

Abstract
Mammalian artificial chromosomes (MACs) provide a means to introduce large payloads of genetic information into the cell in an autonomously replicating, non-integrating format. Unique among MACs, the mammalian satellite DNA-based Artificial Chromosome Expression (ACE) can be reproducibly generated de novo in cell lines of different species and readily purified from the host cells' chromosomes. Purified mammalian ACEs can then be re-introduced into a variety of recipient cell lines where they have been stably maintained for extended periods in the absence of selective pressure. In order to extend the utility of ACEs, we have established the ACE System, a versatile and flexible platform for the reliable engineering of ACEs. The ACE System includes a Platform ACE, containing >50 recombination acceptor sites, that can carry single or multiple copies of genes of interest using specially designed targeting vectors (ATV) and a site-specific integrase (ACE Integrase). Using this approach, specific loading of one or two gene targets has been achieved in LMTK− and CHO cells. The use of the ACE System for biological engineering of eukaryotic cells, including mammalian cells, with applications in biopharmaceutical production, transgenesis and gene-based cell therapy is discussed.
INTRODUCTION
Although plasmid and viral gene delivery systems have been used successfully to introduce genes of interest into mammalian cell lines and transgenic animals, they are limited with regard to the amount of foreign DNA sequence that can be delivered. For applications where cloning and delivery of large genomic loci are desired, such as fragments containing long-range genetic elements required for appropriate regulation of gene expression, developmentally regulated multi-gene loci, or multiple copies of two or more genes in fixed stoichiometry, plasmid or viral vectors may be inadequate. In order to circumvent these limitations, a second generation of bacterial (1,2) and yeast (3,4) chromosomal-based cloning vectors has been developed, with carrying capacities for yeast artificial chromosomes in excess of 1 Mb (5). However, an additional limitation that the earlier versions of these vector systems share with conventional plasmid vectors is that they require integration into host mammalian chromosomes for stable maintenance. Such integrations are most often random and the sites of integrations may have profound and unpredictable effects on the expression of introduced genes (6). In addition, such random integrations may result in inactivation or change in regulation of host genes (7). To allow for episomal maintenance in mammalian cells, BAC and YAC constructs have been retrofitted with modified Epstein–Barr virus origins of replication (8–11). This approach is not without concern, as there may be safety issues associated with the co-expression of transacting factors required for their function (12,13). Moreover, while these vectors can be maintained for long periods in cultured cells in vitro, their variable copy number may result in unpredictable gene expression levels.
Technologies that can accommodate large DNA payloads and do not require integration into the host genome for long-term stable maintenance would be advantageous, both in terms of having a more predictable gene expression and non-interference in host cell functions. Such systems would also be of great utility in gene therapy applications where safety and stability considerations are paramount. Recently there have been efforts to generate prototype mammalian artificial chromosomes (MACs) that encompass these features. Recent progress with this technology has been extensively reviewed (14,15).
Compared to traditional methodologies, MACs offer significant advantages for cellular protein production, animal transgenesis and gene-based cell therapy applications on account of their high carrying capacity and ability to self-replicate without relying on integration into the host genome. Despite the numerous advantages of MAC technology, systematic limitations have precluded any widespread implementation. These limitations include the requirement for de novo chromosome synthesis for each individual application, the inability to shuttle MACs easily across various cell types and the inability to precisely engineer gene targets onto the artificial chromosome. For broad applicability of MAC technology, all of these limitations must be addressed.
Previously, it has been demonstrated that large satellite DNA-based artificial chromosomes, termed SATACs (SATellite DNA-based Artificial Chromosomes), can readily be generated de novo in a variety of host cell backgrounds (16–22). The Artificial Chromosome Expression (ACE), a SATAC developed for use as a gene delivery and expression system, has unique features that set it apart from other artificial chromosomes. By virtue of their size and composition, ACEs can be purified to virtual homogeneity by high-speed flow cytometry (23) and then transferred to a variety of different cell types (e.g. cell lines, primary cells and stem cells) using optimized transfection methodologies (24–26). Additionally, ACEs can be introduced into embryos (27–29) via a modified microinjection procedure to generate transgenic animals that are then able to transmit the ACE through their germ line to progeny for multiple generations (27,29).
To further expand the utility of the ACE, the ACE System was developed as a means to rapidly and reliably load sequences onto an ACE. An ACE that is pre-engineered to contain multiple site-specific recombination acceptor sites (Platform ACE) can be loaded with heterologous gene sequences using a mutant lambda integrase (ACE Integrase) in combination with a targeting vector [ACE Targeting Vector (ATV)] harboring a recombination donor site and the sequence/gene(s) of interest. After loading of targeting vectors onto the Platform ACE, the chromosome retains all the ACE characteristics listed above, most notably the inherent stability and ease of purification and transfer. Generation of the ACE System components is described in this paper, and examples are provided of cell lines expressing a therapeutic protein for biopharmaceutical manufacturing or gene-based cell therapy.
MATERIALS AND METHODS
Cultured cell lines
Cells were cultured at 37°C in 5% C02 in a humidified incubator and were routinely passaged every 3–4 days, just prior to reaching confluence. Cells were harvested by aspirating the medium, washing the monolayer with sterile Dulbecco's phosphate buffered saline (dPBS; Invitrogen) and incubating in the presence of a small amount of 0.05% trypsin–EDTA (Invitrogen) at 37°C for 5 min. After the cells began to lift, the trypsin–EDTA was quenched by the addition of an excess of complete medium. The cell concentration was determined using an automated Coulter Z1 Particle Counter (Beckman Coulter) and was diluted to 1:10 with additional fresh complete medium and replated onto 10 cm dishes.
LMTK− cells were obtained from ATCC (CCL-1.3) and maintained as monolayer cultures in 10 cm plastic culture dishes (BD Falcon) in complete LMTK− medium consisting of 90% Dulbecco's minimal essential medium (DMEM; Invitrogen), 10% FBS (Cansera). LMTK− derived CHR1 cells, containing the Platform ACE, were maintained as described above in complete LMTK− growth medium supplemented with 5 μg/ml puromycin (Sigma).
CHO-DG44 cells were obtained from Dr Lawrence Chasin (Columbia University) and were maintained as monolayer cultures in 10 cm dishes in complete DG44 medium consisting of 95% alpha modification of minimum essential medium plus ribonucleosides, deoxyribonucleosides (MEM alpha, ribonucleosides and deoxyribonucleosides; Invitrogen) and 5% FBS. The DG44 derived line ChY1 containing the Platform ACE was maintained as described above, in complete DG44 medium supplemented with 5 μg/ml puromycin.
CHO-S cells (Invitrogen) were routinely maintained as a suspension culture in spinner or shake flasks. Cells were seeded at a density of 2 × 105 cells per ml in a chemically defined protein-free medium (CD-CHO, Invitrogen) supplemented to 8 mM glutamine and 10 ml/l hypoxanthine, thymidine concentrate (Invitrogen) and cultured in suspension for 3 to 4 days or until the cell density reached 1 × 106 cells/ml, whereupon the cells were passaged by dilution back to 2 × 105 cells/ml in a new flask. Prior to transfection, cells were adapted to adherent growth by seeding onto plastic tissue culture dish in a medium consisting of DMEM supplemented with 10% FBS, 8 mM glutamine and 1× non-essential amino acids (Invitrogen), as well as selective antibiotic if required. Adherent cells were passaged essentially as described above for CHO-DG44. CHO-S derived lines, which were maintained under adherent growth conditions, could be readily readapted to serum free suspension growth by substitution back to CD-CHO medium. The CHO-S derived cell line containing the Platform ACE, ChC2, was maintained as described above, except in a medium supplemented with 5 μg/ml puromycin.
DT40 chicken bursal lymphoma cells were obtained from ATCC (CRL-2111) and maintained in 10 cm dishes in a medium consisting of DMEM supplemented with 10% FBS and 2 mM l-glutamine.
Plasmid construction
Unless otherwise indicated, all enzymes used for restriction and modification of DNA samples were obtained from New England Biolabs.
pCXLamIntWT, pCXLamIntR and pCXLamIntROK
The lambda integrase gene was amplified by PCR from bacteriophage lambda DNA (cI857 ind 1 Sam7, New England Biolabs) using the LamInt primer pair (see Supplementary Material, Primer Table.pdf). The 1 kb amplified fragment was then digested with EcoRI, gel-purified and cloned into the EcoRI site of pUC19 to yield the plasmid pUC19LamInt. A point mutation was introduced into the lambda integrase coding sequence at amino acid 174 (E174R) using the QuickChange Site Directed Mutagenesis kit (Stratagene) and the mutagenizing primers INT174R1 (5′-CGTCAGCCGTAAGTCTTGATCTCCTTACTCTAGATTTTGCTGCGCG-3′) and INT174R2 (5′- CGCGCAGCAAAATCTAGAGTAAGGAGATCAAGACTTACGGCTGACG-3′) to generate the plasmid pUC19LamIntR. In parallel, pUC19LamInt was digested with EcoRI to release the coding sequence of the wild-type integrase. The mammalian expression vector pCX-EGFP (30) was a generous gift from M. Okabe (Osaka University). It was digested with EcoRI in order to permit replacement of the eGFP sequence with firstly, the IntR fragment, yielding the plasmid pCXLamIntR, and also with the wild type integrase fragment, yielding the plasmid pCXLamIntWT. In order to provide an optimized Kozak sequence, the plasmid pCXLamIntR was modified by PCR amplification of the IntR coding sequences using the primers INTR(OK)1 (5′-TGGAATTCGCCGCCACCATGGGAAGAAGGCGAAGTCATGAG-3′) and INTR(OK)2 (5′-CAGCCTGCACCTGAGGAGTG-3′). The amplified fragment was digested with EcoRI, gel-purified and cloned into the EcoR1 site of pCX in place of eGFP, to generate pCXLamIntROK.
