen of naïve mice (Figure 1A, 1B left). However, the absolute number of CD48++ CD4+ T cells was greatest in the inguinal LN (iLN) and spleen (Figure 1B, right). Foxp3+ regulatory T cells (Treg) in the spleen had slightly higher CD48 expression than Foxp3− CD4+ T cells (Tconv) in naïve mice, and CD48 expression increased on both populations during EAE (Figure 1C). All splenic CD48++ Tconv were also CD44+, and included many Ki67+ and α4 integrinhi cells (Figure 1D), consistent with previous descriptions of CD48 upregulation on activated T cells (16, 17). CD48 expression increased slightly on CD11c+MHCII+ dendritic cells but decreased on CD11b+MHCII+ macrophages and did not differ on B cells, CD8+ T cells, or NK cells in the spleens of mice with EAE compared to naïve mice (Figure S1A). These data suggest that CD48 upregulation during MOG35-55/CFA-induced EAE is a feature of activated CD4+ T cells.
Figure 1. CD48 expression increases on activated CD4+ T cells during EAE.

A-F WT mice were immunized with MOG35-55/CFA to induce EAE. Spleen, LN and CNS (brain and spinal cord) were collected at the peak of disease, and analyzed by flow cytometry. WT or CD48−/− spleens from naïve mice were used as controls. A. CD48 expression on CD3+CD4+ cells from the spleen of naïve CD48−/− and WT mice, and spleen and CNS of WT mice with EAE. B. Percent (left) and number (right) of CD48++ of CD4+CD3+. C. Representative staining (left) and percentages (right) of CD48++ Foxp3− and Foxp3+ T cells in the spleen. D. CD48 co-expression with CD44, Ki67 and α4 integrin. E. Representative CD48 and intracellular cytokine expression in CD3+CD4+Foxp3- cells isolated from CNS and spleen at peak EAE and restimulated with PMA/ionomycin. F. Percentages of cytokine-producing cells among CD48++ and CD48+ cells, based on gates in E. G-H. WT Thy1.1 mice were given 2D2 Thy1.2+ CD4+ T cells i.v., immunized with MOG35-55/CFA, and 10 days later CD48++ and CD48+ 2D2 were purified by cell sorting. G. Representative gating for cell sorting. H. Number of CD4+ T cells (left) and cytokines in supernatants (right) after restimulation in vitro with MOG35-55 for 3 days. Representative of 3 independent experiments with 4-6 immunized mice and one naïve control (A-D), 3 independent experiments with 4-5 mice (E-F), and 4 independent experiments with 4-5 biological replicates (G-H).
Similarly, stimulation of naïve CD4+ T cells with anti-CD3 and anti-CD28 in vitro led to upregulation of CD48 in a dose-dependent manner (Figure S1B). In mixed cultures of MOG-specific 2D2 TCR transgenic CD4+ T cells and OVA-specific OTII TCR transgenic CD4+ T cells, 2D2 CD4+ T cells upregulated CD48 in cultures stimulated with MOG35-55 or anti-CD3, but not with OVA peptide (Figure S1C). To investigate antigen-specific CD48 upregulation in vivo, we transferred 2D2 CD4+ T cells to WT mice and immunized recipients with MOG35-55/CFA. In unimmunized recipients, CD48 expression was similar on 2D2 and recipient CD4+ T cells in the spleen (Figure S1D, left), whereas in immunized mice the percentage of splenic CD48++ 2D2 cells was increased compared to recipient CD4+ T cells, and correlated with increased percentages of CD44+ and Ki67+ 2D2 CD4+ T cells (Figure S1D, right). These findings suggest that elevated CD48 expression is a feature of antigen-activated CD4+ effector T cells (Teff) in vitro and in vivo.
To investigate functional differences between CD48+ and CD48++ CD4+ T cells, we assessed their cytokine production at the peak of EAE. Among Tconv cells in the spleen, GM-CSF+, IFN-γ+, IL-17A+ and IL-10+ CD4+ T cells were predominantly CD48++, while IL-2+ cells were primarily CD48+ (Figure 1E-F). In the CNS, GM-CSF+, IFN-γ+, IL-17A+ and IL-2+ cells were predominantly CD48++, although some IL-2+ cells had intermediate CD48 expression and IL-10+ cells were a mix of CD48+ and CD48++. We also transferred 2D2 CD4+ T cells into congenic mice, immunized with MOG35-55/CFA, and ten days later sorted CD48++ and CD48+ 2D2 cells from recipients (Figure 1G) and restimulated these populations with MOG35-55 in vitro. CD48++ 2D2 produced IL-17A, IFN-γ, GM-CSF, TNF-α, and IL-6, whereas CD48+ 2D2 produced IL-2 (Figure 1H). There were no significant differences in IL-10 or IL-4 production, or in numbers of 2D2 CD4+ T cells at day 3 of culture. Thus, high CD48 expression identified a distinct functional CD4+ T cell population producing high levels of pathogenic cytokines during EAE.
Anti-CD48 mAb attenuates EAE
The increased expression of CD48 on activated CD4+ T cells producing pathogenic cytokines at peak EAE, together with the costimulatory function of CD48, led us to investigate the consequences of modulating CD48 during EAE using an anti-CD48 mAb. Consistent with prior reports (21, 27), anti-CD48 mAb did not deplete lymphocytes in naïve WT mice (Figure S2A-B), but resulted in downregulation of CD48 surface expression (data not shown) and changes in lymphocyte distribution (Figure S2C-E), including a 33% reduction in splenic CD8+ T cells and a 60% reduction in splenic NK cells but no changes in splenic CD4+ T cells. Anti-CD48 treatment initiated before EAE induction had no significant effect on clinical disease (Figure S3A). In contrast, EAE was dramatically attenuated in mice that received anti-CD48 after EAE induction but prior to disease onset (Figure 2A). These mice had reduced incidence (Table I), delayed onset (Figure 2B), and diminished severity of EAE (Figure 2C). Histological analyses at the onset and peak of disease (days 12 and 15 post-immunization, respectively) revealed few lesions in the CNS of anti-CD48 treated mice, while control mice had numerous lesions in both the brain and spinal cord (Table II, Figure S3B). Cellular analysis showed that anti-CD48 treated mice had fewer CD45hi cells in the CNS compared to control mice at peak of disease (Figure 2D), and a 90% reduction in CD4+ T cells in the CNS (Figure 2E). The percentages of Tregs among CD4+ T cells were similar in the spleen, but lower in the CNS of anti-CD48 treated mice compared to controls (Figure 2F).
Figure 2. Anti-CD48 mAb attenuates MOG35-55-induced EAE.

