

Abstract
A new nanocarrier is developed for the passage of gatifloxacin through the blood–brain barrier to treat central nervous system tuberculosis. Gatifloxacin nanoparticles were prepared by nanoprecipitation using poly(lactic-co-glycolic acid) (PLGA) 502 and polysorbate 80 or Labrafil as surface modifiers. The evaluation of in vivo blood–brain barrier transport was carried out in male Wistar rats using rhodamine-loaded PLGA nanoparticles prepared with and without the surface modifiers. At 30 and 60 minutes after administration, nanoparticle biodistribution into the brain (hippocampus and cortex), lungs, and liver was studied. The results obtained from the cerebral cortex and hippocampus showed that functionalization of rhodamine nanoparticles significantly increased their passage into the central nervous system. At 60 minutes, rhodamine concentrations decreased in both the lungs and the liver but were still high in the cerebral cortex. To distinguish the effect between the surfactants, gatifloxacin-loaded PLGA nanoparticles were prepared. The best results corresponded to the formulation prepared with polysorbate 80 with regard to encapsulation efficiency (28.2%), particle size (176.5 nm), and ζ-potential (−20.1 mV), thereby resulting in a promising drug delivery system to treat cerebral tuberculosis.
Keywords: polysorbate 80, Labrafil, gatifloxacin, nanoparticles, targeting, blood–brain barrier, brain delivery
Introduction
Tuberculosis (tubercle bacillus [TB]) is a pathology present mainly in the lungs but can also develop as extrapulmonary infection in the central nervous system (CNS) or elsewhere in the body. In the last few years, the epidemiology of TB has been altered, due to the emergence of HIV infection. TB of the CNS represents ~1% of the disease worldwide, is devastating and often fatal, and affects primarily young children. The mortality is extraordinarily high, with severe neurological sequelae. Two major forms of CNS TB include meningitis, which accounts for 0.5%–1% of all TB disease, and intracranial tuberculomas, which represent ~40% of brain tumors.1
The blood–brain barrier (BBB) is an effective barrier for most drugs, thereby hindering effective treatment, as in the case of CNS TB. The possibility of crossing this barrier is restrictive, due to the absence of paracellular and transcellular channels, with just a few substances being able to get across this barrier by means of diffusion or using receptor- or carrier-mediated transport. Moreover, the luminal and abluminal membranes of the brain capillary endothelium are separated by ~200 nm of endothelial cytoplasm, limiting the passage of drugs. Therefore, the BBB protects the brain from toxic substances but restricts the entrance of many therapeutically important drugs.2
Treatment of CNS TB is complex, due to increasing incidence of multidrug-resistant (MDR), extensively DR, and totally DR strains of Mycobacterium tuberculosis. MDR-TB is defined as TB caused by M. tuberculosis that is resistant to at least isoniazid and rifampicin. Resistance phenomena represent a major threat to effective therapeutic control of TB. For instance, in 2014 approximately 480,000 cases of MDR-TB were described, of which 3.3% corresponded to new cases and 20% to previously treated cases.3 One of the mechanisms involved in MDR-TB is drug efflux mediated by permeability of P-glycoprotein (P-gp), which prevents cellular drug accumulation into the brain, thereby reducing the efficacy of treatment. Efforts to reduce MDR have been studied using nonionic surfactants, such as Tween, Pluronic, d-α-tocopheryl, polyethylene glycol (PEG), and Brij, due to their potential actions in reducing cellular drug efflux.4
New-generation fluoroquinolones are second-line anti-TB drugs that have proven to be very efficient when treating MDR-TB. Within the quinolones, gatifloxacin (Gat) is a new compound twice as active as levofloxacin; however, it exhibits lower access to the CNS.5,6 Gat is of high interest, due to its low incidence of drug resistance, being a substrate of P-gp.7,8 For this reason, the administration of Gat with agents able to inhibit P-gp efflux together with the use of therapeutic systems able to increase its access to the CNS would benefit improvement in the therapeutic outcome of Gat therapy.
Nanotechnology can be a promising strategy when targeting CNS diseases, such as TB. In fact, one of the most important challenges of pharmaceutical technology is to develop efficient transport systems able to carry drugs across the BBB. Several studies have shown that poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NPs) of sizes around 250 nm can cross the BBB.9–11 However, the percentage of particles able to pass the barrier is low. To overcome this problem, several approaches are under investigation, eg, coating NPs with surfactants or surface-modifier agents with specific ligands, as in the case of polysorbate 80 NPs.12,13 Moreover, therapeutic NPs have emerged as an innovative and promising option to target P-gp-mediated multidrug resistance.14
Taking all these into consideration, we developed Gat-loaded PLGA NPs able to cross the BBB for the treatment of CNS TB. Moreover, we prepared and evaluated in vivo PLGA NPs functionalized with two different surfactants (polysorbate 80 and Labrafil M1944CS) as surface modifiers, in order to evaluate their passage through the BBB. The biodistribution of the NPs was studied in the liver and lungs, which are very representative organs for CNS TB.