Recombination acceptor site plasmid pSV40attPPuro
The SV40 promoter was removed from the plasmid pPUR (Clontech) as a PvuII and StuI fragment, and inserted into the EcoICRI (Promega) site of pNEB193 (New England Biolabs) by blunt end ligation to generate the plasmid pNEB193SV40. The 282 bp recombination acceptor site attP was PCR amplified from lambda phage DNA template (cI857 ind 1 Sam 7) using the attP primer set (see Supplementary Material, Primer Table.pdf). The amplified fragment was then gel-purified and blunt end cloned into SmaI linearized pNEB193SV40 to yield the plasmid p193SV40attP (sense). The attP sense orientation was identified by the presence of the unique NdeI site 25 bp from the 5′ end of the attP site. The puromycin resistance gene was removed from the plasmid pPUR (Clontech) with AgeI and BamHI, 5′-overhangs were filled in with Klenow polymerase and it was cloned into the filled-in AscI site of the p193SV40attP (sense) plasmid to generate pSV40attPPuro.
ATVs
To test whether engineered plasmids can be introduced site-specifically onto the Platform ACE, two types of targeting vectors were generated, pBLAS-GFP and pZeo-EPO (Figure 1). To generate the cHS4-core tandem arrays, which are part of these vectors, the 1.2 kb cHS4 region was first amplified from DNA extracted from the chicken bursal lymphoma cell line, DT40, with cHS4 primer set, which in turn was used as the template for the amplification of the 237 bp cHS4-core element using the cHS4-core primer set (see Supplementary Material, Primer Table.pdf). The cHS4-core DNA fragments were ligated to generate arrays, which were then cloned, and a DNA sequence encoding six tandem cHS4-core elements was used in subsequent constructions.
Figure 1.

ATVs. Schematic of the ATVs used to load genes of interest onto the Platform ACE. Both vectors encode a gene of interest cloned between a CX promoter and the rabbit β-globin poly(A) signal, with these three elements flanked on both sides by six tandem copies of the chicken β-globin cHS4-core elements. Also encoded in both vectors is an attB recombination donor site adjacent to a promoterless selectable marker. (See Materials and Methods for details). In (A), the pBLAS-GFP ATV encodes the humanized hrGFP and the promoterless blasticidinR genes. The 96 bp deletion at the distal end of the CX promoter attenuates hrGFP gene expression. In (B), the pZeo-EPO ATV encodes the huEPO and the promoterless zeomycinR genes.
pBLAS-GFP (Figure 1A) encodes humanized Renilla green fluorescent protein (hrGFP, Stratagene) expressed from the CX promoter (taken from pCX-EGFP) with the rabbit β-globin poly(A) signal (from pCX-EGFP), all of which are flanked on either side with a tandem array of six cHS4 core elements (31). The vector encodes a promoterless blasticidin resistance gene (BACPAC Resource Center, Children's Hospital Oakland Research Institute, Oakland, CA) with a SV40 poly(A) signal cloned just downstream of the attB site, which was reconstructed by annealing the attB primer set (see Supplementary Material, Primer Table.pdf). A 96 bp XbaI–EcoRI deletion was made in the distal portion of the CX promoter (at the junction of the chicken β-actin promoter and the 3′-flanking rabbit β-globin gene sequence), attenuating hrGFP gene expression.
For the construction of pZeo-EPO (Figure 1B), the 2.1 kb genomic erythropoietin (EPO) coding sequence was generated by PCR from a human placental genomic DNA library (Clontech) using the EPO primer set (see Supplementary Material, Primer Table.pdf). The amplified fragment was cloned downstream of the complete CX promoter and upstream of the rabbit β-globin poly(A) sequences, flanked on either side with a tandem array of six cHS4-core elements. In this construct, there is an attB recognition site adjacent to a promoterless gene for zeomycin resistance (zeoR, Invitrogen) with an SV40 poly(A) signal sequence. The zeoR gene confers resistance to both Zeocin (Invitrogen) and Bleocin (CalBiochem).
Transfection
For generation of the Platform ACE, LMTK− cells (passage 6) were seeded into two 6 cm dishes (BD Falcon) at 7.5 × 105 cells per dish. Calcium phosphate transfection was carried out 1 day after seeding as described previously (17), with a mixture of 18 μg of cosmid pFK161 (encoding an 8.2 kb murine rDNA sequence) (17) and 2 μg pSV40attPPuro, linearized with the restriction endonucleases ClaI and ScaI, respectively. After 24 h, the transfected cells underwent a glycerol shock treatment, and then were split into 15 cm dishes (one 6 cm dish into three 15 cm dishes) as described above. Three days later, when the cells were ∼70% confluent, the culture medium was replaced with selection medium, consisting of complete LMTK− medium plus 5 μg/ml puromycin. Selection medium was changed three times a week. Forty-eight puromycin-resistant colonies were ring cloned from the dishes and transferred to individual wells of 24-well tissue culture dishes (BD Falcon). When cells in the wells neared confluence, they were harvested using trypsin treatment and each clonal cell suspension was distributed between three equivalent wells on three separate 24-well plates. Clones from one set of plates were analyzed by fluorescent in situ hybridization (FISH) (see below), while clones in the remaining replicate plates were frozen in situ as per the following procedure: medium was removed from the near confluent wells, the wells rinsed twice with 1 ml aliquots of trypsin–EDTA and incubated for 3 min at 37°C. Freezing medium (0.5 ml DMEM, 20% FBS and 10% DMSO) was then added to the dissociated cells, and the multi-well dishes were sealed with two sterile CRYOtags (Gordon Technologies) then transferred to a −80°C freezer. When stored in this fashion, cells retained viability for up to 3 months post-freezing. Wells containing candidate clones identified by FISH were subsequently thawed and expanded for further characterization.
For targeted integration on the CHR1 Platform ACE, transfections were carried out as follows. One day prior to transfection, CHR1 cells were seeded at 3.5 × 105 cells per well of a 6-well culture dish. On the day of transfection, for each well transfected, 2.5 μg of ATV and 2.5 μg of ACE Integrase expression vector were mixed with a 6-fold equivalent of manufacturer's suggested quantity of ExGen500 (Fermentas) and incubated with cells according to the manufacturer's instructions. Each transfected well was subsequently expanded to two 15 cm culture dishes 24 h post-transfection. Selection medium containing 3.0 μg/ml blasticidin or 500 μg/ml Zeocin (depending on version of ATV used) was added the following day (i.e. 48 h post-transfection) and was changed every 2–3 days subsequently. After 14 days in selection medium, drug-resistant colonies were harvested via ring cloning and further expanded for analysis.
For targeted integration onto the ChY1 Platform ACE cell line, 1 day prior to transfection, the cells were plated at a density of 0.4 × 106 cells per well of a 6-well culture dish. Three hours prior to transfection, medium was aspirated and replaced with serum free medium. For each well transfected, 1 μg of the ATV and 1 μg of ACE Integrase expression vector were complexed with LipofectAMINE PLUS reagent (Invitrogen) and the complex was incubated with cells according to manufacturer's recommendations. Each transfected well was subsequently expanded to two 15 cm culture dishes 24 h post-transfection. Selection medium containing 3 μg/ml blasticidin or 90 μg/ml Bleocin (depending on version of ATV used) was added the following day (i.e. 48 h post-transfection) and was changed every 2–3 days subsequently. After 14 days incubation under selection, drug-resistant colonies were harvested via cloning ring and further expanded for analysis.
Transfer of the Platform ACE into CHO-DG44 cells and CHO-S cells was carried out by a lipid-based transfection technique essentially as described previously (24,25). ACE transfer colonies were selected in appropriate growth medium supplemented with 5 μg/ml puromycin, isolated using cloning rings, then expanded for analysis and liquid N2 storage.
Flow cytometry
Purification of Platform ACE chromosomes from CHR1 cells was performed essentially as described (23) except that the cells were blocked in 1 μg/ml colchicine for 16 h prior to metaphase chromosome harvest and chromosomes were stained in 2.5 μg/ml Hoechst 33258 (Sigma) and 50 μg/ml chromomycin A3 (Sigma), prior to sorting. Platform ACEs were sorted on a FACSVantage SE flow cytometer (Becton Dickinson Immunocytometry Systems) equipped with two Innova 306 lasers (Coherent). The harvested chromosomes were held in sample-handling assembly during sorting and the isolated ACEs were held in a sample-collection assembly during collection. Both assemblies were maintained at 4°C by the circulation of chilled polyethylene glycol using a RTE-111 Refrigerated Bath Circulator (NESLAB). For analysis of GFP expression, a flow cytometer based assay was used. Briefly, the transfected cells were excited by a 488 nm tuned argon laser at 200 mW, and GFP fluorescence was collected through a 500 EFLP filter. The fluorescence level at which cells were determined to be positive for GFP was set by observing the histogram of negative control cells (untransfected ChY1 cells), such that <1% appeared in the positive GFP expression gate. Cell populations were additionally gated on the basis of side scatter versus forward scatter to exclude cell debris and aggregates. Single cell subcloning was performed using the CloneCyt Integrated Deposition System option (Becton Dickinson Immunocytometry Systems).
PCR and Southern analysis
For the preparation of genomic DNA from small numbers of cells, the DNAEASY kit (Qiagen) was used. Briefly, 1–2 × 106 cells were harvested and DNA was prepared according to the manufacturer's recommendations. For larger DNA preps (>2 × 106 cells), the FlexiGENE kit (Qiagen) was used according to the manufacturer's recommendations.
For PCR analysis of targeted integrations, a 20 μl reaction mix was set up for each sample consisting of: 1× HotStar Taq (Qiagen) reaction buffer, 200 μM dNTPs, 0.75 μM forward and reverse primers, 0.5 U of HotStar Taq and 100 ng of genomic DNA. PCR was carried out using the following conditions in a PTC200 DNA Engine Thermocycler (MJ Research): Cycle 1: 95°C for 15 min; Cycles 2–38: 95°C for 30 s, 60°C for 30 s and 72°C for 75 s; Cycle 39: 72°C for 10 min. Blasticidin or zeomycin specific primer sets were used for screening respective clones (see Supplementary Material, Primer Table.pdf).