A-F WT mice were immunized with MOG35-55/CFA, and treated with anti-CD48 or isotype control (cIgG) on days 6, 9 and 12 after immunization. A. Mean clinical scores, ±SEM. B. Day of EAE onset for mice that developed EAE. C. Peak clinical scores for all mice; bar represents median. D-F. CNS and spleen were collected for flow cytometric analysis at the peak of disease. Numbers of CD45+ cells (D) and CD3+CD4+ T cells (E) in the CNS. F. Percentages of Foxp3+ of CD4+CD3+ T cells in CNS and spleen. G-H. WT mice were immunized with MOG35-55/CFA, and anti-CD48 or cIgG treatment was started on the day of EAE onset in each mouse. G. Mean clinical scores, ±SEM, relative to day of disease onset. H. Peak clinical scores; bar represents median. I-J. 2D2 CD4+ T cells were purified by cell sorting and transferred into Rag1−/− mice. Recipients were immunized with MOG35-55/CFA, and given anti-CD48 or cIgG on days 4, 7 and 10. I. Mean clinical scores ±SEM. J. Day of EAE onset. Representative of 6 independent experiments with 4-6 mice per group (A-C), 3 independent experiments with 4-5 mice per group and one naïve control (D-F), 3 independent experiments with 4-8 mice per group (G-H), or 3 independent experiments with 4-6 mice per group (I-J). Gray arrows in A, G and I indicate antibody treatments (Tx). Dots in B, C, H and J represent individual mice.
Table I.
Anti-CD48 mAb limits incidence and severity of EAE in WT mice
| Treatment | cIgG | Anti-CD48 | |
|---|---|---|---|
| During 15 day observation period | Incidence | 9/10 | 0/10*** |
| Median peak score | 2 | NA | |
| Mean day of onset | 12.6 ± 0.4 | NA | |
| During 21 day observation period | Incidence | 15/15 | 8/15** |
| Median peak score | 3 | 1.5** | |
| Mean day of onset | 12.4 ± 0.2 | 17.6 ± 0.6**** | |
WT mice were immunized for EAE as described, and treated with cIgG or anti-CD48 on days 6, 9 and 12 after immunization. Clinical disease was monitored for 15 or 21 days. Data are pooled from 5 independent experiments with 5 mice per group per experiment. Median peak scores and mean day of onset are for sick mice only. NA, not applicable.
* = p<0.05
p<0.01
p<0.001
p<0.0001.
Table II.
Anti-CD48 mAb limits histologic EAE
| Treatment: | cIgG | anti-CD48 |
|---|---|---|
| Number of lesions in brain | ||
| Parenchyma | 39.8 ± 14.2 | 2.4 ± 1.0 * |
| Meninges | 41.6 ± 9.1 | 2.8 ± 1.1 ** |
| Number of lesions in spinal cord | ||
| Parenchyma | 80.4 ± 18.9 | 0 ± 0 ** |
| Meninges | 89.9 ± 14.0 | 0.8 ± 0.5 *** |
| Clinical incidence | 5/5 | 0/5 ** |
| Day of onset | 13.4 ± 0.2 | NA |
| Maximum clinical score | 2 | NA |
WT mice were immunized for EAE as described, and treated with cIgG or anti-CD48 on days 6, 9 and 12 after immunization. Mice were sacrificed on day 15, and brain and spinal cord were collected for histology. Data are mean ±SEM, except clinical score is median. NA, not applicable.
p<0.05
p<0.01
p<0.001.
We also examined whether anti-CD48 could attenuate EAE therapeutically. Antibody administration starting on the day of EAE onset reduced clinical scores throughout the course of treatment (Figure 2G, H). When anti-CD48 treatment was initiated later during EAE when all mice had clinical scores of 2 or greater, resolution of disease was modestly increased compared to controls (Figure S3C-D). Collectively, these data indicate that anti-CD48 can significantly attenuate EAE in WT mice, with the extent of clinical benefit depending on the timing of treatment.
B cells, CD8 T cells, and NK cells are not required for anti-CD48-mediated attenuation of EAE
B cells and CD8 T cells are not critical for EAE, but can modulate disease (28-30). To determine whether these cells were required for the ability of anti-CD48 to attenuate EAE, we transferred purified 2D2 CD4+ T cells into Rag1−/− mice and induced EAE by MOG35-55/CFA immunization. Anti-CD48 treatment delayed EAE onset and reduced disease severity in recipients (Figure 2I, J), with the protective effects diminishing after the end of treatment. NK cells can modulate EAE (31, 32) and CD48 can regulate NK cell function. However, NK cell depletion in WT mice did not alter the therapeutic effects of anti-CD48 on EAE (Figure S4A-D). When we transferred 2D2 CD4+ T cells into Il2rγc−/−Rag2−/− mice (which lack NK, T and B cells) and induced EAE with MOG35-55/CFA, anti-CD48 treatment delayed EAE onset and limited 2D2 CD4+ T cell accumulation in the CNS but did not significantly reduce peak clinical scores (Figure S4E-H). Collectively, these data indicate that B cells, CD8 T cells and NK cells are not essential for the ability of anti-CD48 to attenuate EAE.
Anti-CD48 limits antigen-specific cytokine production in the spleen
Because CD4+ T cells are critical for EAE, and were reduced in the CNS of anti-CD48 treated mice, we hypothesized that anti-CD48 reduced the generation and/or function of pathogenic MOG-specific CD4+ T cells. To test this, we immunized mice with MOG35-55/CFA and gave anti-CD48 or cIgG 6 and 9 days later, and examined cytokine production by CD4+ T cells from the spleen and iLN on day 12 after EAE induction, by restimulating spleen and LN cells with MOG35-55 in vitro. There were no differences in cytokines in the iLN, but splenocytes from anti-CD48 treated mice produced less IFN-γ, TNF-α and IL-17A compared to controls (Figure 3A), despite similar percentages of CD4+ T cells (Figure 3B) and Tregs (Figure 3C) in the starting cultures.
Figure 3. Anti-CD48 reduces antigen-specific cytokine production in the spleen but not the draining LN.

A-C WT mice were immunized with MOG35-55/CFA, and given anti-CD48 or cIgG 6 and 9 days later. Spleen and iLN were collected on day 12. A. Cytokines in supernatants from spleen (top) or iLN (bottom) cells restimulated with the indicated concentrations of MOG35-55 for 3 days. Percentages of CD4+ of total cells (B) and Foxp3+ of CD4+ (C) in the iLN and spleen ex vivo. D-F. WT mice were given 2D2 Thy1.1+ CD4+ T cells i.v., immunized with MOG35-55/CFA, and treated with anti-CD48 or cIgG 4 and 6 days later. Spleen, iLN and peripheral blood were collected on day 8 and restimulated with PMA/ionomycin. D. Representative intracellular cytokine staining in splenic 2D2 Thy1.1+ CD4+ T cells. E. Percentages of cytokine-positive cells among 2D2 cells in the spleen, iLN and blood, gated as in D. F. Percentages of cytokine-positive cells among recipient Thy1.1− CD4+ cells. Representative of 2 independent experiments with 5 mice per group (A-C), and 4 independent experiments with 3-7 mice per group (D-F).