Materials and methods
Rhodamine (Rh)-123 was supplied by Sigma-Aldrich (St Louis, MO, USA) and Gat sesquihydrate (1-cyclopropyl-6-fluoro-8-methoxy-7-[3-methylpiperazin-1-yl]-4-oxo-1,4-dihydroquinoline-3-carboxylic acid sesquihydrate) by Santa Cruz Biotechnology (Dallas, TX, USA). PLGA with a ratio of 50:50 PLGA (Resomer® RG 502) was purchased from Evonik (Essen, Germany). Polysorbate 80 was obtained from PanReac (Barcelona, Spain). Labrafil® M 1944 CS (PEG-5 oleate FDA IIG), was supplied by Gattefossé (Lyon, France). Polyvinyl alcohol (PVA; molecular weight 30,000–70,000 Da) was obtained from Sigma-Aldrich. Others reagents used were of analytical grade and provided by Merck (Darmstadt, Germany). Distilled and deionized water (Milli-Q; EMD Millipore, Billerica, MA, USA) was used in the preparation of all buffers and solutions.
Preparation and characterization of NPs
Rhodamine-loaded PLGA NPs
Rh-loaded PLGA NPs (NPR1; Table 1) were prepared by nanoprecipitation using an acetone–water system. Briefly, 50 mg of PLGA and 2.5 mg Rh were dissolved in 4 mL acetone and mixed by vortexing. This mixture was added dropwise into 12 mL of 1% PVA under continuous stirring for 15 minutes. The resulting suspension was then evaporated in a rotary evaporator (Rotavapor-R; Büchi Labortechnik AG, Flawil, Switzerland) to remove acetone completely (2 hours, 25°C, and 70 mbar). The NP suspension obtained was then washed with distilled water three times and centrifuged (Avanti J-301; Beckman Coulter, Brea, CA, USA) at 15,000 rpm for 30 minutes to remove PVA. Finally, the dispersed solution was freeze-dried for 24 hours with sucrose as cryoprotectant (Flexi-Dry MP™; FTS Systems, Stone Ridge, NY, USA).
Table 1.
Nanoparticle formulations developed
| Formulation | Rhodamine (mg) |
Gatifloxacin (mg) |
Polysorbate 80 (%) |
Labrafil (mg) |
|---|---|---|---|---|
| NPR1 | 2.5 | – | – | – |
| NPR2 | 2.5 | – | 1 | – |
| NPR3 | 2.5 | – | – | 3.5 |
| NPB1 | – | – | – | – |
| NPB2 | – | – | 1 | – |
| NPB3 | – | – | – | 3.5 |
| NPG1 | – | 5 | – | – |
| NPG2 | – | 5 | 1 | – |
| NPG3 | – | 5 | – | 3.5 |
Rh-loaded PLGA–polysorbate 80 NPs were prepared by the nanoprecipitation method indicated previously using 12 mL of 0.5% PVA and 0.5% polysorbate 80 as the external phase (NPR2; Table 1). All NP formulations were prepared in triplicate. Rh-loaded PLGA–Labrafil NPs were prepared using the same protocol, but incorporating 3.5 mg Labrafil into the inner phase of the emulsion (NPR3). When Labrafil was incorporated, the desiccation process was performed under vacuum.
Gatifloxacin-loaded PLGA NPs
Gat-loaded PLGA NPs, Gat-loaded PLGA–polysorbate 80 NPs, and Gat-loaded PLGA–Labrafil NPs were prepared using the same procedure but incorporating 5 mg of Gat into the inner phase. Table 1 shows the different formulations prepared. In all cases, blank NPs were prepared.
Morphology and size of NPs
Shape and surface morphology of NPs were analyzed by scanning electron microscopy (SEM; JEM 6335F; JEOL, Tokyo, Japan). Samples were coated with a thin layer of colloidal gold applied in a cathodic vacuum evaporator before observation by SEM at 20 kV. Mean diameter and size distribution of NPs was determined using a Zetatrac Ultra (Microtac, Montgomeryville, PA, USA) at 25°C. NPs were suspended in water. The average diameter is expressed as volume diameter, and the size distribution is graphically represented by volume-distribution curves. These measurements were performed for all the formulations prepared.
Process yield and encapsulation efficiency
Process yield was calculated as the ratio between the total weight of NPs obtained and the total weight of drug, polymer, and other excipients added. Gat-encapsulation efficiency (EE%) was determined by weighing 10 mg of NPs, which were dissolved in 1 mL Cl2CH2. Then, ethanol (15 mL) was added and centrifuged for 5 minutes at 5,000 rpm. This procedure was repeated five times to extract Gat completely. Then, samples were filtered through 0.45 µm filters and the Gat content of each formulation quantified by high-performance liquid chromatography (HPLC).
For HPLC quantification, the instrumentation used was a Series 200 chromatograph equipped with a 1740 HP computer and a 235C diode-array detector (PerkinElmer, Waltham, MA, USA) used at 295 nm. The HPLC system utilized a C18 Mediterranea Sea chromatographic column (250×4 mm, particle size 5 µm; Teknokroma, Barcelona, Spain). The mobile phase consisted of acetonitrile:water (80:20, v:v) with 0.3% trimethylamine. The pH was adjusted to 3.3 with phosphoric acid. The flow rate was 1 mL/min. Data were collected and processed using TurboChrom Navigator version 6.1.1 (PerkinElmer). All analyses were performed at 25°C±0.5°C, and each determination was carried out in triplicate. EE% of Gat was calculated as the ratio between the drug content in the NPs and the amount of drug used for their preparation. To calculate EE, the amount of excipients included in the polymeric matrix was taken into account. In the case of polysorbate, we determined the theoretical amount of polysorbate 80 present in the internal phase that would remain in the formulation. From its solubility in acetone (10 g/100 mL) and water (5 g/100 mL), its water–acetone partition coefficient was estimated (Kr=0.5).15
Rh EE was determined by adding 5 mg NPs of each formulation to 6 mL Cl2CH. The solution was centrifuged for 20 minutes at 10,000 rpm, the supernatant filtered (0.45 µm filters), and appropriated dilutions made with Cl2CH2. Rh was analyzed by spectroscopy fluorescence (λex 351 nm, λem 578 nm, Varian Cary Eclipse fluorescence spectrophotometer; Agilent Technologies, Santa Clara, CA, USA).