For PCR generation of FISH probes, the following 50 μl reaction mix was used: 1 ng plasmid DNA template, 1× Taq HotStar buffer (Qiagen); 200 μM each of dATP, dCTP and dGTP, 130 μM of dTTP, 70 μM biotin-16-UTP or DIG-11-UTP (Roche Biosciences), 400 nM forward and reverse primers and 2.5 U of Taq HotStar (Qiagen). PCR was carried out using the following conditions: Cycle 1: 94°C for 15 min; Cycles 2–30: 94°C for 30 s, 55–60°C for 30 s and 72°C for 30 s; Cycle 31: 72°C for 10 min. (See Supplementary Material, Primer Table.pdf, for primer sets used in FISH probe generation.)
PCR generation of the Southern probe for the Chinese hamster metallothionein-1 gene (chMT-1; GenBank accession no. D10551) was carried out essentially as described above for the generation of FISH probes, except that 200 μM of dTTP was used in place of biotin-16-UTP or DIG-11-UTP. For chMT-1 primer set details, see Supplementary Material, Primer Table.pdf.
Southern analysis to determine copy numbers of recombination acceptor sites was carried out as described previously (17). Briefly, genomic DNA samples of CHR1, ChY1 or ChC2 Platform ACE containing cell lines (5–10 μg) were digested with appropriate restriction enzymes and equal amounts of digested DNA were fractionated using agarose gel electrophoresis (0.8–0.9% agarose in TBE). Digested DNA samples from untransfected cell lines were included as negative controls. To estimate copy number, plasmids containing the recombination acceptor site were diluted in LMTK− or CHO genomic DNA to levels equivalent to single or multiple copies, and then digested, size fractionated along side DNA from ACE-containing cells. Fractionated DNA was transferred onto a nylon membrane (HyBond N+, Amersham) according to the manufacturer's instructions.
Restriction enzyme fragments of plasmids used for hybridization probes were fractionated on preparative agarose gels, eluted from gel slices using Qiaex ll resin (Qiagen) and labeled by cross-linking with alkaline phosphatase (AlkPhos Direct, Amersham). Hybridization and subsequent stringency washes were carried out according to manufacturer's directions. Hybridization of bound probe was detected by incubating the membrane with chemiluminescent alkaline phosphatase substrate (CDPStar, Amersham) and signal was captured by exposing the membrane to X-ray film (BioMAX Film, Kodak). To estimate loading variability, membranes were stripped and re-hybridized with a PCR-generated probe to the hamster chMT-1 gene.
FISH analysis
Conventional single-color and two-color FISH analysis and high-resolution FISH were carried out essentially as described previously (16,32) using PCR-generated probes from pSAT-1 (mouse major satellite DNA repeat probe) (33), pMKC104 (mouse minor satellite DNA repeat probe) (33), pSV40attPPuro (attP and SV40attP recombination acceptor site probes) (see Supplementary Material, Primer Table.pdf) or using nick translated probes generated from pSV40attPPuro (recombination acceptor site probe), pFK161 (murine rDNA probe), pBLAS-GFP (hrGFP probe), and pZeo-EPO (huEPO probe). In some cases, TSA signal amplification (NEN) was carried out prior to visualization according to the manufacturer's directions. For detection of telomere sequences, mitotic spreads were hybridized with a commercially obtained peptide nucleic acid probe (Dako) according to the manufacturer's protocols. Microscopy was performed using a Zeiss Axioplan 2 photomicroscope under epi-fluorescent illumination and 100× oil immersion objective, or an Olympus AH-2 photomicroscope equipped for epi-fluorescent illumination. Digital images were acquired using either Quips Genetics Workstation (Vysis) or Genus workstation (Applied Imaging).
ELISA determination of EPO expression
An enzyme-linked immunosorbent assay (ELISA) to estimate EPO titer was carried out as follows: EPO capture antibody (monoclonal mouse anti-human EPO, R&D Systems) was diluted 1:200 in phosphate-buffered saline (PBS). An aliquot of 75 μl of diluted capture antibody was added per well of a 96-well ELISA plate (Nunc) and incubated overnight at 4°C. Coated wells were washed four times with PBST (PBS and 0.15% Tween-20). An aliquot of 300 μl of blocking buffer (PBS and 1% BSA) was added to each well and incubated for 1 h at 37°C. Blocked wells were then washed four times with PBST.
Samples consisting of either recombinant huEPO (for standard curve generation, 250 IU/ml, Roche Diagnostics) or conditioned medium from transfected cell lines were diluted in growth medium then added to coated wells and incubated for 1 h at 37°C, followed by four washes with PBST. An aliquot of 75 μl of polyclonal rabbit anti-human EPO antibody (R&D Systems) diluted 1:250 in dilution buffer (PBS +0.5% BSA and 0.01% Tween-20) was added to each well and incubated for 1 h at 37°C followed by 4 washes with PBST. An aliquot of 75 μl of alkaline phosphatase conjugated goat anti-rabbit antibody (Jackson ImmunoResearch) diluted 1:4000 in dilution buffer was added to each well and incubated for 1 h at 37°C, followed by four washes with PBST. An aliquot of 75 μl of 1 mg/ml p-nitrophenylphosphate (Sigma) in substrate buffer (1 M diethanolamine, 0.5 mM MgCl2, pH 9.8) was added to each well and incubated in the dark for 15–60 min, at which point the reactions were stopped by the addition of 100 μl of 3 M NaOH. The optical density (405–490 nm) of sample wells was determined using a plate reader (Biotek 800), and EPO concentrations of samples were interpolated from the standard curve.
For determination of EPO-specific productivity, EPO-expressing primary transfectants were plated at a density of 105 cell/2 ml growth medium (without antibiotic)/well of 6-well culture plates (BD Falcon). For each transfectant at 24, 48, 72 and 96 h post-plating, samples of medium were harvested from wells for ELISA determination and cells were harvested from corresponding wells for cell count determination. The medium was not changed during the course of the experiment.
Specific productivity (Qp) was determined for each time interval according to the following formula
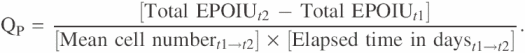
The specific productivity over the entire 4 days was determined from the average of the four intervals. The units of Qp are IU/106 cells*day, where 1 IU equals 0.01 μg of recombinant EPO.
Implantation of NOD-scid mice with EPO-secreting cell lines
NOD-scid mice (NOD.CB17-Prkdcscid/J, Jackson Laboratory) were treated according to the protocols approved by the University of British Columbia Animal Use Committee. Mice were subcutaneously injected in the right dorsal area with 0.4 ml PBS containing either 5 × 106 Y1F5 cells or 1, 5 or 10 × 106 14-1-36P cells (see Table 1). Mice were euthanized at the end of the experiment. Blood samples were collected via the saphenous vein (34) or from the tail vein. At any given time, ∼200 μl of blood was sampled. Mice were sampled twice (7 days apart) before implantation and sampled two times every 7 days post-implantation. Two micro-capillary tubes (100 μl) were filled with blood, sealed and centrifuged for 5 min in a Micro-Hematocrit Centrifuge (Model DSC-030MH, Unico). The hematocrits were determined by using a hematocrit reader provided by the manufacturer. Data are presented as the mean hematocrits for each set of mice. The paired two-sample means t-test (two-tailed, unequal variance) was used to determine the probability (P-value) that the sample means were equal. The means were considered significantly different if the P ≤ 0.05. Statistical analyses were carried out with Microsoft EXCEL version 2000.
Table 1. In vivo biological responses in NOD-scid mice with ACE-modified CHO cells.
| Cell line engrafted | HuEPO secretion (IU/106 cells*day) | No. of cells engrafted | Peak hematocrits (%) | n | P-value |
|---|---|---|---|---|---|
| Y1F5 | 0 | 5 × 106 | 49.2 ± 1.5 | 3 | — |
| 14-1-36P | 595 | 1 × 106 | 70.2 ± 4.3 | 3 | 0.015 |
| 5 × 106 | 69.2 ± 3.1 | 3 | 0.002 | ||
| 10 × 106 | 71.7 ± 3.5 | 3 | 0.002 |
RESULTS
ACEs are generated by the SATAC process, i.e. amplification-induced centromere formation, and have been shown to be non-integrating and fully functional chromosomes that are maintained stably, side by side with host cell chromosomes (32). Because of the unique base composition afforded by their heterochromatic nature, ACEs can easily be purified to virtual homogeneity by dual laser high-speed flow cytometry (23) and can be readily transferred to a number of heterologous cell lines (24–26). Moreover, due to the known paucity of genes within the pericentric heterochromatin comprising the ACE, introduction of the ACE into foreign cells or organisms should not result in altered gene dosages. Preliminary demonstration of the safety and stability of ACEs has been provided by the generation of ACE-transgenic mouse lines, where the ACE was transmitted to multiple offspring over a number of generations without any apparent phenotypic changes or adverse events (27,29).
In order to expand the utility and usability of ACEs, a new version was envisaged, one that would retain the desirable characteristics described above but would also provide a platform for the simple, reproducible and specific introduction of genes of interest along with all elements required for their control and optimized expression. This second-generation ACE is referred to as the Platform ACE and is part of a system (the ACE System), components of which are described in Figure 2. These components include: the Platform ACE containing multiple attP (35,36) recombination acceptor sites (Figure 2A); a modified DNA integrase derived from lambda integrase (37), termed herein as ACE Integrase, which can catalyze the site-specific integration of genes of interest onto the Platform ACE (Figure 2B); and an ATV, which serves to shuttle genes of interest onto the Platform ACE in a site-specific manner (Figure 2C). As can be seen by the configuration of the Platform ACE acceptor sites and ATV donor sites, the system is designed to allow for a strong selection of targeted clones. The Platform ACE attP acceptor site is interposed between the SV40 promoter and coding sequence of the puromycin resistance (puroR) gene, only marginally attenuating the expression of the puroR gene (data not shown) and permitting the use of puromycin resistance to select cell lines during initial generation of the Platform ACE and for ACE transfer protocols (see below). If the promoterless secondary drug-selectable marker on the ATV (zeoR, blastR or hygroR) integrates correctly via recombination between attB and attP sites, it will become activated by virtue of its integrated position downstream of the acceptor site's SV40 promoter. This is illustrated in Figure 2D, which shows a schematic representation of an integration site on a Platform ACE into which a gene of interest has been inserted. Thus, one can enrich for the correctly targeted event using the secondary drug selection system.
Figure 2.