To determine if this was due to fewer antigen-specific cytokine-producing cells or reduced cytokine production on a per cell basis, we transferred 2D2 CD4+ T cells to wild type mice, immunized with MOG35-55/CFA, administered anti-CD48 or cIgG on days 4 and 7 and examined cytokine production by intracellular staining on day 8 post-immunization. There were no differences in the MFIs of cytokine-producing 2D2 cells (Figure 3D), but anti-CD48 treated mice had lower percentages of IFN-γ+, IL-17A+ and GM-CSF+ 2D2 CD4+ T cells in the spleen, compared to controls (Figure 3D, E). There were very few splenic IL-10+ 2D2 CD4+ T cells, and no differences between control and anti-CD48 treated mice in percentages of IL-2+ 2D2 cells (data not shown) or cytokine-producing 2D2 cells in the iLNs (Figure 3E). Cytokine production by recipient CD4+ T cells in control and anti-CD48 treated mice were largely unchanged (Figure 3F). These findings suggest that anti-CD48 treatment during EAE reduces the percentage of antigen-specific cytokine-producing CD4+ T cells in the spleen before disease onset, but does not affect cytokine production at the site of T cell priming in the iLN.
Anti-CD48 attenuates EAE during the effector phase of disease
To distinguish effects of anti-CD48 on T cell differentiation and effector function, we next asked if anti-CD48 could limit EAE induced by adoptively transferred Th1-polarized 2D2 cells (33). This approach circumvents potential effects of anti-CD48 on CD4+ T cell differentiation. Anti-CD48 administration after adoptive transfer but prior to disease onset resulted in significantly attenuated EAE (Figure 4A), decreased peak clinical scores (Figure 4B), and reduced 2D2 CD4+ T cell accumulation in the CNS at the peak of disease (Figure 4C, left), compared to controls. To exclude failed adoptive transfers, we investigated whether 2D2 CD4+ T cells were detectable in the spleens of all recipients, and found that 2D2 CD4+ T cells were abundant in the spleens of anti-CD48 treated mice (Figure 4C, right). The overall cellularity of spleens was higher in disease-free mice compared to mice with clinical EAE (data not shown), consistent with our prior experience. The proportion of splenic Tregs was not altered in anti-CD48-treated mice (Figure 4D), but the percentage of Tregs among CD4+ T cells in the CNS was lower in anti-CD48 treated mice compared to controls (Figure 4D), although very few CD4+ T cells could be recovered from the CNS of disease-free mice. These data indicate that anti-CD48 can attenuate EAE induced by adoptive transfer of myelin-reactive CD4+ Teff.
Figure 4. Anti-CD48 attenuates EAE and limits Teff cell numbers during the effector phase of disease.

A-D 2D2 Th1 CD4+ Teff (Thy1.1) cells were transferred i.p. to WT recipients, and anti-CD48 or cIgG was given on days 4, 7 and 10. CNS and spleen were collected on day 13 for analysis by flow cytometry. A. Mean clinical scores, ±SEM. B. Peak clinical scores for all mice; bar represents median. C. Number of 2D2 CD4+Thy1.1+ T cells in the CNS (left) and spleen (right). D. Percentages of Foxp3+ of recipient CD4+Thy1.1− T cells in CNS and spleen. E-M. 2D2 Th1 CD4+ Teff (Thy1.1) were transferred i.v. into WT recipients that received anti-CD48 or cIgG on day 3 and EdU on day 4. Spleen, iLN and blood were collected on day 5 for analysis by flow cytometry. 2D2 and recipient CD4+ T cells were gated as Thy1.1+ CD4+ and Thy1.1− CD4+, respectively. E. Experimental setup. F. Representative CD48, CD44 and Ki67 expression on 2D2 and recipient CD4+ T cells in control mice. G. Cells were restimulated in vitro with PMA/ionomycin. Representative IFN-γ staining (left) and percentages of IFN-γ+ (right) of 2D2 CD4+ cells. Number of 2D2 (H) and IFN-γ+ 2D2 (I) CD4+ T cells. J. Representative Ki67 expression (left) and percentages of Ki67+ (right) 2D2 CD4+ cells. K. Representative EdU staining (left) and percentages of EdU+ (right) 2D2 CD4+ cells. L. Numbers of recipient CD4+ T cells. M. Percentages of Foxp3+ of recipient CD4+ T cells. Representative of 3 independent experiments with 5-7 mice per group. Gray arrows in A indicate antibody treatments (Tx). Dots in B, C and L represent individual mice. NA, not applicable.
Anti-CD48 limits CD4+ Teff numbers and proliferation in vivo
Because cellular differences at the peak of EAE could be confounded by the inherent differences between sick and healthy mice, we next examined a time point prior to disease onset to investigate how anti-CD48 affects 2D2 CD4+ Teff cells. We gave anti-CD48 on day 3 and analyzed cells on day 5 after adoptive transfer of 2D2 Th1 cells into wild type recipients (Figure 4E). Splenic 2D2 CD4+ Teff were predominantly CD48++, CD44+ and Ki67+, consistent with their activated phenotype (Figure 4F). There were no differences in the percentages of IFN-γ+ 2D2 CD4+ Teff in control and anti-CD48 treated mice (Figure 4G). However, anti-CD48 treated mice had significantly fewer 2D2 CD4+ T cells in the spleen, blood and LNs (Figure 4H), resulting in greatly reduced numbers of IFN-γ-producing 2D2 CD4+ Teff compared to controls at this early time point (Figure 4I). The percentages of Ki67+ (Figure 4J) and EdU+ (Figure 4K) 2D2 CD4+ Teff also were decreased in anti-CD48 treated mice, suggesting that anti-CD48 limited proliferation of Teff in vivo. These changes were antigen-specific, as the numbers of recipient CD4+ T cells were not altered (Figure 4L). The percentages of Tregs were similar in mice given anti-CD48 or cIgG (Figure 4M), resulting in increased Treg:2D2 ratios in anti-CD48 treated mice (data not shown). We examined whether increased cell death might contribute to reduced cell numbers, but there was not increased apoptosis in anti-CD48 treated mice as assessed by Annexin V staining in 2D2 CD4+ T cells, and only a trend towards increased FITCZ-VAD-FMK (a measure of active caspases), compared to controls (data not shown). Thus, 2D2 CD4+ Teff numbers and proliferation are reduced in anti-CD48 treated mice prior to onset of EAE, resulting in fewer IFN-γ-producing myelin-reactive CD4+ Teff. This suggests that anti-CD48 attenuates EAE by limiting the number of pathogenic CD4+ Teff at early phases such that clinical disease is mild or not detectable.
CD48 expression on CD4+ T cells is required for anti-CD48-mediated attenuation of EAE
To determine the cell types responsible for the effects of anti-CD48 on limiting 2D2 CD4+ Teff proliferation, we next evaluated the effects of anti-CD48 on WT and CD48−/− 2D2 Th1 CD4+ T cells restimulated in vitro with MOG35-55 and either WT or CD48−/− APCs. Anti-CD48 reduced proliferation of both WT and CD48−/− 2D2 CD4+ Teff restimulated with WT APCs, as assessed by dilution of CellTrace Violet (Figure 5A-B). However, anti-CD48 had a minimal effect on proliferation of WT 2D2 CD4+ Teff restimulated with CD48−/− APCs, and only at low MOG35-55 concentrations (Figure 5A). This suggests that anti-CD48 limits proliferation primarily by blocking CD48 on APCs, and to a lesser extent by blocking CD48 on T cells. This is consistent with a costimulatory role for CD48 (9), and indicates that anti-CD48 can decrease expansion of myelin-specific CD4+ Teff in vitro.