ζ-Potential
ζ-Potential measurements were obtained by laser-Doppler anemometry using a Zetasizer (Malvern Instruments, Malvern, UK). Measurements were carried out at 25°C in an aqueous solution using an effective voltage of 150 V. Briefly, 5 mg of each lyophilized NP formulation was weighed, placed in a flask, diluted with 50 mL of distilled water, and kept in a sonicator for 5 minutes. The aqueous dispersions of NPs were then introduced into a capillary cell (Malvern Instruments) for ζ-potential measurements. All formulations were analyzed in triplicate.
In vitro release studies
In vitro release of Gat from NPs was carried out in a heated bath (WB22; Memmert GmbH, Schwabach, Germany) at 37°C±0.2°C and constant shaking at 100 rpm. NPs (10 mg) were suspended in 3 mL phosphate-buffered saline (PBS) at pH 7.4. At regular time intervals, 2.5 mL of the PBS was withdrawn, centrifuged at 15,000 rpm, filtered through 0.45 nm filters, and Gat content quantified by the HPLC method described before. Formulations NPG1–NPG3 were analyzed in triplicate.
Biodistribution of rhodamine NPs
Male Wistar rats (Harlan Laboratories, Indianapolis, IN, USA) weighing 250–300 g were housed and maintained in a room at 22°C±2°C with automatic light cycles (12-hour light/dark) and with food and water consumption ad libitum. All animal procedures were approved by the ethics committee of the Universidad Complutense de Madrid (Spain) and conducted according to European Community Council Directive 010/63/UE. Efforts were made to minimize the number of animals used and their suffering.
The following animal groups were established in the study: group 1 (G1), control group, animals (n=6) receiving Rh (1.3 mg/kg) in saline; group 2 (G2), animals (n=6) receiving an amount equivalent to 1.3 mg/kg Rh encapsulated within formulation NPR1 suspended in saline; group 3 (G3), animals (n=6) receiving an amount equivalent to 1.3 mg/kg Rh encapsulated within formulation NPR2 (prepared with 0.5% polysorbate 80) suspended in saline; and group 4 (G4), animals (n=6) receiving an amount equivalent to 1.3 mg/kg Rh encapsulated within formulation NPR3 (prepared with Labrafil) suspended in saline.
Animals were anesthetized with isoflurane, and injection of the corresponding formulation (0.5 mL) was performed in the tail vein by means of a 25 G syringe. At 30 or 60 minutes after administration, animals were killed and brains, lungs, and livers removed.
In vivo evaluation of NPs transport through the BBB
Rat brain coronal sections (25 µm thickness) were obtained by means of a cryostat (CM1850; Leica Microsystems, Wetzlar, Germany). Brain sections were mounted on Superfrost Plus glass slides (Thermo Fisher Scientific, Waltham, MA, USA) and stored at −80°C until analysis. For visualization, brain sections were fixed for 10 minutes in PBS (pH 7.4) containing 4% formaldehyde. After two washes in PBS (pH 7.4, 1 minute each), the slides were rinsed in a solution containing 0.0001% DAPI (Sigma-Aldrich), which forms fluorescent complexes with natural double-stranded DNA.
Afterward, the slices were dehydrated by submerging them in increasing concentrations of ethanol (70%, 95% and 100%, 1 minute each). Once dehydrated, the slides were rinsed with xylene for 2 minutes. Finally, DPX mounting medium (Sigma-Aldrich) was poured onto the brain sections and coverslipped. Samples were kept horizontally in the dark for 24 hours to harden the mounting medium before manipulation.
Brain sections were observed with fluorescence microscopy (IX51; Olympus Corporation, Tokyo, Japan). For this, three sections per rat and region (cerebral cortex and hippocampus) and 40 sections of each region per rat were analyzed. All images were captured under the same conditions of exposure time and gain. Tetramethylrhodamine isothiocyanate and blue/cyan filters were used to visualize Rh and DAPI labeling, respectively. Brain slices were analyzed at 460 nm excitation wavelength and 500 nm emission fluorescence. The images were digitally captured and processed with ImageJ software version 1.46r.
Quantification of rhodamine fluorescence in the brain
The fluorescence intensity of Rh encapsulated within NPs was analyzed by confocal microscopy (Olympus IX51) at 540 nm excitation wavelength and 570 nm emission fluorescence. All micrographs obtained by confocal microscopy were analyzed with ImageJ version 1.47 for Rh quantification. Data are expressed as averages of fluorescence intensity measured in each image.
Biodistribution of rhodamine and rhodamine NPs in lungs and liver
Lungs and livers were weighed and homogenized with known volumes of CH2Cl2 to disrupt the NPs and in order to extract all Rh content. After 4 hours at room temperature, the extracts were centrifuged at 2,000 g for 10 minutes. Rh-fluorescence intensity was measured using the Varian Cary Eclipse (λex 351 nm, λem 578 nm). The extraction efficiency of Rh from the organs was determined using spiked known amounts of Rh. Extracts of Rh-free tissues were used as control (autofluorescence of body tissue). The amount of Rh distributed in tissues was calculated based on standard curves and expressed as amount of Rh per gram of tissue. Data (mean ± standard deviation) were analyzed by one-way analysis of variance, followed by Student’s t-test. Differences were considered significant at P<0.05.