Components of the Platform ACE System. (A) Platform ACE is a murine artificial chromosome pre-engineered to contain multiple recombination acceptor attP sites. (B) ACE Integrase is based on the enzyme lambda integrase, which has been modified as described in the text. The modification renders the integrase functionally independent of bacterial host cell factors and capable of operating in a mammalian context. In the Platform ACE system, an ACE Integrase expression vector is co-transfected with a targeting vector (see below) into a cell line harboring the Platform ACE. The transiently expressed ACE Integrase then catalyses the integration of the targeting vector onto the Platform ACE. (C) The ATV is a plasmid-based shuttle vector that conveys a gene(s) of interest onto the Platform ACE by means of targeted recombination between the recombination acceptor attP sites present on the Platform ACE and the recombination donor attB site of the ATV, catalyzed by the ACE Integrase. The presence of a promoterless antibiotic resistance gene downstream of the attB donor site allows for selection of targeted integration events. (D) A representation of a ‘loaded’ recombination acceptor site on the Platform ACE. Note that the targeted integration of the targeting vector has resulted in the activation of the promoterless antibiotic resistance gene by virtue of its in frame insertion downstream of the SV40 promoter present in the ACE acceptor site. (E) Southern-blot analysis of Platform ACE. Genomic DNA was hybridized with a labeled probe encoding the SV40 promoter and attP site. Lanes 1–3: copy number controls: 50 copies (lane 1), 100 copies (lane 2), 250 copies (lane 3); LMTK− negative control (without ACE, Lane 4) and CHR1 cell line (containing Platform ACE, Lane 5). See Materials and Methods for details.
Generation of the platform ACE
The amplification process that is central to the formation of an ACE (22) implies that multiple copies of recombination acceptor sites would be created on a Platform ACE. Full utilization of these multiple sites required a specific and vectorial method of gene insertion, catalyzed by a DNA recombinase that would permit sequential insertion, but not excision of genes in the available sites of a Platform ACE. One such enzyme, lambda integrase, catalyses recombination between distinct attP and attB target sequences found in phage and bacterial DNA, respectively, resulting in the integration of the phage into the bacterial genome, now flanked by distinct attR and attL sites. This reaction additionally requires the interaction of specific host-derived factors with attP to allow the integration reaction to proceed. Of note is that attR and attL sites are not themselves appropriate targets for further integrative reactions with attP or attB sequences. While lambda integrase can catalyze recombination between attR and attL sites, resulting in excision of the intervening phage sequence, this latter reaction cannot proceed without interaction of a unique phage-encoded accessory factor, Xis (37). This inherent asymmetry of action afforded by the lambda integrase and associated attB/attP recombination provides the basis for the novel site-specific integration system used to introduce genes of interest onto a Platform ACE (described more fully below).
The Platform ACE was generated as described in Materials and Methods and according to the previously described protocols (16–22). Briefly, 48 puromycin-resistant clones were screened by FISH to identify those that had undergone the large-scale amplification that entails the first step in ACE formation. One clone, B5-19, while heterogeneous, was found to contain a small ACE-like chromosome in 24% of spreads examined, as well as a smaller number of dicentric chromosomes containing a highly amplified segment between centromeres (data not shown). In order to obtain a more homogeneous clone, single cell subcloning by flow cytometry was carried out on B5-19 cells. Of the 117 single cell subclones examined by FISH, only one, CHR1, was found to contain the ACE in most of the spreads examined (>90%).
In order to determine the number of recombination acceptor sites present on the CHR1 ACE, Southern-blot analysis was carried out on genomic DNA isolated from the CHR1 cell line. As can be seen in Figure 2E, there are 50–100 copies of the acceptor site on the ACE, confirming the highly amplified structure revealed by FISH. An extensive analysis of the CHR1 ACE is shown in Figure 3. As can be seen in the various FISH images the ACE is acrocentric, with mouse major satellite sequences, a marker for pericentric heterochromatin, predominantly localized to centromeric regions (Figure 3A and C). Mouse minor satellite sequences, related to the human centromeric alphoid satellite DNA (33), are also found on the ACE and localized to the centromeric region (Figure 3F). The recombination acceptor sites (pSV40attPPuro) are localized in the long arms of the chromosome and appear to be highly amplified (Figure 3A, E and F); high-resolution FISH analysis on partially decondensed ACE metaphase chromosomes reveals three to four large blocks of hybridizing sequences corresponding to the recombination acceptor sites (Figure 3E). The rDNA sequences of cosmid pFK161 co-localize with the acceptor sites in the chromosome arms (Figure 3E), consistent with their co-integration and subsequent co-amplification during generation of the ACE. High-resolution FISH analysis confirms the co-localization of most rDNA sequences and recombination acceptor sites on the long arms but also reveals some regions of rDNA and recombination acceptor sites that are not co-localized (Figure 3E). Both short and long arms of the ACE have also maintained and/or acquired telomeres at their distal tips (Figure 3D). Taken together, these data indicate that in addition to the multiple recombination acceptor sites, the CHR1 ACE contains the basic functional constituents of a typical mammalian chromosome: a centromere embedded in pericentric heterochromatin and telomeric sequences, which protect the chromosome ends. Thus, the CHR1 ACE was selected as a candidate Platform ACE.
Figure 3.

FISH analyses of CHR1 Platform ACE Cell Line. Mitotic spreads of the CHR1 Platform ACE cell line (derived from murine LMTK− cells). The arrows indicate single Platform ACEs. (A) Mixture of probes: biotin-labeled attP (FITC signal) PCR amplimer and DIG-labeled mouse major satellite DNA repeat PCR amplimer (rhodamine signal). attP labeling was further enhanced by TSA treatment. (B) Probe is a biotin-labeled SV40attP PCR amplimer (FITC signal). (C) Mixture of probes: biotin-labeled pFK161 (rDNA; FITC signal) was generated by nick translation and DIG-labeled mouse major satellite DNA repeat (rhodamine signal) PCR amplimer. (D) Mixture of probes: a biotin-labeled telomere peptide nucleic acid probe (FITC signal), commercially prepared (Dako) and DIG-labeled pSV40attPPuro (rhodamine signal) generated by nick translation. Note that telomere probe signal is apparent at the ends of both short and long arms of the Platform ACE. (E) Mixture of probes: biotin-labeled pSV40attPPuro probe (FITC signal) and a DIG-labeled pFK161 (rDNA DNA fragment) probe (rhodamine signal), both probes were generated by nick translation. High-resolution FISH performed on spreads obtained from nuclei treated with Hoechst dye to allow for partial de-condensation of metaphase chromosomes. Note that while both probes label the long arm of the Platform ACE selectively they are not entirely coincident in all regions. (F) Mixture of probes: biotin-labeled mouse minor satellite DNA repeat (FITC signal) PCR amplimer and a DIG-labeled pSV40attPpuro probe (rhodamine signal) generated by nick translation.
Generation and testing of the ACE integrase
As described above, in its normal context the lambda integrase requires bacterial host cell encoded factors that are thought to assist the formation of the ternary complex between the lambda integrase, the lambda phage attP recombination donor site and the bacterial genome's attB recombination acceptor site (37). For such a system to operate in a mammalian context, the requirement for host cell factors must be overcome. It has been shown that mutation of the glutamine residue at position 174 of the lambda integrase (Int) gene to lysine (Int-H mutant) removes the requirement for host factors not only in bacterial recombination (38,39) but also in mammalian cells (40–42). A mutant lambda integrase was generated with a similar Int mutation, E174R, and is henceforth referred to as the ACE Integrase. The gene encoding the ACE Integrase was subcloned into a mammalian expression vector, producing pCXLamIntR. Anticipating that increasing the transcription efficiency would increase the ACE Integrase expression in mammalian cells, the pCXLamIntR plasmid was modified by the substitution of an optimized mammalian Kozak sequence (43,44), generating pCXLamIntROK, as described in Materials and Methods.
In order to test the ability of the ACE Integrase to catalyze site-specific integration of attB sequences into the attP sites of the Platform ACE, the following experiment was carried out (see Supplementary Material, Specificity of ACE Integrase.pdf). Targeting vectors were constructed, which contain a promoterless blastR gene downstream of an attB site either in sense orientation (pattBBSR) or antisense orientation (pBSRattB). Each was co-transfected into the CHR1 cell line with either the pCXLamIntWT (wild-type lambda integrase) or the pCXLamIntR (ACE Integrase) expression vector. As expected, the only targeting vector to produce appreciable quantities of blasticidin-resistant colonies was pattBBSR, since only the orientation with blastR gene 3′ of the attB sequence could be inserted in the correct orientation downstream of the SV40 promoter to allow for drug selection. In addition, only the ACE Integrase proved efficient at catalyzing this integrative event, suggesting that ACE Integrase could function autonomously in a mammalian context, as has been reported for Int-H and other similar mutants (40–42). To verify the genotype of the blasticidin-resistant colonies, 21 were selected for genomic DNA analysis by PCR using primer pairs that would amplify across the attR junction formed by a targeted integration. All of the colonies gave rise to a PCR amplified product of 961 bp, the size predicted for the fragment spanning the attR junction. Additionally, sequence analysis of all the 21 PCR products corresponded exactly to that expected for targeted integration (data not shown). Southern-blot analysis of blasticidin-resistant clones confirmed the presence of the unique junction fragment encoding the attR junction as well as lack of other random integration events in the clones analyzed. The Southern-blot analysis data also demonstrate that it is possible to integrate multiple copies of the targeting vector in a single round of targeting, since clones containing 1–15 copies were obtained. Taken together, these data demonstrate that the ACE Integrase catalyzes site-specific integrations of an attB-containing targeting vector into the attP sites located on the Platform ACE in the context of a LMTK− cell line. While it is possible that functionally analogous mammalian nuclear factors can substitute for bacterial host-derived factors, the observation that wild-type integrase does not catalyze significant levels of site specific integration in the LMTK− background argues against this likelihood. These data also demonstrate the ability of the ACE Integrase to catalyze intermolecular recombinations where at least one target sequence is embedded in chromatin. In this latter respect the ACE Integrase resembles the phiC3I phage integrase, which has also been shown to catalyze intermolecular recombination in a mammalian context (45,46).