Figure 5. Anti-CD48 mAb limits proliferation and attenuates EAE through interactions with CD48 on distinct cell populations.

A-B. Representative CellTrace Violet dilution in Th1-polarized 2D2 (A) or CD48−/− 2D2 (B) CD4+ T cells restimulated in vitro with TCRα−/− (top rows) or CD48−/− TCRα−/− (bottom rows) splenocytes and MOG35-55 plus 10μg/mL cIgG or anti-CD48 for 3 days. C-F. 2D2 (C-D) or CD48−/− 2D2 (E-F) CD4+ T cells were purified by cell sorting and transferred to CD48−/− Rag−/− or Rag−/− recipients, respectively. Recipients were immunized with MOG35-55/CFA, and treated with anti-CD48 or cIgG on days 4, 7 and 10. C, E Mean clinical scores, ±SEM. E, F Number of 2D2 CD4+Thy1.1+ T cells in the CNS (left) and spleen (right). Representative of 5 independent experiments (A), 2 independent experiments (B), 4 independent experiments with 3-5 mice per group (C-D), 5 independent experiments with 4-6 mice per group (E-F). Gray arrows in C and E indicate antibody treatments (Tx). Dots in D and F represent individual mice.
We hypothesized that the ability of anti-CD48 to attenuate EAE would similarly rely on CD48 expression on APCs. We have found that CD48−/− mice can develop EAE (data not shown). Therefore, to test our hypothesis we transferred WT 2D2 CD4+ T cells into CD48−/− Rag1−/− mice, immunized with MOG35-55/CFA, and treated with anti-CD48 prior to disease onset. Surprisingly, anti-CD48 was able to completely prevent EAE in these mice (Figure 5C), limiting the number of 2D2 CD4+ T cells in both the CNS and spleen when compared to controls (Figure 5D). In contrast, when we transferred CD48−/− 2D2 CD4+ T cells to Rag1−/− mice and immunized with MOG35-55/CFA, anti-CD48 did not attenuate EAE (Figure 5E), nor limit CD4+ T cell accumulation in the CNS (Figure 5F). Similarly, anti-CD48 attenuated 2D2 Th1 adoptive transfer EAE in CD48−/− recipients, but not CD48−/− 2D2 Th1 adoptive transfer EAE in WT recipients (data not shown). Collectively, these findings demonstrate that CD48 expression on CD4+ T cells is required for anti-CD48-mediated attenuation of EAE, and implicate a mechanism of action other than limiting proliferation.
An FcγR blocking mAb limits the effects of anti-CD48 on 2D2 CD4+ Teff numbers in vivo
Although the anti-CD48 mAb that we used is not reported to be depleting, we considered that FcγR-mediated effects of anti-CD48 might be involved in limiting the number of Teff in vivo. To investigate this possibility, we gave an anti-FcγRII/FcγRIII blocking antibody (anti-FcγR) along with anti-CD48 to WT recipients of 2D2 Th1 CD4+ Teff, and examined cells in the spleen on day 5 after transfer (Figure 6A). Anti-CD48 did not reduce the numbers of total 2D2 (Figure 6B) or IFN-γ+ 2D2 CD4+ Teff (Figure 6C) in mice given anti-FcγR, in contrast to controls, suggesting that anti-CD48 decreases 2D2 CD4+ Teff numbers in an FcγR-dependent fashion. However, mice treated with anti-CD48 plus anti-FcγR had reduced percentages of Ki67+ (Figure 6D) and EdU+ (data not shown) 2D2 CD4+ Teff, compared to mice that received anti-FcγR only, suggesting the effects of anti-CD48 on proliferation are FcγR-independent. 2D2 CD4+ Teff in anti-CD48-treated mice had decreased percentages of α4 integrinhi (Figure 6E) and increased percentages of CD69+ (Figure 6F) cells compared to controls, irrespective of anti-FcγR treatment. Taken together, these results suggest that reduced Teff numbers in the spleens of anti-CD48-treated mice are due at least in part to an FcγR-dependent mechanism, but that anti-CD48 also has FcγR-independent effects on T cells.
Figure 6. An FcγR blocking antibody limits the effect of anti-CD48 on 2D2 Th1 CD4+ Teff cells in vivo.
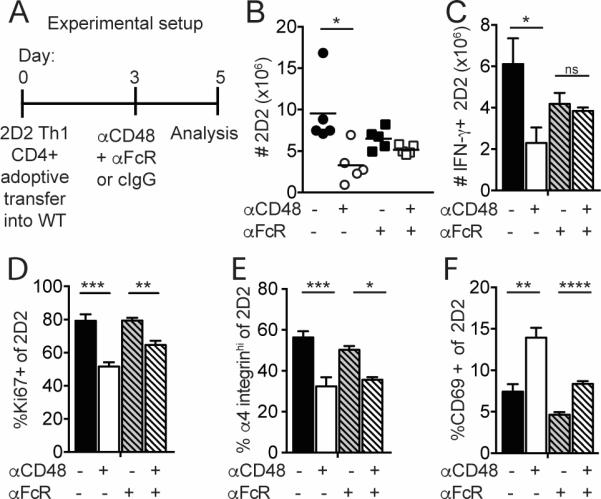
2D2 Th1 CD4+ Teff (Thy1.1+) cells were transferred i.v. into WT recipients that received anti-CD48 or cIgG plus anti-CD16/CD32 or isotype control on day 3. Spleen was collected on day 5 for analysis by flow cytometry as in Figure 5. A. Experimental setup. B. Numbers of 2D2 CD4+ in the spleen. C. Numbers of IFN-γ+ 2D2 cells after restimulation with PMA/ionomycin. Percentages of Ki67+ (D), α4-integrinhi (E) and CD69+ (F) of 2D2 CD4+ T cells. Representative of 4 independent experiments with 4-5 mice per group. Dots in B represent individual mice.
FcγRs contribute to anti-CD48-mediated attenuation of EAE
To assess the role of FcγRs in anti-CD48-mediated EAE amelioration, we co-administered anti-FcγR and anti-CD48 to mice during EAE. While mice given only anti-CD48 did not develop EAE, clinical EAE was somewhat attenuated in mice treated with anti-CD48 plus anti-FcγR (Figure 7A), compared to mice given only anti-FcγR. However, there were no significant differences in the day of onset (Figure 7B), peak clinical scores (Figure 7C), number of CD4+ T cells in the CNS (Figure 7D), or percentage of Tregs among CD4+ T cells in the CNS or spleen (Figure 7E) in mice given anti-CD48 plus anti-FcγR compared to mice given only anti-FcγR.
Figure 7. Fcγ receptors contribute to anti-CD48-mediated attenuation of EAE.