Cytotoxicity studies
In order to study neuronal viability, DAPI-labeled cells at the level of the hippocampus were quantified after administration of Rh NP formulations. DAPI is a fluorescent dye that selectively binds to double-stranded DNA of survival cells. Brain sections were observed with fluorescence microscopy (Olympus IX51), and neuronal cells were analyzed using ImageJ version 1.46r. The number of neuronal cells was determined at 30 and 60 minutes for all formulations. Data (mean number of neuronal cells ± standard deviation) were analyzed by one-way analysis of variance, followed by Student’s t-test. Differences were considered significant at P<0.05.
Results and discussion
The main objective of this work was to develop the first nanocarrier for Gat consisting of Gat-loaded PLGA NPs destined to facilitate and increase the passage of the drug across the BBB, in order to improve treatment of brain TB. The BBB is a key barrier that limits the access of drugs to the brain. For this, we firstly developed Rh-loaded NPs functionalized by the incorporation of two different surfactants in order to characterize their passage through the BBB and to select the most appropriate formulation from this point of view. As surface modifiers, polysorbate 80 and Labrafil were used (Table 1). Rh is a fluorescent dye utilized to quantify the biodistribution of NPs. It is widely used due to its reduced ability to cross the BBB even if given intravenously.16
Mean particle sizes of Rh NPs were 234.8±4.3 nm, 194.9±5.7 nm, and 237.8±11 nm for formulations NPR1, NPR2, and NPR3, respectively (Table 2). Lower particle sizes were obtained for formulations prepared with polysorbate 80. The reduction of particle size with the addition of polysorbate 80 was in accordance with other work in which a reduction in particle size was obtained when polysorbate 80 was used in the preparation of PLGA NPs.17 With regard to EE, mean values were 55.3%±0.4% (formulation NPR1), 51.3%±0.3% (formulation NPR2), and 60.9%±0.4% (formulation NPR3) (Table 2), which corresponded to drug payloads of 2.63±0.04 mg, 2.42±0.02 mg, and 2.72±0.02 mg/100 mg NPs, respectively.
Table 2.
Characteristics of the nanoparticle formulations prepared
| Formulation | Mean particle size ± standard deviation (nm) | ζ-Potential ± standard deviation (mV) | Encapsulation efficiency ± standard deviation (%) |
|---|---|---|---|
| NPR1 | 234.8±4.3 | – | 55.3±0.4 |
| NPR2 | 194.9±5.7 | – | 51.3±0.3 |
| NPR3 | 237.8±11 | – | 60.9±0.4 |
| NPB1 | 150.5±5.1 | −23±1.3 | – |
| NPB2 | 98.9±9.6 | −19.1±1.1 | – |
| NPB3 | 156.3±6.1 | −17.3±1 | – |
| NPG1 | 176.6±11.6 | −18.6±0.4 | 34.1±0.1 |
| NPG2 | 176.5±2.9 | −20.1±1.1 | 28.2±0.2 |
| NPG3 | 182.9±2.5 | −19.3±0.8 | 10.4±1.1 |
Brain biodistribution studies of Rh-loaded NPs (formulations NPR1, NPR2, and NPR3) and Rh in solution were performed in male Wistar rats. For this, analysis of brain slices (cortex and hippocamppus) was carried out by confocal microscopy at 30 and 60 min with the intensity of fluorescence being measured (Table 3). As an example, Figure 1 shows several microphograps corresponding to brain samples (cortex). After administration of Rh in saline (Figure 1A and 1E) low fluorescence intensity was obtained at 30 and 60 min, indicating the low ability of Rh to cross the BBB. These results are in agreement with other studies in which Rh was used to study the passage of drugs through the BBB.18,19 Increased fluorescence intensity of NPR1, NPR2, and NPR3 was observed in both cortex and hippocamppus (Table 3), thereby confirming their access to the CNS.
Table 3.
Brain biodistribution of rhodamine solution and rhodamine-loaded nanoparticles
| Formulation | Fluorescence intensity, mean ± standard deviation
|
|||
|---|---|---|---|---|
| 30 minutes
|
60 minutes
|
|||
| Hippocampus | Cortex | Hippocampus | Cortex | |
| Rhodamine solution | 13.77±1.39 | 10.78±1.11 | 9.79±1.57 | 3.13±1.53 |
| NPR1 | 19.95±1.69 | 16.99±3.48 | 10.31±1.83 | 9.22±2.05 |
| NPR2 | 29.72±1.05 | 17.56±3.42 | 30.26±2.13 | 31.36±1.95 |
| NPR3 | 29.61±2.45 | 18.09±2.11 | 21.35±1.71 | 26.25±2.83 |
Figure 1.