It has been shown that the Int-H mutant, unlike the wild-type lambda integrase, has the capacity to catalyze excisive recombination reactions, i.e. recombination between attR and attL sites, in the absence of host cell factors in a bacterial context (39). In the mammalian context, Int-H has been shown to efficiently catalyze excisive recombination between targets on episomally maintained plasmids, however, excisive recombination between attL and attR sites integrated on a mammalian chromosome was barely detectable (40–42). In light of the similarity with Int-H, the possibility that ACE Integrase can catalyze excisive events on episomes maintained in mammalian cells cannot be currently eliminated. By the same token, however, it is likely that ACE Integrase shares the Int-H mutant's preference for attB/attP over attR/attL targets when embedded in chromatin. The ACE Integrase preference for integrative recombination is further demonstrated by the double loading experiments described below.
Transfer of Platform ACE from CHR1 LMTK− cells to a CHO cell background
In order to exploit the Platform ACE for high-level expression of secreted proteins, it would be advantageous to maintain the ACE in a cellular context that supports high-level expression, such as CHO cells. While it is technically feasible to generate an ACE in CHO cells (Gy. Hadlaczky, unpublished results), a more efficient approach would be to transfer the pre-existing Platform ACE from the CHR1 cell line to a CHO cell line. Accordingly, the CHR1 Platform ACE was purified by high-speed flow cytometry (Figure 4A) and subsequently transferred to CHO-DG44 [dihydrofolate reductase (DHFR) deficient] and CHO-S (wild-type DHFR) cells using commercially available cationic lipid reagents as described previously (24–26). Puromycin-resistant CHO-DG44 or CHO-S colonies were expanded and metaphase nuclei prepared for FISH analysis (Figure 4B, data not shown). Cell lines analyzed from both sets of transfers reveal intact Platform ACEs as confirmed by the presence of the mouse major satellite sequences and murine rDNA sequences on a small acrocentric chromosome. Note as well that there are no translocations of the Platform ACE onto the host hamster chromosomes. The extensive regions of co-amplified rDNA hybridization (Figure 4B) are reminiscent of those seen on the CHR1 Platform ACE (Figure 3), indicating that these sequences were maintained in the transferred Platform ACE. Southern-blot analysis of genomic DNA from CHR1 and one derived clone each of DG44 and CHO-S (the latter two referred to as ChY1 and ChC2, respectively, Figure 4C) indicates that the copy numbers of the attP sites on the Platform ACE in all three cell lines remain relatively constant, providing further evidence that the process of transfer does not perturb the structure of the Platform ACE. FISH analysis further demonstrates that the ChY1 Platform ACE (See Supplementary Material, FISH Analysis of ChY1.pdf) contains an amplified region of SV40 attP-Puro sequences in the chromosome arms and mouse minor sequences associated with the centromeric regions as was observed for the CHR1 Platform ACE (Figure 3). Thus, the Platform ACE appeared to maintain its overall acrocentric architecture and integrity as well as its major DNA constituents subsequent to its transfer into the CHO background. Similar results were obtained for the ChC2 Platform ACE (data not shown).
Figure 4.
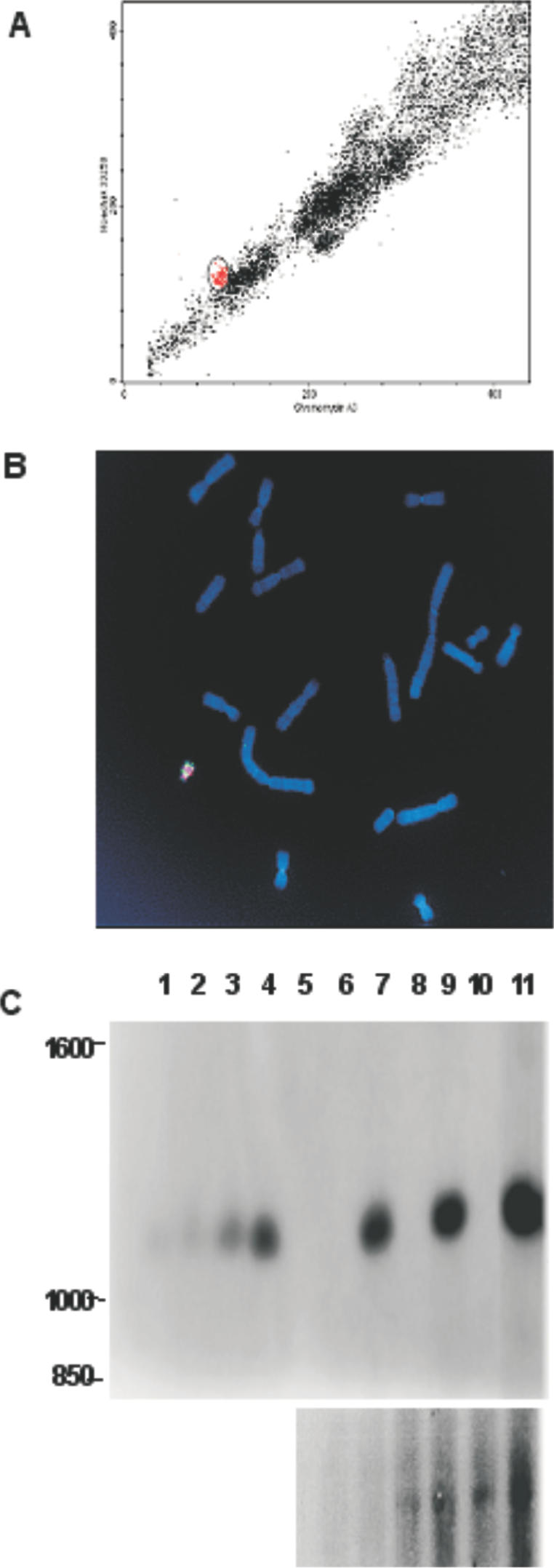
Transfer of the Platform ACE into heterologous cell lines. (A) Flow karyogram of metaphase chromosomes isolated from CHR1 cells and treated with Hoechst 33258 and chromomycin A3 dyes as described in Materials and Methods. The region circled and highlighted in red consists predominantly of the Platform ACE. (B) FISH analysis of the ChY1 cell line. The purified Platform ACE was transferred to CHO-DG44 cells and the resultant puromycin-resistant clone ChY1 was characterized by FISH. Metaphase chromosome spreads were prepared as described and hybridized with a mixture of DIG-labeled mouse major satellite repeat probe (rhodamine) PCR amplimer and biotin-labeled pFK161 probe (rDNA, FITC) generated by nick translation. (C) Southern-blot analysis of ChY1 and ChC2 Platform ACE cell lines. Genomic DNA samples from wildtype cell lines LMTK−, CHO-DG44 and CHO-S, as well as the corresponding Platform ACE cell lines CHR1, ChY1 and ChC2 were digested with NcoI and were fractionated on a 1% agarose gel. These were run along side varying dilutions of NcoI-digested pSV40attPPuro DNA so as to allow for determination of copy number. The fractionated DNA was transferred to nylon membrane by capillary blotting and the resultant membrane was hybridized with a labeled attP probe fragment (upper autoradiograph), then stripped and reprobed with a PCR-labeled hamster metallothionein-1 gene fragment (lower autoradiograph). Lane 1–4: copy number controls; 5 copies (lane 1), 10 copies (lane 2), 25 copies (lane 3), 50 copies (lane 4). Lane 5: empty. Lanes 6–11: cell line DNA; LMTK− (lane 6), CHR1 (lane 7), CHO-DG44 (lane 8), ChY1 (lane 9), CHO-S (lane 10), ChC2 (lane 11). The attP probe hybridizes to the expected 1.1 kb band in the CHR1, ChY1 and ChC2 DNA samples (upper). The hamster metallothionein-1 gene (chMT-1) probe detects the single copy gene and is used as a loading control for the CHO samples (lanes 8–11, lower panel). The hamster metallothionein-1 gene probe does not hybridize to the murine LMTK− derived samples, hence the absence of signal in lanes 6 and 7 (lower). Lanes 6, 8 and 10 are derived from the parental cell lines; lanes 7, 9 and 11 are cell lines derived after Platform ACE transfer. Note that the ChC2 sample (lane 11) is overloaded when compared with the other samples.
In order to assess the stability of the Platform ACE in the CHO background, the Platform ACE containing cell lines ChY1 and CHR1 were maintained in continuous culture for extended periods of time. At specific intervals, metaphase chromosomes of both lines were prepared and examined for the presence of intact Platform ACEs. As can be seen in Figure 5, the Platform ACE was maintained in >90% of the CHR1 cells for at least 2 months and in the ChY1 cell line up to 4 months. Puromycin selection was not required for maintenance of the Platform ACE, as similar percentages of Platform ACE persistence were observed for both CHR1 and ChY1 cells in the presence or absence of antibiotic. These results confirm that the murine chromosome-derived Platform ACE is fully functional as a chromosome in its own right, even in a heterologous cellular context (i.e. after transfer to cells derived from a different species).
Figure 5.

Stability of Platform ACE in LMTK− and CHO backgrounds. CHR1 and ChY1 cell lines were maintained in continuous culture for extended periods of time (up to 60 days for CHR1 cells and 140 days for ChY1 cells), both in the presence (open square, ChY1; closed diamond-CHR1) or absence (closed square, ChY1; open diamond, CHR1) of puromycin. At intermediate times, metaphase nuclei were prepared from each line and subsequently analyzed by FISH. At least 100 metaphase spreads of each line were scored for intact Platform ACEs at each of the times indicated. (FISH analyses were not determined for all cell lines at all time points.)