A-E WT mice were immunized with MOG35-55/CFA and given anti-CD48 or cIgG plus anti-CD16/CD32 or isotype control on days 8, 11 and 14 after immunization. Spleen and CNS were collected on day 17 for analysis by flow cytometry. A. Mean clinical scores, ±SEM. B. Day of EAE onset for mice that developed EAE. C. Peak clinical scores for all mice; bar represents median. D. Number of CD4+ T cells in the CNS. E. Percentages of Foxp3+ of CD4+ T cells. F-J. Fcεr1γ−/− mice were immunized with MOG35-55/CFA, and given anti-CD48 or cIgG on days 7, 10 and 13 after immunization. On day 16, CNS, spleen and iLN were collected for analysis by flow cytometry. F. Mean clinical scores, ±SEM. G. Day of EAE onset. H. Peak clinical scores; bar represents median. I. Numbers of CD4+ T cells in the CNS. J. Percentages of Foxp3+ of CD4+ T cells. Representative of 5 independent experiments with 4-5 mice per group (A-E), and 6 independent experiments with 3-7 mice per group (F-J). Gray arrows in A and F indicate antibody treatments (Tx). Dots in B, C, D, G, H and I represent individual mice.
As a second means to evaluate the role of FcγRs in anti-CD48-mediated EAE attenuation, we administered anti-CD48 to Fcεr1γ−/− mice, which lack the common gamma chain for FcγRs and therefore lack surface expression of FcγR I, III and IV. Although Fcεr1γ−/− mice have delayed onset and severity of MOG35-55/CFA-induced EAE compared to WT mice (34, 35), anti-CD48 failed to attenuate EAE in this strain (Figure 7F). The day of onset was slightly delayed (Figure 7G) and peak scores were slightly lower (Figure 7H) in anti-CD48 treated Fcεr1γ−/− mice compared to controls, but these changes were not significant. Furthermore, the number of CD4+ T cells in the CNS was not reduced in anti-CD48 treated Fcεr1γ−/− mice (Figure 7I), nor were there differences in the percentages of Tregs among CD4+ T cells in the CNS, spleen or iLN (Figure 7J). These results suggest that FcγRs are critical for anti-CD48 mAb to attenuate EAE.
DISCUSSION
The association of CD48 and CD58 loci with risk for MS, combined with the known roles for CD48 in T cell activation and tolerance in mice, led us to examine the role of CD48 in EAE. We identified a subpopulation of CD4+ T cells that highly express CD48 (CD48++) during EAE, with features of pathogenic myelin-reactive T cells. CD48++ CD4+ T cells were enriched in the CNS at the peak of EAE, and included IFN-γ+, GM-CSF+ and IL-17A+ cells. Treatment with anti-CD48 mAb during EAE attenuated disease, limited CD4+ T cell accumulation in the CNS, and reduced the number of MOG-specific cytokine-producing CD4+ Teff in the spleen prior to disease onset. The ability of anti-CD48 to attenuate EAE was critically dependent on CD48 expression on CD4+ T cells, and was abrogated in Fcεr1γ−/− mice and in WT mice treated with anti-CD16/CD32. While CD48 is widely expressed on immune cells, these observations collectively suggest that anti-CD48 exerts its effects on EAE primarily by an FcγR-dependent mechanism, resulting in reduced pathogenic CD4+ T cell numbers mainly through interactions of anti-CD48 with CD48 on CD4+ T cells. Our results also show that high CD48 expression can serve as a biomarker of pathogenic CD4+ T cells during EAE and point to CD48 as a potential target for immunotherapy for T cell-mediated autoimmune diseases.
While little is known about CD48 in human disease, mouse studies have indicated important immunoregulatory functions for this receptor. CD48 on both the T cell and APC contributes to T cell activation (9, 36), and CD48 deficiency on T cells or APCs significantly attenuated disease in a mouse model of colitis (20). In models of allergy and asthma, CD48 expression on eosinophils and mast cells promoted the inflammatory response, and anti-CD48 treatment limited eosinophil accumulation in the lung after antigen rechallenge (27). Furthermore, as the ligand for CD244, CD48 is involved in controlling NK and CD8 T cell cytotoxicity (13), and anti-CD48 treatment can limit transplant rejection in mice (22). Consistent with these roles for CD48, we found that anti-CD48 mAb limited proliferation of CD4+ Teff cells in vivo and in vitro. Anti-CD48 limited proliferation of CD4+ T cells primarily by blocking CD48 on APCs, as indicated by the ability of anti-CD48 to limit proliferation of CD48−/− CD4+ T cells stimulated with WT APCs in vitro, while having minimal effect on proliferation of WT CD4+ T cells stimulated with CD48−/− APCs (Figure 5A). Anti-CD48 attenuated EAE when administered during early phases of disease and reduced CD4+ Teff numbers in an FcγR-dependent fashion.
Our data support several mechanisms by which anti-CD48 attenuates EAE. CD48 expression on CD4+ T cells was necessary for the ability of anti-CD48 mAb to attenuate EAE, and CD48 expression on other cell types was not required, as demonstrated by the ability of anti-CD48 to attenuate EAE mediated by WT CD4+ T cells in CD48−/− mice, but not in WT mice given CD48−/− 2D2 CD4+ T cells. These findings suggest that the anti-proliferative effects of CD48 blockade on APCs are not sufficient to attenuate EAE. FcγRs were critical for EAE amelioration by anti-CD48. At early time points after adoptive transfer of 2D2 CD4+ T cells into WT recipients, anti-CD48 treatment resulted in reduced numbers of 2D2 CD4+ Teff cells, while anti-CD48 combined with anti-CD16/CD32 treatment did not. Furthermore, anti-CD48 failed to attenuate EAE in WT mice given anti-CD16/CD32 or in Fcεr1γ−/− mice. Collectively, these data suggest that anti-CD48 exerts its beneficial effects on EAE by preventing the number of pathogenic CD4+ T cells from exceeding a critical threshold, by interacting with CD48 on CD4+ T cells and with FcγRs.
Intriguingly, the effects of anti-CD48 on CD4+ T cell numbers were most pronounced on pathogenic cytokine-producing CD4+ T cells, and were greater on cells in the spleen and blood than the LNs. We speculate that this could be due to a combination of factors. First, elevated CD48 expression on activated CD4+ T cells may render these cells more susceptible to effects of the mAb. Second, trafficking or tissue localization specific to activated CD4+ T cells may lead to increased encounters with FcγR-expressing cells, as suggested by our observation of reduced cytokine production by cells from the spleen but not the iLN in anti-CD48 treated MOG35-55/CFA-immunized mice (Figure 3A, 3E). CD48++ CD4+ T cells appear to be most susceptible to anti-CD48-mediated elimination after leaving the LN, likely related to encounters with FcγR-expressing cells such as Kupffer cells in the liver or macrophages in the spleen. The lack of a second anti-CD48 mAb clone, together with the downregulation of CD48 after anti-CD48 treatment, prevented us from quantifying the number and localization of CD48++ cells in anti-CD48 treated mice after MOG35-55/CFA immunization. However, when we used cytokine production as a surrogate marker for CD48++, we observed that cytokine-producing 2D2 CD4+ T cells were reduced in the spleen and blood, but not in the iLN, of anti-CD48 treated mice (Figure 3D). Similarly, 2D2 Th1 CD4+ Teff cells (which are uniformly CD48++ cells) (Figure 5C), were diminished in anti-CD48 treated mice on day 5 after adoptive transfer (85% reduction in the blood, 77% reduction in the spleen, and 64% reduction in LNs (Figure 4H), compared to controls), but there was no change in the number of host Treg (Figure 4M) or Tconv cells (Figure 4L). Collectively, these data support a model whereby anti-CD48 exerts its greatest FcγR-dependent effects on circulating cells with high CD48 expression.