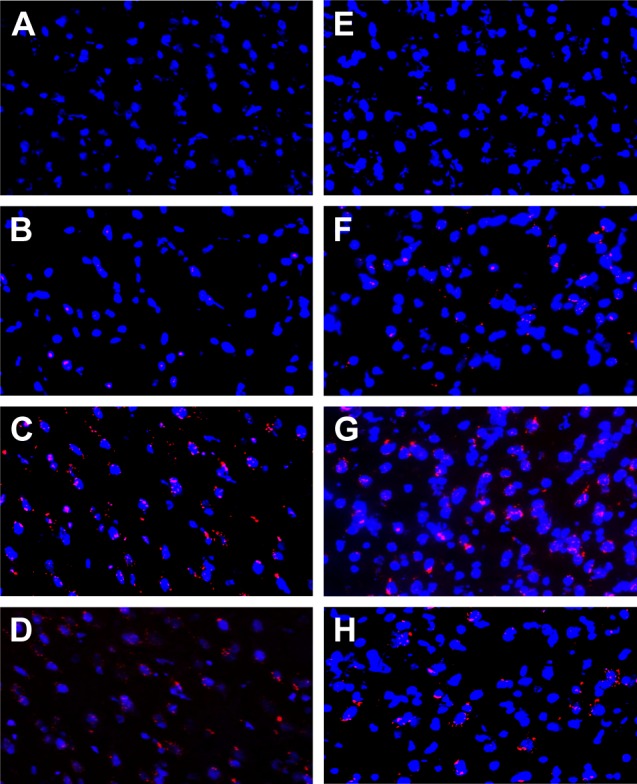
Confocal microscopy images of the cerebral cortex.
Notes: Control (A), NPR1 (B), NPR2 (C), and NPR3 (D) obtained at 30 minutes. Control (E), NPR1 (F), NPR2 (G), and NPR3 (H) obtained at 60 minutes.
On the other hand, fluorescence intensity decreased at 60 minutes for Rh given in solution and for nonfunctionalized Rh-loaded NPs (NPR1). This reduction was more pronounced in the cortex, where a decrease in fluorescence >60% resulted for Rh in saline. This decrease could be explained by the fact that Rh is a substrate for P-gp, thus being ejected from the BBB.4 At 60 minutes, the highest fluorescence intensity values in both hippocampus and cortex were obtained with functionalized NPs (NPR2 and NPR3) when compared to nonfunctionalized NPs (NPR1), indicating that both formulations increased their passage through the BBB (Table 3). This could also be attributed to the potential effects of nonionic surfactants (Tween, Pluronic, d-α-tocopheryl, PEG, and Brij) on reducing cellular drug efflux.4 Moreover, a comparison of functionalized NPs (NPR2 and NPR3) with Rh in solution showed their fluorescence intensity in the cortex was almost tenfold higher. With regard to formulation NPR2, the fluorescence intensity obtained was around threefold higher than that exhibited by formulation NPR1. This significant increase can be attributed to either the release of Rh from NPs or the presence of Rh inside the BBB.
It has been demonstrated that functionalization of NPs with polysorbate 80 enables transport of these nanocarriers across the BBB, with this passage being preferentially mediated by adsorption of ApoA1, ApoB, and/or ApoE onto their surfaces, as described for polysorbate 80-coated polybutylcyanoacrylate NPs.22 These apolipoproteins mediate the interaction with LDL receptors or other receptors of this kind on the BBB followed by the aforementioned brain-uptake processes, where endocytosis is followed by transcytosis.11 Another study showed that polysorbate 80-coated NPs were taken up into human and bovine endothelial cells more rapidly and to a greater extent than conventional nanocarriers.23 On the other hand, some authors have indicated that NPs coated with hydrophilic surfactants do not interact with ApoE, thereby lowering their ability to cross the BBB.24 With Labrafil, a slightly more hydrophobic surfactant than polysorbate 80, NPs are able to cross the BBB, with the mechanism involved still unknown, in spite of its known ability to increase the permeability of biological membranes.25 Our results demonstrate that surface modification of the polymer system with polysorbate 80 or Labrafil facilitates the access of NPs to the CNS, thereby raising interest in the NP formulations developed to improve access to the brain of active ingredients that exhibit limited passage.
PLGA is a polymer widely used in the preparation of microparticles and nanosystems, due to its biodegradable and biocompatible properties. However, toxic effects on cells and tissues have been frequently related to the administration of polymeric NPs.26 When given intravenously, NPs interact with proteins and other molecules present in blood and tissues, leading to a structure whose composition and thickness varies depending on the chemical properties of the surface system used.27 Research has suggested the involvement of NPs in the production of reactive oxygen species, as well as inflammatory processes affecting the BBB and microglia present in the brain parenchyma.28 The small size of NPs appears to increase reactivity and influences their physicochemical properties and interaction with the environment due to the direct relationship between nanometric particle sizes and high surface:volume ratios.
For this, cytotoxicity studies of the formulations prepared were carried out. Figure 2 shows several hippocampus micro-photographs obtained at 30 and 60 minutes for the control solution (Rh in solution) and the three NP formulations tested (NPR1, NPR2, and NPR3). No statistically significant differences were found in cell viability among these formulations. These results show that under our experimental conditions, the number of surviving neurons was not significantly affected by functionalization of NPs with either polysorbate 80 or Labrafil.
Figure 2.