Single and double loading of the ACE in CHO cells
The transfer of the Platform ACE to CHO cells, a cell line commonly used for the expression and production of heterologous secreted proteins, allows for the use of the ACE in a protein expression context. To test the utility of the ChY1 Platform ACE cell line and its ability to support targeted integration via the system described above, an ATV, pBLAS-GFP (Figure 1A), was constructed to insert a GFP expression cassette consisting of a CX promoter linked to the hrGFP coding sequence, onto the Platform ACE. In order to mitigate against the known repressive effect of heterochromatin on gene expression, the entire hrGFP transcription unit was flanked with six head-to-tail repeats of a segment of the chicken β-globin LCR hypersensitive site 4 (cHS4), which had been shown to act as a chromatin domain boundary element (31). As before, a promoterless blastR gene enables the detection of targeted events by incubating the transfected cells in blasticidin-supplemented media. Moreover, the insertion event would result in the generation of novel flanking attR and attL sites, which could be screened by PCR to detect targeted integration. In the experiment described in Figure 6A–C, 9 of the 11 blasticidin-resistant clones were found to be GFP positive and, when analyzed by PCR, found to contain the expected attR junction (Figure 6A). One primary transfectant from this experiment, Y1F5, was chosen for further investigation. As can be seen in Figure 6B, virtually all the cells of the Y1F5 transfectant (97%) expressed GFP and at a level which allowed for visual detection under the fluorescence microscope as well as by flow cytometry. When metaphase spreads of Y1F5 cells were analyzed by FISH using probes directed against both mouse major satellite and the pBLAS-GFP (Figure 6C), the integrity of the Platform ACE was clearly maintained, and ATV integrations could only be detected on the Platform ACE (i.e. no integrations of the ATV could be detected on host CHO chromosomes).
Figure 6.

Targeted integration of hrGFP onto Platform ACE. ATV pBLAS-GFP was co-transfected with the ACE Integrase expression vector pCXlamIntROK into ChY1 cells. Correct integration of the pBLAS-GFP into the Platform ACE acceptor site(s) would result in activation of the blastR gene. Transfected cells were placed in selective medium (blasticidin supplemented) and drug-resistant colonies were analyzed as described below (panels A–C). A huEPO ATV, pZeo-EPO was co-transfected with the ACE Integrase expression vector pCXlamIntROK into the Y1F5 clone, obtained from the above transfection. Correct integration of pZeo-EPO into the Platform ACE acceptor site(s) would result in activation of the zeoR gene. Transfected cells were placed in selective medium (Bleocin supplemented) and drug-resistant colonies were analyzed as described below (panels D–G). (A) DNA was prepared from blasticidin-resistant clones and analyzed by PCR for the presence of an attR site, indicative of targeted integration of the pBLAS-GFP onto the attP acceptor site(s) of the Platform ACE. The blasticidin primers (See Supplementary Material, Primer Table.pdf) will amplify a 961 bp fragment spanning the attR junction of a targeted integration. PCR reactions were analyzed by agarose gel electrophoresis: Analyzed clones (lanes 1–10), ChY1 negative control (lane 11), positive control (lane 12). On the basis of positive PCR signal, 9 out of the 10 clones analyzed had undergone targeted integration of the pBLAS-GFP ATV onto the Platform ACE. Clone Y1F5 (lane 9) was selected for double loading. (B) Clone Y1F5 (panel A, lane 9) was harvested and analyzed by flow cytometry for GFP fluorescence as described in Materials and Methods. As can be seen, 98% of cells are expressing GFP. (C) Metaphase chromosome spreads prepared from Clone Y1F5 were hybridized with a mixture of DIG-labeled mouse major satellite DNA repeat PCR amplimer probe (rhodamine) and biotin-labeled hrGFP targeting vector, pBLAS-GFP, (FITC) generated by nick translation. Note that the Platform ACE is labeled with both probes and, by contrast, neither probe labeled the native CHO chromosomes. (D) DNA was prepared from Bleocin-resistant clones and analyzed by PCR for the presence of an attR site, indicative of targeted integration of the pZeo-EPO onto the attP acceptor site(s) of the Platform ACE. The zeomycin primers (see Supplementary Material, Primer Table.pdf) will amplify a 697 bp fragment spanning the attR junction of a targeted integration event. PCR reactions were analyzed by agarose gel electrophoresis: Analyzed clones (lanes 1–24), positive control (lane 25), negative control (lane 26). On the basis of positive PCR signal, 20 out of 24 clones analyzed had undergone targeted integration of the pZeo-EPO ATV onto the Platform ACE. Clone 14-1-36P (lane 16) was selected for further analysis. (E) Clone 14-1-36P (panel D, lane 16) was harvested and analyzed by flow cytometry for GFP fluorescence as described in Materials and Methods. As can be seen, after a second round of gene loading >98% of cells still expressed GFP. (F) Metaphase chromosome spreads prepared from Clone 14-1-36P were hybridized with a mixture of DIG-labeled mouse major satellite repeat probe (rhodamine) PCR amplimer and biotin-labeled huEPO targeting vector, pZeo-EPO, (FITC) generated by nick translation. Note that the Platform ACE is labeled with both probes and, by contrast, neither probe labeled the native CHO chromosomes. (G) The mean huEPO productivity was determined for selected clones as described in Materials and Methods.
Southern-blot analyses suggest that in a typical targeting reaction, <30% of the available recombination donor sites were occupied by inserted genes (data not shown). In order to test the feasibility of incorporating additional genes onto unoccupied sites of a previously targeted Platform ACE, a second ATV was generated, pZeo-EPO (Figure 1B), designed to insert a human erythropoietin (huEPO) gene. This vector was similar in design to pBLAS-GFP, except for the genetic payload (huEPO) and selectable marker (in this case zeoR, which confers resistance to Zeocin and Bleocin). After co-transfection with pCXLamIntROK into Y1F5 cells, 24 Bleocin-resistant colonies were obtained. Out of these 24 colonies, 20 were found to contain the expected 697 bp PCR fragment indicative of the attR junction (Figure 6D) and all retained GFP fluorescence. One double-loaded transfectant cell line, 14-1-36P, was expanded and analyzed for GFP expression by fluorescent cell cytometry and for ACE structure by FISH using probes directed against mouse major satellite and pZeo-EPO. As can be seen in Figure 6E, >98% of 14-1-36P cells expressed GFP, with a fluorescent intensity similar to that observed for the parental cells (Figure 6B). As was the case for the GFP targeting, the integrity of the 14-1-36P ACE was unaffected by the second round of targeting and huEPO–DNA integrations could only be detected on the Platform ACE (Figure 6F).
EPO expression from primary transfectants was determined by ELISA of conditioned medium at the 96-well stage. Those transfectants that expressed at the highest levels were expanded and specific productivity of EPO secretion was determined as described in Materials and Methods. Specific productivities for four of nine assayed transfectants were >400 IU/106 cells*day (Figure 6G), which compares well with the EPO levels obtained from cell lines generated via conventional methods that rely on multiple rounds of gene amplification (47). Moreover, these determinations were performed on cells maintained in static adherent culture under low-density growth conditions. No attempts were made to optimize expression levels by adapting cells into high-density suspension growth or by selecting higher expressers by single cell subcloning. These results demonstrate that the Platform ACE will support high levels of gene expression and that the multiple acceptor sites on the Platform ACE are amenable to multiple rounds of gene loading. As subsequent rounds of loading do not appear to alter the expression of genes integrated in prior rounds, double loading can be envisioned as a method for either modifying an existing Platform ACE to express a new product in addition to the original one, or as a method to augment the copy number of the original targeted gene.
In vivo expression of ACE-derived recombinant human EPO
To demonstrate the feasibility of using the ACE System to deliver prospective genes for gene-based cell therapy applications, the following experiment was performed. NOD-scid mice were engrafted with either Y1F5 or 14-1-36P cells and maintained for a period of 3 weeks, during which hematocrits were monitored weekly. As can be seen from Table 1, mice engrafted with 14-1-36P cells showed significantly elevated peak hematocrit levels relative to those observed with engrafted Y1F5 control cells, strongly indicating that the effect observed in these mice was due to the systemic action of huEPO expressed from the grafts.
DISCUSSION
The ACE System, consisting of a Platform ATVs and ACE Integrase, was developed as a modular platform that would enable a wide-ranging variety of new phenotypes to be introduced into individual eukaryotic cells or multi-cellular organisms, essentially permitting the biological ‘re-engineering’ of said organism. The ability of the ACE System to engender such changes lies primarily in the flexibility of the Platform ACE artificial chromosome, into which multiple genes of interest can be specifically introduced by the concerted action of specially designed ATVs and ACE Integrase. The properties of the ACE Integrase allow for multiple copies of a single gene or several genes to be transferred onto the Platform ACE, in a chromosomal environment conducive to reproducible gene expression. Once so modified by introduction of heterologous genetic material, the Platform ACE can be easily purified and efficiently transferred into numerous cell types without species restriction. When transferred into heterologous lines, the Platform ACE is stably maintained side by side with host chromosomes and its genetic payload can be exploited according to the application.
Of note is that the ACE System also provides the first example of an intermolecular recombination catalyzed by bacteriophage lambda integrase expressed in mammalian cells. Previously, it has been shown that engineered bacteriophage lambda integrases can catalyze intramolecular recombination between transfected plasmid substrates and between appropriate DNA sequences (attB/attP and attR/attL) flanking drug-resistant marker genes within the chromosomes of gene targeted mouse embryonic stems cells (40–42). The results from the present study demonstrate that the ACE Integrase (λIntR mutant lambda integrase) can efficiently promote intermolecular recombination between a donor vector (ATV) and the Platform ACE chromosome residing in a cell. While other members of the tyrosine family of recombinases, such as Cre and FLP, and the serine integrase φC31 have been used to promote intermolecular recombination (48), the ACE Integrase provides a useful alternative to the current repertoire of recombinases applicable to mammalian cell engineering.
A key component of the ACE System, the Platform ACE chromosome, was generated by induced amplification of pericentric heterochromatic regions of an existing mouse chromosome, resulting in transient formation of a dicentric chromosome, followed by breakage to yield a smaller fragment that was stably maintained by the host cell (16–22). Consistent with its mode of generation, the Platform ACE was found to contain amplified regions of mouse major satellite DNA, a marker for pericentric heterochromatin (33) as well as mouse minor satellite DNA, a marker for centromeric regions in all mouse chromosomes except Y (49). While the function of mouse minor satellite may not be well understood (49–51), its presence and restricted localization proximal to the mouse major repeats and primary constriction on the Platform ACE are reflective of normal chromosome structure and function. Indeed, artificial chromosomes generated by the SATAC process have been used to generate healthy transgenic founder mice and several generations of healthy transgenic offspring (27,29), suggesting that altered gene dosage and/or deregulated expression of co-amplified genes is not an inherent property of ACE artificial chromosomes, and attesting to their functionality and stability. Platform ACEs were also stably maintained in >90% of spreads from both the LMTK− derived CHR1 cell line over at least 60 population doublings, and the CHO-derived ChY1 cell line over more than 120 population doublings, with or without antibiotic selection. These data confirm that the Platform ACE retained full chromosomal functionality and was able to replicate and segregate accurately among daughter cells, even after transfer between species (24–26).