The enhanced effect on CD48++ cell numbers in the blood is reminiscent of the effects of other depleting antibodies. Recently, Montalvao et al. reported that Kupffer cells in the liver contribute to B cell elimination by the anti-CD20 mAb rituxan, resulting in accelerated and enhanced depletion of B cells from the blood and liver compared to spleen and LNs (37). Although further studies are needed to identify key cell populations involved in anti-CD48-mediated reduction of CD4+ Teff cells in EAE, such a mechanism for anti-CD48-mediated depletion is consistent with our observations. Notably, anti-CD2 mAb treatment in the Lewis rat model of EAE attenuated disease and reduced the number of circulating T cells (38), and these effects were thought to be due to depletion of T cells (39) and/or suppression of activation (40). This parallels our studies of anti-CD48 in EAE in mice, and suggests that targeting this pathway in MS with depleting and/or blocking mAb merits further investigation.
We were surprised to find that an antibody targeting a widely expressed surface antigen could lead to selective cell reduction, especially considering that other groups had found that this anti-CD48 mAb does not cause depletion of lymphocytes. However, other antibodies are described to eliminate cells with high antigen expression preferentially. For example, the anti-CD25 mAb (clone PC61) eliminates Tregs, which highly express CD25, more than Tconv (41). Further studies evaluating the effect of anti-CD48 on all cell types, in additional disease models, will be critical to evaluate the therapeutic applications of anti-CD48 antibodies. Although we found that B cells and CD8+ T cells were not required for anti-CD48 to attenuate EAE, and that anti-CD48 had its most profound effect during EAE on CD48++ CD4+ T cells, anti-CD48 is known to affect other cell subsets in other contexts.
Importantly, depleting and blocking effects of anti-CD48 are not mutually exclusive, and both may contribute to EAE attenuation. Although anti-CD48 could attenuate EAE independent of T cell priming, this does not exclude a role for anti-CD48 in priming of CD4+ T cells in the MOG35-55/CFA immunization model of EAE. We observed changes in α4 integrin and CD69 expression on 2D2 Th1 CD4+ Teff after combined anti-CD48 and anti-CD16/CD32 treatment (Figure 6E-F), suggesting that anti-CD48 may alter Teff cell activation independent of FcγRs. In addition, when we gave a single dose of anti-CD48 at the peak of EAE, we observed a reduction in Ki67+ cells among CD4+ T cells in the CNS two days later (data not shown), suggesting that anti-CD48 also might limit proliferation in the CNS directly, and/or reduce entry of activated Ki67+ CD4+ T cells into the inflamed CNS, even though the clinical effects of treatment during peak disease were mild (Figure S3). Thus, it appears that anti-CD48 may act at multiple locations and at multiple time points during EAE. The generation of non-depleting anti-CD48 blocking mAbs is needed to enable temporal dissection of CD48 function in EAE without the confounding factor of FcγR-mediated effects.
Despite constitutive expression of CD48 on hematopoietic cells, our studies reinforce that CD48 expression is dynamically modulated on T cells and suggest that elevated CD48 expression may serve as a biomarker for functional differences in Teff. We found that CD48++ CD4+ T cells were a subset of CD44+ cells during EAE, and that this CD48++ population contained the majority of inflammatory cytokine-producing cells, suggesting that CD48++ CD4+ T cells are a unique subset of activated T cells. Increased CD48 expression also has been described on T cells during LCMV infection (16) and on CD4+ T cells undergoing proliferation in vivo and in vitro (17), but functional differences between CD48++ and CD48+ cells were not examined. We have begun to compare transcriptional differences between CD48++ and CD48+ CD4+ T cells and found that CD48 is upregulated at a transcriptional level in CD48++ cells, along with genes associated with pathogenic T cell responses in EAE including Bhlhe40 (42), Rorα (43), and Id2 (44). These transcriptional analyses confirm our observations that CD48++ CD4+ T cells are highly activated, but do not correlate specifically with Th1 or Th17 transcriptional signatures (McArdel, unpublished data). Consistent with these findings, CD48 surface expression did not differ between Th1 and Th17 CD4+ T cells differentiated in vitro (data not shown). While CD48 upregulation can be induced in vitro with antigen or anti-CD3 plus anti-CD28 stimulation (Figure S1), further work is needed to understand how CD48 expression is regulated in vivo. It is not yet clear if or how elevated CD48 expression itself influences T cell function. CD48 reduces the threshold for T cell activation in a cell intrinsic manner, via its association with LAT and CD2 (9-11), so it is plausible that increased CD48 surface expression modulates the threshold for re-activation of Teff cells. Understanding the regulation and function of elevated CD48 expression may aid in understanding the characteristics of CD48++ CD4+ T cells.
While CD48 function has not been examined in human autoimmune diseases, studies of CD2 and CD58 suggest that the CD48:CD2:CD58 pathway is important in autoimmunity. In humans, CD58 is widely expressed on both hematopoietic and epithelial cells (12), and is a risk allele in MS. Further work is needed to understand how CD58 is linked to immune dysregulation. CD2 is a therapeutic target for immunosuppression. The CD58-Ig fusion protein, alefacept, was found to specifically reduce effector memory T cells in the blood and reduced disease symptoms in patients with psoriasis (45) and type 1 diabetes (46). Our findings give impetus to the investigation of CD48 in MS.
In summary, our studies demonstrate that CD48 is highly expressed on pathogenic CD4+ T cells during EAE and point to CD48 as a potential therapeutic target for autoimmune diseases. During EAE, CD48 cell surface expression increased on both encephalitogenic CD4+ T cells and Tregs. However, treatment with anti-CD48 during EAE selectively reduced CD4+ Teff, but not Tregs, and dramatically attenuated disease. Our studies, together with the associations of CD48 and CD58 with susceptibility to MS, support further investigation of the CD48:CD2:CD58 pathway in MS and autoimmunity.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Justin Trombley for technical assistance, Baolin Chang, Xiaohui He and Huiping Zhang for mouse colony maintenance, Sarah Hillman for administrative support, and Dr. Peter Sage for thoughtful discussion. Because of space restrictions, we were able to cite only a fraction of the relevant literature and we apologize to colleagues whose contributions may not be acknowledged.
Footnotes
This work was supported by NIH grant PO1 AI065687.
Abbreviations used: MS, multiple sclerosis; APC, antigen presenting cell; EAE, experimental autoimmune encephalomyelitis; WT, wild type; MOG35-55, myelin oligodendrocyte glycoprotein 35-55 peptide; EdU, 5-ethynyl-2’-deoxyuridine; LN, lymph node; iLN, inguinal LN; Treg, T regulatory cell; Tconv, T conventional cell; Teff, T effector cell.