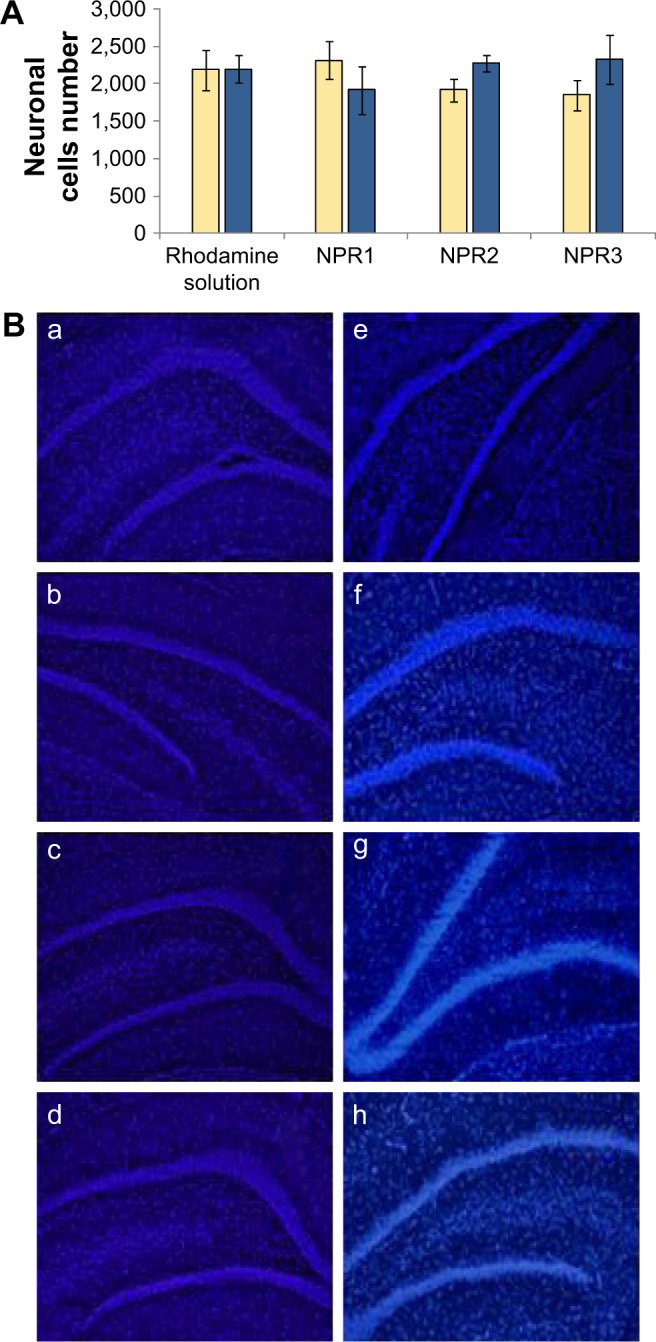
Neuronal cells corresponding to control and treated animal groups (A). Hippocampus sections obtained for control and treatment groups (B): images obtained at 30 minutes for control group treated with Rhodamine solution (a), group trated with formulation NPR1 (b), group treated with formulation NPR2 (c), and group treated with formulation NPR3 (d). Imaged obtained at 60 minutes for control group (e), group treated with formulation NPR1 (f), group treated with formulation NPR2 (g), and group treated with formulation NPR3 (h).
Biodistribution of NPs in liver and lungs was expressed as the amount of Rh per gram of tissue in order to compensate for animal variability (Figure 3). At both times assayed (30 and 60 minutes), Rh concentrations in lungs were higher than in livers for all three formulations (NPR1, NPR2, and NPR3). At 30 minutes, Rh concentrations in lungs were almost threefold higher than those found in livers for formulation NPR2, which was coated with polysorbate 80 (12.93 vs 4.31 µg Rh/g tissue). This greater biodistribution of NPs in the lungs would also be required to target M. tuberculosis, which is mainly present in the airways, and could be explained by the fact that polysorbate 80 is an ionic surfactant with hydrophilic characteristics that prevents opsonization and thus its identification by the mononuclear phagocyte system. For formulations NPR1 and NPR3, this ratio was 2.1- and 2.4-fold, respectively. After 60 minutes, Rh concentrations markedly decreased in both organs for the three formulations assayed (Figure 3). In summary, after 60 minutes, concentrations of Rh decreased in both organs increased in the brain (Table 3). This can only be attributed to either increased passage of NPs across the BBB or the inhibiting effect caused by the surfactants on P-gp efflux.
Figure 3.

Mean concentrations of rhodamine (Rh) ± standard deviation.
Notes: In the liver and lungs at 30 minutes (A) and 60 minutes (B) after administration of Rh-PLGA NPs. NPR1, Rh-PLGA NPs; NPR2, Rh-PLGA NPs prepared with polysorbate 80; NPR3, Rh-PLGA NPs prepared with Labrafil.
Abbreviations: PLGA, poly(lactic-co-glycolic acid); NPs, nanoparticles.
Taking into consideration that both functionalized formulations – NPR2 and NPR3 – showed improved biodistribution to the brain, further studies were carried out in order to distinguish between them. For this purpose, several Gat-loaded PLGA NPs were prepared without surface modifier (NPG1) and with two surfactants – polysorbate 80 (NPG2) and Labrafil (NPG3) – used to increase Gat access to the brain (Table 1). Gat-loaded PLGA NPs were characterized in terms of morphology, size, ζ-potential, EE, and in vitro release.
SEM images of Gat-loaded PLGA NPs (Figure 4) and blank NPs (data not shown) revealed that all formulations were spherical. Previous studies have shown that spherical particles moving through a vessel do not deviate from its streamline motion unless experiencing an external force; however, in the case of aspheric particles, segregation can occur.29 Mean particle sizes for formulations NPG1, NPG2, and NPG3 were 176.6±11.6 nm, 176.5±2.9 nm, and 182.9±2.5 nm, respectively. Span values of Gat-loaded PLGA NPs ranged between 0.7 and 1.07, with PI <0.38, thereby reflecting a slight polydispersion in particle sizes, which indicates that the method employed for the preparation of NPs results in monodisperse distributions. This is particularly important when considering the passage of NPs through the BBB. Studies have revealed out that NPs coated with polysorbate 80 and with average particle size of about 250 nm are able to cross the BBB.20,21
Figure 4.

Scanning electron microscopy images and mean in vitro release profiles of Gat.