The ACE System relies on a flexible engineering approach whereby a Platform ACE is generated and engineered to contain pre-existing recombination acceptor sites for targeted insertion of heterologous genes. Although the targeted approach to introducing genes of interest onto a MAC equipped with a single loxP site has already been demonstrated in the literature (52–55), the ACE System has further extended the utility of artificial chromosomes as gene delivery systems through the properties of the ACE Integrase. Thus, if not all sites of the Platform ACE are loaded in a single targeting reaction, then a second round of gene targeting can be performed to fill additional sites, with little or no impact on previously loaded sites. This can be performed with the same gene as initially loaded, to increase its copy number, or if desired, a different gene of interest can be inserted in subsequent rounds of loading.
In these experiments, the ACE System was used to generate cell lines that displayed high levels of expression from loaded copies of the huEPO gene. In fact many of the lines so generated expressed EPO at commercially relevant levels, supporting the assertion that the environment comprising the integrated ATV on the Platform ACE provides an appropriate context for high-level gene expression. The multiplicity of available loading sites on the Platform ACE also allow for increased numbers of incorporated gene copies without resorting to gene amplification strategies. The ability to purify the Platform ACE and transfer it to multiple host cell types opens up the possibility of testing various cell types for a variety of applications. Furthermore, loaded Platform ACEs already expressing a gene of interest can be further modified by inserting genes that may improve the host cell growth characteristics (56) or post-translational modifications of target proteins (57).
As with other MAC technologies, ACEs may have numerous advantages over conventional transgenic animal technologies; in particular, the ability to introduce very large DNA sequences into a phenotypically inert and non-integrating autonomously replicating artificial chromosome (53–55,58,59). ACEs offer additional advantages and flexibility for MAC-mediated transgenesis in that ACEs can be purified to near homogeneity and directly microinjected into the pronuclei of oocytes. For example, we have previously reported the generation of transgenic mice carrying an ACE (27). In these studies, ACEs have been shown to be extremely stable and passed through four generations of progeny with high fidelity (27,29).
The versatility of the ACE System for gene delivery and gene expression in mammalian cell lines, including stem cells and primary cells (24–26), suggests that it may be utilized in gene-based cell therapy applications. For example, a gene encoding a therapeutic protein can be cloned onto the Platform ACE, resultant cell lines tested for appropriate levels of gene expression and then the engineered Platform ACE can be transferred into recipient cells for implantation. A proof of concept for such a paradigm was provided by the engraftment of NOD-scid mice with huEPO-loaded ChY1 cells. The multi-loading capability of the Platform ACE can be further exploited by introducing genes that enable identification and tracing of the engrafting cells, or that can alter the survivability of the implanted cells in response to external stimulus (e.g. suicide genes). Such modification provides an additional measure of safety for the recipient while also allowing the action of the implant to be terminated when no longer required.
In summary, the ACE System, comprising a Platform ACE artificial chromosome as well as ACE Integrase and ATVs, enables the rapid and rational engineering of mammalian artificial chromosomes. This system further extends the utility of MACs by combining their inherent advantages (i.e. stability and large carrying capacity) with the ease of purification and transferability of ACEs. In summary, the ACE System represents a unique engineering technology that facilitates the rapid and reproducible generation of modified mammalian cells or animals with desired phenotypic characteristics.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We acknowledge the expert technical assistance of Sandra Babich and Adele Telenius. We thank Katalin Fodor, Tünde Praznovszky and Barnabas Szakal for providing unpublished data. We also thank Joseph Zendegui, Shafique Fidai, Andor Udvardy and Imre Cserpan, for their valuable scientific insights during the course of this work as well as Shamila Gorjian and Helen Zeitler for their help and guidance in the preparation of the manuscript.
REFERENCES
- 1.Sternberg N. (1990) Bacteriophage P1 cloning system for the isolation, amplification, and recovery of DNA fragments as large as 100 kilobase pairs. Proc. Natl Acad. Sci. USA, 87, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ioannou P.A., Amemiya,C.T., Garnes,J., Kroisel,P.M., Shizuya,H., Chen,C., Batzer,M.A. and DeJong,P.J. (1994) A new bacteriophage P1-derived vector for the propagation of large human DNA fragments. Nature Genet., 6, 84–89. [DOI] [PubMed] [Google Scholar]
- 3.Murray A.W. and Szostak,J.W. (1983) Construction of artificial chromosomes in yeast. Nature, 305, 189–193. [DOI] [PubMed] [Google Scholar]
- 4.Burke D.T., Carle,G.F. and Olson,M.V. (1987) Cloning of large segments of exogenous DNA into yeast by means of artificial chromosome vectors. Science, 236, 806–812. [DOI] [PubMed] [Google Scholar]
- 5.Chumakov I.M., Rigault,P., Le,Gall.I., Bellanne-Chantelot,C., Billault,A., Guillou,S., Soularue,P., Guasconi,G., Poullier,E., Gros,I. et al. (1995) A YAC contig map of the human genome. Nature, 377, 175–297. [DOI] [PubMed] [Google Scholar]
- 6.Al Shawi R., Kinnaird,J., Burke,J. and Bishop,J.O. (1990) Expression of a foreign gene in a line of transgenic mice is modulated by a chromosomal position effect. Mol. Cell. Biol., 10, 1192–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costantini F., Radice,G., Lee,J.L., Chada,K.K., Perry,W. and Son,H.J. (1989) Insertional mutations in transgenic mice. Prog. Nucleic Acid Res. Mol. Biol., 36, 159–169. [DOI] [PubMed] [Google Scholar]
- 8.Calos M. (1996) The potential of extrachromosomal replicating vectors for gene therapy. Trends Genet., 12, 463–466. [DOI] [PubMed] [Google Scholar]
- 9.Kelleher Z.T., Fu,H., Livanos,E., Wendelburg,B., Gulino,S. and Vos,J.M. (1998) Epstein–Barr-based episomal chromosomes shuttle 100 kb of self-replicating circular human DNA in mouse cells. Nat. Biotechnol., 16, 762–768. [DOI] [PubMed] [Google Scholar]
- 10.Wade-Martins R., Frampton,J. and James,M.R. (1999) Long-term stability of large insert genomic DNA episomal shuttle vectors in human cells. Nucleic Acids Res., 27, 1674–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tolmachova T., Simpson,K. and Huxley,C. (1999) Analysis of a YAC with human telomeres and oriP from Epstein–Barr virus in yeast and 293 cells. Nucleic Acids Res., 27, 3736–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson J.B., Bell,J.L. and Levine,A.J. (1996) Expression of Epstein–Barr virus nuclear antigen-1 induces B cell neoplasia in transgenic mice. EMBO. J., 15, 3117–3126. [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson J.B. and Levine,A.J. (1992) The oncogenic potential of Epstein–Barr virus nuclear antigen 1 in transgenic mice. Curr. Top. Microbiol. Immunol., 182, 375–384. [DOI] [PubMed] [Google Scholar]
- 14.Lipps H.J., Jenke,A.C.W., Nehlsen.K., Scinteie,M.F., Stehle,I.M. and Bode,J. (2003) Chromosome-based vectors for gene therapy. Gene, 304, 23–33. [DOI] [PubMed] [Google Scholar]
- 15.Lim H.N. and Farr,C.J. (2004) Chromosome-based vectors for mammalian cells: an overview. Methods Mol. Biol., 240, 167–186. [DOI] [PubMed] [Google Scholar]
- 16.Csonka E., Cserpan,I., Fodor,K., Hollo,G., Katona,R., Kereso,J., Praznovszky,T., Szakal,B., Telenius,A., de Jong,G. et al. (2000) Novel generation of human satellite DNA-based artificial chromosomes in mammalian cells. J. Cell. Sci., 113, 3207–3216. [DOI] [PubMed] [Google Scholar]
- 17.Stewart S., MacDonald,N., Perkins,E., de Jong,G., Perez,C. and Lindenbaum,M. (2002) Retrofitting of a satellite repeat DNA-based murine artificial chromosome (ACEs) to contain loxP recombination sites. Gene Ther., 9, 719–723. [DOI] [PubMed] [Google Scholar]
- 18.Hollo G., Kereso,J., Praznovszky,T., Cserpan,I., Fodor,K., Katona,R., Csonka,E., Fatyol,K., Szeles,A., Szalay,A.A. et al. (1996) Evidence for a megareplicon covering megabases of centromeric chromosome segments. Chromosome Res., 4, 240–247. [DOI] [PubMed] [Google Scholar]
- 19.Kereso J., Praznovszky,T., Cserpan,I., Fodor,K., Katona,R., Csonka,E., Fatyol,K., Hollo,G., Szeles,A., Ross,A.R. et al. (1996) De novo chromosome formations by large-scale amplification of the centromeric region of mouse chromosomes. Chromosome Res., 4, 226–239. [DOI] [PubMed] [Google Scholar]