REFERENCES
- 1.International Multiple Sclerosis Genetics, C. Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, de Bakker PI, Gabriel SB, Mirel DB, Ivinson AJ, Pericak-Vance MA, Gregory SG, Rioux JD, McCauley JL, Haines JL, Barcellos LF, Cree B, Oksenberg JR, Hauser SL. Risk alleles for multiple sclerosis identified by a genomewide study. The New England journal of medicine. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 2.International Multiple Sclerosis Genetics, C. Network-based multiple sclerosis pathway analysis with GWAS data from 15,000 cases and 30,000 controls. American journal of human genetics. 2013;92:854–865. doi: 10.1016/j.ajhg.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Compston A, Coles A. Multiple sclerosis. Lancet. 2002;359:1221–1231. doi: 10.1016/S0140-6736(02)08220-X. [DOI] [PubMed] [Google Scholar]
- 4.De Jager PL, Baecher-Allan C, Maier LM, Arthur AT, Ottoboni L, Barcellos L, McCauley JL, Sawcer S, Goris A, Saarela J, Yelensky R, Price A, Leppa V, Patterson N, de Bakker PI, Tran D, Aubin C, Pobywajlo S, Rossin E, Hu X, Ashley CW, Choy E, Rioux JD, Pericak-Vance MA, Ivinson A, Booth DR, Stewart GJ, Palotie A, Peltonen L, Dubois B, Haines JL, Weiner HL, Compston A, Hauser SL, Daly MJ, Reich D, Oksenberg JR, Hafler DA. The role of the CD58 locus in multiple sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5264–5269. doi: 10.1073/pnas.0813310106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arthur AT, Armati PJ, Bye C, Southern MSGC, Heard RN, Stewart GJ, Pollard JD, Booth DR. Genes implicated in multiple sclerosis pathogenesis from consilience of genotyping and expression profiles in relapse and remission. BMC medical genetics. 2008;9:17. doi: 10.1186/1471-2350-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaplan AJ, Chavin KD, Yagita H, Sandrin MS, Qin LH, Lin J, Lindenmayer G, Bromberg JS. Production and characterization of soluble and transmembrane murine CD2. Demonstration that CD48 is a ligand for CD2 and that CD48 adhesion is regulated by CD2. Journal of immunology. 1993;151:4022–4032. [PubMed] [Google Scholar]
- 7.Brown MH, Boles K, van der Merwe PA, Kumar V, Mathew PA, Barclay AN. 2B4, the natural killer and T cell immunoglobulin superfamily surface protein, is a ligand for CD48. The Journal of experimental medicine. 1998;188:2083–2090. doi: 10.1084/jem.188.11.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milstein O, Tseng SY, Starr T, Llodra J, Nans A, Liu M, Wild MK, van der Merwe PA, Stokes DL, Reisner Y, Dustin ML. Nanoscale increases in CD2-CD48-mediated intermembrane spacing decrease adhesion and reorganize the immunological synapse. The Journal of biological chemistry. 2008;283:34414–34422. doi: 10.1074/jbc.M804756200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez-Cabrero J, Wise CJ, Latchman Y, Freeman GJ, Sharpe AH, Reiser H. CD48-deficient mice have a pronounced defect in CD4(+) T cell activation. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:1019–1023. doi: 10.1073/pnas.96.3.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moran M, Miceli MC. Engagement of GPI-linked CD48 contributes to TCR signals and cytoskeletal reorganization: a role for lipid rafts in T cell activation. Immunity. 1998;9:787–796. doi: 10.1016/s1074-7613(00)80644-5. [DOI] [PubMed] [Google Scholar]
- 11.Muhammad A, Schiller HB, Forster F, Eckerstorfer P, Geyeregger R, Leksa V, Zlabinger GJ, Sibilia M, Sonnleitner A, Paster W, Stockinger H. Sequential cooperation of CD2 and CD48 in the buildup of the early TCR signalosome. Journal of immunology. 2009;182:7672–7680. doi: 10.4049/jimmunol.0800691. [DOI] [PubMed] [Google Scholar]
- 12.Springer TA, Dustin ML, Kishimoto TK, Marlin SD. The lymphocyte function-associated LFA-1, CD2, and LFA-3 molecules: cell adhesion receptors of the immune system. Annual review of immunology. 1987;5:223–252. doi: 10.1146/annurev.iy.05.040187.001255. [DOI] [PubMed] [Google Scholar]
- 13.Waggoner SN, Kumar V. Evolving role of 2B4/CD244 in T and NK cell responses during virus infection. Frontiers in immunology. 2012;3:377. doi: 10.3389/fimmu.2012.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elishmereni M, Levi-Schaffer F. CD48: A co-stimulatory receptor of immunity. The international journal of biochemistry & cell biology. 2011;43:25–28. doi: 10.1016/j.biocel.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 15.McArdel SL, Terhorst C, Sharpe AH. Roles of CD48 in regulating immunity and tolerance. Clinical immunology. 2016;164:10–20. doi: 10.1016/j.clim.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature. 2012;481:394–398. doi: 10.1038/nature10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fazekas de St Groth B, Smith AL, Higgins CA. T cell activation: in vivo veritas. Immunology and cell biology. 2004;82:260–268. doi: 10.1111/j.0818-9641.2004.01243.x. [DOI] [PubMed] [Google Scholar]
- 18.Keszei M, Latchman YE, Vanguri VK, Brown DR, Detre C, Morra M, Arancibia-Carcamo CV, Paul E, Calpe S, Castro W, Wang N, Terhorst C, Sharpe AH. Auto-antibody production and glomerulonephritis in congenic Slamf1−/− and Slamf2−/− [B6.129] but not in Slamf1−/− and Slamf2−/− [BALB/c.129] mice. International immunology. 2011;23:149–158. doi: 10.1093/intimm/dxq465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koh AE, Njoroge SW, Feliu M, Cook A, Selig MK, Latchman YE, Sharpe AH, Colvin RB, Paul E. The SLAM family member CD48 (Slamf2) protects lupus-prone mice from autoimmune nephritis. Journal of autoimmunity. 2011;37:48–57. doi: 10.1016/j.jaut.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abadia-Molina AC, Ji H, Faubion WA, Julien A, Latchman Y, Yagita H, Sharpe A, Bhan AK, Terhorst C. CD48 controls T-cell and antigen-presenting cell functions in experimental colitis. Gastroenterology. 2006;130:424–434. doi: 10.1053/j.gastro.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 21.Chavin KD, Qin L, Lin J, Woodward J, Baliga P, Kato K, Yagita H, Bromberg JS. Anti-CD48 (murine CD2 ligand) mAbs suppress cell mediated immunity in vivo. International immunology. 1994;6:701–709. doi: 10.1093/intimm/6.5.701. [DOI] [PubMed] [Google Scholar]