Notes: From Gat-loaded PLGA NPs (NPG1) (A), Gat-loaded PLGA NPs prepared with polysorbate 80 (NPG2) (B), and Gat-loaded PLGA NPs prepared with Labrafil (NPG3) (C).
Abbreviations: Gat, gatifloxacin; PLGA, poly(lactic-co-glycolic acid); NPs, nanoparticles.
For all formulations, ζ-potential was negative, probably due to the presence of ionized carboxyl groups on the surface of NPs. For blank NPs (NPB1), a value of −23±1.3 mV was measured, similar to that described in the literature for NPs prepared with Resomer 502 PLGA (Table 2).30 This value decreased significantly when NPs were functionalized with either polysorbate 80 (NPB2, −19.1±1.1 mV) or Labrafil (NPB3, −17.3±1 mV). This modification of ζ-potential confirms the presence of an agent (surfactant) in the polymeric matrix. Encapsulation of Gat significantly diminished ζ-potential when compared with blank NPs (−23±1.3 mV). With regard to ζ-potential, the presence of Gat showed higher influence than that of surfactants. Research has indicated that positive ζ-potential values improve the passage of NPs across the BBB, though exhibiting stronger immune response.31 Furthermore, high and negative ζ-potential values prevent aggregation phenomena. It has been reported that a stable nanosuspension, mainly stabilized by electrostatic repulsion, exhibited ζ-potential values near to −30 mV, thereby facilitating its admnistration.32 In our case, it can be assumed that our NP formulations would probably have remained long enough in the bloodstream to be able to cross the BBB.
For all the formulations prepared, mean process yield ranged from 43.1%±4.5% to 66.1%±2.1%. EE values were 34.1%±0.12%, 28.2%±0.2%, and 10.4%±1.1% for formulations NPG1, NPG2, and NPG3, respectively (Table 2). Gat is a compound with low solubility in several nonpolar solvents, thereby exhibiting low affinity for the polymeric matrix. Moreover, the incorporation of Labrafil significantly reduces its EE, probably related to the lower solubility of the drug in the presence of the surfactant.
In vitro release of Gat from NPs was studied at normal blood pH (7.4). Gat release from all formulations exhibited a biphasic profile in which a rapid release occurred within the first 10 hours (Figure 4). This rapid release could be explained by both the physicochemical characteristics of Gat, which exhibits greater affinity for the PBS medium than for the polymer matrix, and the small particle sizes obtained. Moreover, the system has a high surface area:volume ratio, which favors very rapid release.33,34 Though this rapid release of Gat provides short temporal control of drug release, the main purpose of our delivery system is to reach its site of action and cross the BBB, a purpose that can be reasonably achieved, taking into consideration the rapid access of NPs to the brain. Considering that at 30 minutes, >80% of Gat was still encapsulated within the NPs, this amount would be enough to exert its therapeutic action in the CNS.
Conclusion
PLGA NPs prepared with two surfactants (Labrafil and polysorbate 80) showed improved passage across the BBB than PLGA NPs, thereby resulting in adequate carriers to deliver drugs into the CNS. EE of Gat in PLGA NPs functionalized with Labrafil was low. For this reason, this nanosystem could only be adequate for the encapsulation of drug substances with high affinity for this polymeric matrix. However, Gat-loaded PLGA NPs functionalized with polysorbate 80 presented higher EE and adequate size distribution, being thus a promising drug-delivery system to treat CNS TB.
Acknowledgments
This work was partially supported by the Complutense University UCM research group (910,939). The authors thank Centro de Microscopía Electrónica Luis Bru (CAI, UCM), and the Unidad de Cartografía Cerebral (Instituto Pluridisciplinar, UCM) for their technical assistance.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Kulchavenya E. Extrapulmonary tuberculosis: are statistical reports accurate? Ther Adv Infect Dis. 2014;2(2):61–70. doi: 10.1177/2049936114528173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37(1):13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization . Global Tuberculosis Report. Geneva: WHO; 2015. [Google Scholar]
- 4.Chen HH, Huang WC, Chiang WH, et al. pH-responsive therapeutic solid lipid nanoparticles for reducing P-glycoprotein-mediated drug efflux of multidrug resistant cancer cells. Int J Nanomedicine. 2015;10:5035–5048. doi: 10.2147/IJN.S86053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saravolatz LD, Leggett J. Gatifloxacin, gemifloxacin, and moxifloxacin: the role of 3 newer fluoroquinolones. Clin Infect Dis. 2003;37(9):1210–1215. doi: 10.1086/378809. [DOI] [PubMed] [Google Scholar]