- 20.Hadlaczky G., Praznovszky,T., Cserpan,I., Kereso,J., Peterfy,M., Kelemen,I., Atalay,E., Szeles,A., Szelei,J., Tubak,V. et al. (1991) Centromere formation in mouse cells co-transformed with human DNA and a dominant marker gene. Proc. Natl Acad. Sci. USA, 88, 8106–8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Praznovszky T., Kereso,J., Tubak,V., Cserpan,I., Fatyol,K. and Hadlaczky,G. (1991) De novo chromosome formation in rodent cells. Proc. Natl Acad. Sci. USA, 88, 11042–11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hadlaczky G. (2001) Satellite DNA-based artificial chromosomes for use in gene therapy. Curr. Opin. Mol. Ther., 3, 125–132. [PubMed] [Google Scholar]
- 23.de Jong G., Telenius,A.H., Telenius,H., Perez,C.F., Drayer,J.I. and Hadlaczky,G. (1999) Mammalian artificial chromosome pilot production facility: large-scale isolation of functional satellite DNA-based artificial chromosomes. Cytometry, 35, 129–133. [PubMed] [Google Scholar]
- 24.de Jong G., Telenius,A., Vanderbyl,S., Meitz,A. and Drayer,J. (2001) Efficient in vitro transfer of a 60 Mb mammalian artificial chromosome into murine and hamster cells using cationic lipids and dendrimers. Chromosome Res., 9, 475–485. [DOI] [PubMed] [Google Scholar]
- 25.Vanderbyl S., MacDonald,N. and de Jong,G. (2001) A flow cytometry technique for measuring chromosome-mediated gene transfer. Cytometry, 44, 100–105. [DOI] [PubMed] [Google Scholar]
- 26.Vanderbyl S., MacDonald,G.N., Sidhu,S., Gung,L., Telenius,A., Perez,C. and Perkins,E. (2004) Transfer and stable transgene expression of a mammalian artificial chromosome into bone marrow-derived human mesenchymal stem cells. Stem Cells, 22, 324–333. [DOI] [PubMed] [Google Scholar]
- 27.Co D.O., Borowski,A.H., Leung,J.D., van der Kaa,J., Hengst,S., Platenburg,G.J., Pieper,F.R., Perez,C.F., Jirik,F.R. and Drayer,J.I. (2000) Generation of transgenic mice and germline transmission of a mammalian artificial chromosome introduced into embryos by pronuclear microinjection. Chromosome Res., 8, 183–191. [DOI] [PubMed] [Google Scholar]
- 28.Wang B., Lazaris,A., Lindenbaum,M., Stewart,S., Co,D.O., Perez,C.F., Drayer,J.I. and Karatzas,C.N. (2001) Expression of a reporter gene after microinjection of mammalian artificial chromosomes into pronuclei of bovine zygotes. Mol. Reprod. Dev., 60, 433–438. [DOI] [PubMed] [Google Scholar]
- 29.Monteith D.P., Leung,J.D., Borowski,A.H., Co,D.O., Praznovszky,T., Jirik,F.R., Hadlaczky,G. and Perez,C.F. (2004) Pronuclear microinjection of purified artificial chromosomes for generation of transgenic mice: pick-and-inject technique. Methods Mol. Biol., 240, 227–242. [DOI] [PubMed] [Google Scholar]
- 30.Kato M., Yamanouchi,K., Ikawa,M., Okabe,M., Naito,K. and Tojo,H. (1999) Efficient selection of transgenic mouse embryos using EGFP as a marker gene. Mol. Reprod. Dev., 54, 43–48. [DOI] [PubMed] [Google Scholar]
- 31.Chung J.H., Bell,A.C. and Felsenfeld,G. (1997) Characterization of the chicken beta-globin insulator. Proc. Natl Acad. Sci. USA, 94, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Telenius H., Szeles,A., Kereso,J., Csonka,E., Praznovszky,T., Imreh,S., Maxwell,A., Perez,C.F., Drayer,J.I. and Hadlaczky,G. (1999) Stability of a functional murine satellite DNA-based artificial chromosome across mammalian species. Chromosome Res., 7, 3–7. [DOI] [PubMed] [Google Scholar]
- 33.Wong A.K. and Rattner,J.B. (1988) Sequence organization and cytological localization of the minor satellite of mouse. Nucleic Acids Res., 16, 11645–11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hem A., Smith,A.J. and Solberg,P. (1998) Saphenous vein puncture for blood sampling of the mouse, rat, hamster, gerbil, guinea pig, ferret and mink. Lab. Anim., 32, 364–368. [DOI] [PubMed] [Google Scholar]
- 35.Bauer C.E., Gardner,J.F. and Gumport,R.I. (1985) Extent of sequence homology required for bacteriophage lambda site-specific recombination. J. Mol. Biol., 181, 187–197. [DOI] [PubMed] [Google Scholar]
- 36.Ross W., Landy,A., Kikuchi,Y. and Nash,H. (1979) Interaction of int protein with specific sites on lambda att DNA. Cell, 18, 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landy A. (1989) Dynamic, structural, and regulatory aspects of lambda site-specific recombination. Annu. Rev. Biochem., 58, 913–949. [DOI] [PubMed] [Google Scholar]
- 38.Lange-Gustafson B.J. and Nash,H.A. (1984) Purification and properties of Int-h, a variant protein involved in site-specific recombination of bacteriophage lambda. J. Biol. Chem., 259, 12724–12732. [PubMed] [Google Scholar]
- 39.Christ N. and Droge,P. (1999) Alterations in the directionality of lambda site-specific recombination catalyzed by mutant integrases in vivo. J. Mol. Biol., 288, 825–836. [DOI] [PubMed] [Google Scholar]
- 40.Lorbach E., Christ,N., Schwikardi,M. and Droge,P. (2000) Site-specific recombination in human cells catalyzed by phage lambda integrase mutants. J. Mol. Biol., 296, 1175–1181. [DOI] [PubMed] [Google Scholar]
- 41.Christ N., Corona,T. and Droge,P. (2002) Site-specific recombination in eukaryotic cells mediated by mutant λ integrases: implications for synaptic complex formation and the reactivity of episomal DNA segments. J. Mol. Biol., 319, 305–314. [DOI] [PubMed] [Google Scholar]
- 42.Christ N. and Droge,P. (2002) Genetic manipulation of mouse embryonic stem cells by mutant λ integrase. Genesis, 32, 203–208. [DOI] [PubMed] [Google Scholar]
- 43.Kozak M. (1984) Point mutations close to the AUG initiator codon affect the efficiency of translation of rat pre-proinsulin in vivo. Nature, 308, 241–246. [DOI] [PubMed] [Google Scholar]
- 44.Kozak M. (1986) Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell, 44, 283–292. [DOI] [PubMed] [Google Scholar]
- 45.Thyagarajan B., Olivares,E.C., Hollis,R.P., Ginsburg,D.S. and Calos,M.P. (2001) Site-specific genomic integration in mammalian cells mediated by phage phiC31 integrase. Mol. Cell Biol., 21, 3926–3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belteki G., Gertsenstein,M., Ow,D.W. and Nagy,A. (2003) Site-specific cassette exchange and germline transmission with mouse ES cells expressing phiC31 integrase. Nat. Biotechnol., 21, 321–324. [DOI] [PubMed] [Google Scholar]
- 47.Egrie J. (1990) The cloning and production of recombinant human erythropoietin. Pharmacotherapy, 10, 3S–8S. [PubMed] [Google Scholar]
- 48.Groth A.C. and Calos,M.P. (2004) Phage Integrases: biology and applications. J. Mol. Biol., 335, 667–678. [DOI] [PubMed] [Google Scholar]
- 49.Broccoli D., Miller,O.J. and Miller,D.A. (1990) Relationship of mouse minor satellite DNA to centromere activity. Cytogenet. Cell Genet., 54, 182–186. [DOI] [PubMed] [Google Scholar]
- 50.Vig B.K. and Richards,B.T. (1992) Formation of primary constriction and heterochromatin in mouse does not require minor satellite DNA. Exp. Cell Res., 201, 292–298. [DOI] [PubMed] [Google Scholar]
- 51.Vig B.K., Latour,D. and Frankovich,J. (1994) Dissociation of minor satellite from the centromere in mouse. J. Cell. Sci., 107, 3091–3095. [DOI] [PubMed] [Google Scholar]
- 52.Moralli D., Vagnarellli,P., Bensi,M., De Carli,L. and Raimondi,E. (2001) Insertion of a loxP site in a size-reduced human accessory chromosome. Cytogenet Cell Genet., 94, 113–120. [DOI] [PubMed] [Google Scholar]
- 53.Voet T., Vermeesch,J., Carens,A., Durr,J., Labaere,C., Duhamel,H., David,G. and Marynen,P. (2001) Efficient male and female germline transmission of a human chromosomal vector in mice. Genome Res., 11, 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Voet T., Schoenmakers,E., Carpentier,S., Labaere,C. and Marynen,P. (2003) Controlled transgene dosage and PAC-mediated transgenesis in mice using a chromosomal vector. Genomics, 82, 596–605. [DOI] [PubMed] [Google Scholar]
- 55.Kuroiwa Y., Tomizuka,K. and Ishida,I. (2004) Use of natural and artificial chromosome vectors for animal transgenesis. Methods Mol. Biol., 240, 207–225. [DOI] [PubMed] [Google Scholar]
- 56.Meents H., Enenkel,B., Werner,R.G. and Fussenegger,M. (2002) p27Kip1-mediated controlled proliferation technology increases constitutive sICAM production in CHO-DUKX adapted for growth in suspension and serum-free media. Biotechnol. Bioeng., 79, 619–627. [DOI] [PubMed] [Google Scholar]
- 57.Bragonzi A., Distefano,G., Buckberry,L.D., Acerbis,G., Foglieni,C., Lamotte,D., Campi,G., Marc,A., Soria,M.R., Jenkins,N. et al. (2000) A new Chinese hamster ovary cell line expressing alpha 2,6-sialyltransferase used as universal host for the production of human-like sialylated recombinant glycoproteins. Biochem. Biophys. Acta, 1474, 273–282. [DOI] [PubMed] [Google Scholar]
- 58.Mee P.J., Shen,M.H., Smith,A.G. and Brown,W.R. (2003) An unpaired mouse centromere passes consistently through male meiosis and does not significantly compromise spermatogenesis. Chromosoma, 112, 183–189. [DOI] [PubMed] [Google Scholar]
- 59.Kazuki Y., Kimuram,M., Nishigaki,R., Kai,Y., Abe,S., Okita,C., Shirayoshi,Y., Schulz,T.C., Tomizuka,K., Hanaoka,K. et al. (2004) Human chromosome 21q22.2-qter carries a gene(s) responsible for downregulation of mlc2a and PEBP in Down syndrome model mice. Biochem. Biophys. Res. Commun., 317, 491–499. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.