- 22.Qin L, Chavin KD, Lin J, Yagita H, Bromberg JS. Anti-CD2 receptor and anti-CD2 ligand (CD48) antibodies synergize to prolong allograft survival. The Journal of experimental medicine. 1994;179:341–346. doi: 10.1084/jem.179.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson AP, Harp CT, Noronha A, Miller SD. The experimental autoimmune encephalomyelitis (EAE) model of MS: utility for understanding disease pathophysiology and treatment. Handbook of clinical neurology. 2014;122:173–189. doi: 10.1016/B978-0-444-52001-2.00008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. The Journal of experimental medicine. 2003;197:1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 26.Sobel RA, Tuohy VK, Lu ZJ, Laursen RA, Lees MB. Acute experimental allergic encephalomyelitis in SJL/J mice induced by a synthetic peptide of myelin proteolipid protein. Journal of neuropathology and experimental neurology. 1990;49:468–479. doi: 10.1097/00005072-199009000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Munitz A, Bachelet I, Finkelman FD, Rothenberg ME, Levi-Schaffer F. CD48 is critically involved in allergic eosinophilic airway inflammation. American journal of respiratory and critical care medicine. 2007;175:911–918. doi: 10.1164/rccm.200605-695OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krishnamoorthy G, Lassmann H, Wekerle H, Holz A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. The Journal of clinical investigation. 2006;116:2385–2392. doi: 10.1172/JCI28330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bettini M, Rosenthal K, Evavold BD. Pathogenic MOG-reactive CD8+ T cells require MOG-reactive CD4+ T cells for sustained CNS inflammation during chronic EAE. Journal of neuroimmunology. 2009;213:60–68. doi: 10.1016/j.jneuroim.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mann MK, Ray A, Basu S, Karp CL, Dittel BN. Pathogenic and regulatory roles for B cells in experimental autoimmune encephalomyelitis. Autoimmunity. 2012;45:388–399. doi: 10.3109/08916934.2012.665523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcia JA, Pino PA, Mizutani M, Cardona SM, Charo IF, Ransohoff RM, Forsthuber TG, Cardona AE. Regulation of adaptive immunity by the fractalkine receptor during autoimmune inflammation. Journal of immunology. 2013;191:1063–1072. doi: 10.4049/jimmunol.1300040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hao J, Liu R, Piao W, Zhou Q, Vollmer TL, Campagnolo DI, Xiang R, La Cava A, Van Kaer L, Shi FD. Central nervous system (CNS)-resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. The Journal of experimental medicine. 2010;207:1907–1921. doi: 10.1084/jem.20092749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jager A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. Journal of immunology. 2009;183:7169–7177. doi: 10.4049/jimmunol.0901906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szalai AJ, Hu X, Raman C, Barnum SR. Requirement of the Fc receptor common gamma-chain for gamma delta T cell-mediated promotion of murine experimental autoimmune encephalomyelitis. European journal of immunology. 2005;35:3487–3492. doi: 10.1002/eji.200535285. [DOI] [PubMed] [Google Scholar]
- 35.Breij EC, Heijnen P, Vloet R, Saito T, van de Winkel JG, Dijkstra CD, Amor S, Verbeek S. The FcRgamma chain is not essential for induction of experimental allergic encephalomyelitis (EAE) or anti-myelin antibody-mediated exacerbation of EAE. Journal of neuropathology and experimental neurology. 2005;64:304–311. doi: 10.1093/jnen/64.4.304. [DOI] [PubMed] [Google Scholar]
- 36.Latchman Y, Reiser H. Enhanced murine CD4+ T cell responses induced by the CD2 ligand CD48. European journal of immunology. 1998;28:4325–4331. doi: 10.1002/(SICI)1521-4141(199812)28:12<4325::AID-IMMU4325>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 37.Montalvao F, Garcia Z, Celli S, Breart B, Deguine J, Van Rooijen N, Bousso P. The mechanism of anti-CD20-mediated B cell depletion revealed by intravital imaging. The Journal of clinical investigation. 2013;123:5098–5103. doi: 10.1172/JCI70972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jung S, Toyka K, Hartung HP. Suppression of experimental autoimmune encephalomyelitis in Lewis rats by antibodies against CD2. European journal of immunology. 1995;25:1391–1398. doi: 10.1002/eji.1830250538. [DOI] [PubMed] [Google Scholar]
- 39.Hoffmann JC, Herklotz C, Zeidler H, Bayer B, Westermann J. Anti-CD2 (OX34) MoAb treatment of adjuvant arthritic rats: attenuation of established arthritis, selective depletion of CD4+ T cells, and CD2 down-modulation. Clinical and experimental immunology. 1997;110:63–71. doi: 10.1046/j.1365-2249.1997.4881385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sido B, Otto G, Zimmermann R, Muller P, Meuer SC, Dengler TJ. Modulation of the CD2 receptor and not disruption of the CD2/CD48 interaction is the principal action of CD2-mediated immunosuppression in the rat. Cellular immunology. 1997;182:57–67. doi: 10.1006/cimm.1997.1204. [DOI] [PubMed] [Google Scholar]
- 41.Setiady YY, Coccia JA, Park PU. In vivo depletion of CD4+FOXP3+ Treg cells by the PC61 anti-CD25 monoclonal antibody is mediated by FcgammaRIII+ phagocytes. European journal of immunology. 2010;40:780–786. doi: 10.1002/eji.200939613. [DOI] [PubMed] [Google Scholar]
- 42.Lin CC, Bradstreet TR, Schwarzkopf EA, Jarjour NN, Chou C, Archambault AS, Sim J, Zinselmeyer BH, Carrero JA, Wu GF, Taneja R, Artyomov MN, Russell JH, Edelson BT. IL-1-induced Bhlhe40 identifies pathogenic T helper cells in a model of autoimmune neuroinflammation. The Journal of experimental medicine. 2016;213:251–271. doi: 10.1084/jem.20150568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, Watowich SS, Tian Q, Jetten AM, Dong C. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin YY, Jones-Mason ME, Inoue M, Lasorella A, Iavarone A, Li QJ, Shinohara ML, Zhuang Y. Transcriptional regulator Id2 is required for the CD4 T cell immune response in the development of experimental autoimmune encephalomyelitis. Journal of immunology. 2012;189:1400–1405. doi: 10.4049/jimmunol.1200491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chamian F, Lin SL, Lee E, Kikuchi T, Gilleaudeau P, Sullivan-Whalen M, Cardinale I, Khatcherian A, Novitskaya I, Wittkowski KM, Krueger JG, Lowes MA. Alefacept (anti-CD2) causes a selective reduction in circulating effector memory T cells (Tem) and relative preservation of central memory T cells (Tcm) in psoriasis. Journal of translational medicine. 2007;5:27. doi: 10.1186/1479-5876-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rigby MR, Harris KM, Pinckney A, DiMeglio LA, Rendell MS, Felner EI, Dostou JM, Gitelman SE, Griffin KJ, Tsalikian E, Gottlieb PA, Greenbaum CJ, Sherry NA, Moore WV, Monzavi R, Willi SM, Raskin P, Keyes-Elstein L, Long SA, Kanaparthi S, Lim N, Phippard D, Soppe CL, Fitzgibbon ML, McNamara J, Nepom GT, Ehlers MR. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. The Journal of clinical investigation. 2015;125:3285–3296. doi: 10.1172/JCI81722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.