- 6.Thwaites GE, Bhavnani SM, Chau TT, et al. Randomized pharmacokinetic and pharmacodynamic comparison of fluoroquinolones for tuberculous meningitis. Antimicrob Agents Chemother. 2011;55(7):3244–3253. doi: 10.1128/AAC.00064-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Z, Lu J, Wang Y, Pang Y, Zhao Y. Prevalence and molecular characterization of fluoroquinolone-resistant Mycobacterium tuberculosis isolates in China. Antimicrob Agents Chemother. 2014;58(1):364–369. doi: 10.1128/AAC.01228-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwatra D, Vadlapatla RK, Vadlapudi AD, Pal D, Mitra AK. Epidemiology and antimicrobial resistance of staphylococci isolated from different infectious diseases. Braz J Microbiol. 2010;41(2):333–344. doi: 10.1590/S1517-838220100002000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hillaireau H, Couvreu P. Nanocarriers entry into the cell: relevance to drug delivery cell. Mol Life Sci. 2009;60(17):2873–2896. doi: 10.1007/s00018-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suchlfart S, Gelperina S, Kreuter J. Transport of drug across the blood-brain barrier by nanoparticles. J Control Release. 2012;161(2):264–273. doi: 10.1016/j.jconrel.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 11.Kreuter J. Mechanism of polymeric nanoparticles-based drug transport across the blood-brain barrier (BBB) J Microencapsul. 2013;30(1):49–54. doi: 10.3109/02652048.2012.692491. [DOI] [PubMed] [Google Scholar]
- 12.Tosi G, Costantino L, Rivasi F, et al. Targeting the central nervous system: in vivo experiments with peptide-derivatized nanoparticles loaded with loperamide and rhodamine-123. J Control Release. 2007;122(1):1–9. doi: 10.1016/j.jconrel.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 13.Kreuter J. Drug delivery to the central nervous system by polymeric nanoparticles: what do we know? Adv Drug Deliv Rev. 2014;71:2–14. doi: 10.1016/j.addr.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Q, Li F. Combating P-glycoprotein-mediated multidrug resistance using therapeutic nanoparticles. Curr Pharm Des. 2013;19(37):6655–6666. doi: 10.2174/1381612811319370009. [DOI] [PubMed] [Google Scholar]
- 15.Vera M, Barcia E, Negro S, et al. New celecoxib multiparticulate systems to improve glioblastoma treatment. Int J Pharm. 2014;473(1–2):518–527. doi: 10.1016/j.ijpharm.2014.07.028. [DOI] [PubMed] [Google Scholar]
- 16.Fazil M, Muhammad S, Haque S, et al. Development and evaluation of rivastigmine loaded chitosan nanoparticles for brain targeting. Eur J Pharm Sci. 2012;47(1):6–15. doi: 10.1016/j.ejps.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 17.Kohei T, Sahori F, Hiromitsu Y, Yoshiaki K. Hybrid-modified poly(D,L-lactide-co-glycolide) nanospheres for a novel cellular drug delivery system. Int J Pharm. 2010;392(1–2):311–313. doi: 10.1016/j.ijpharm.2010.03.042. [DOI] [PubMed] [Google Scholar]
- 18.Martins S, Tho I, Reimold I, et al. Brain delivery of camptothecin by means of solid lipid nanoparticles: formulation design, in vitro and in vivo studies. Int J Pharm. 2012;439(1–2):49–62. doi: 10.1016/j.ijpharm.2012.09.054. [DOI] [PubMed] [Google Scholar]
- 19.Tosi G, Vergoni AV, Ruozi B, et al. Sialic acid and glycopeptides conjugated PLGA nanoparticles for central nervous system targeting: in vivo pharmacological evidence and biodistribution. J Control Release. 2010;145(1):49–57. doi: 10.1016/j.jconrel.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 20.Kreuter J, Ramge P, Petrov V, et al. Direct evidence that polysorbate-80-coated poly(butylcyanoacrylate) nanoparticles deliver drugs to the CNS via specific mechanisms requiring prior binding of drug to the nanoparticles. Pharm Res. 2003;20(3):409–416. doi: 10.1023/a:1022604120952. [DOI] [PubMed] [Google Scholar]
- 21.Alyautdin RN, Reichel A, Löbenberg R, Ramge P, Kreuter J, Begley DJ. Interaction of poly(butylcyanoacrylate) nanoparticles with the blood-brain barrier in vivo and in vitro. J Drug Target. 2001;9(3):209–221. doi: 10.3109/10611860108997929. [DOI] [PubMed] [Google Scholar]
- 22.Ramge P, Unger RE, Oltrogge JB, et al. Polysorbate-80 coating enhances uptake of polybutylcyanoacrylate (PBCA)-nanoparticles by human and bovine primary brain capillary endothelial cells. Eur J Neurosci. 2000;12(6):1931–1940. doi: 10.1046/j.1460-9568.2000.00078.x. [DOI] [PubMed] [Google Scholar]
- 23.Sempf K, Arrey T, Gelperina S, et al. Adsorption of plasma proteins on uncoated PLGA nanoparticles. Eur J Pharm Biopharm. 2013;85(1):53–60. doi: 10.1016/j.ejpb.2012.11.030. [DOI] [PubMed] [Google Scholar]
- 24.Kreuter J, Petrov VE, Kharkevich DA, Alyautdin RN. Influence of the type of surfactant on the analgesic effects induced by the peptide dalargin after its delivery across the blood-brain barrier using surfactant-coated nanoparticles. J Control Release. 1997;49(1):81–87. [Google Scholar]
- 25.Karamanidou T, Karidi K, Bourganis V, Kontonikola K, Kammona O, Kiparissides C. Effective incorporation of insulin in mucus permeating self-nanoemulsifying drug delivery systems. Eur J Pharm Biopharm. 2015;95(Pt A):223–229. doi: 10.1016/j.ejpb.2015.04.013. [DOI] [PubMed] [Google Scholar]
- 26.Kolter M, Ott M, Hauer C, Reimold I, Fricker G. Nanotoxicity of poly(n-butylcyano-acrylate) nanoparticles at the blood-brain barrier, in human whole blood and in vivo. J Control Release. 2015;197(10):165–179. doi: 10.1016/j.jconrel.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 27.Monopoli MP, Aberg C, Salvati A, Dawson KA. Biomolecular coronas provide the biological identity of nanosized materials. Nat Nanotechnol. 2012;7(12):779–786. doi: 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
- 28.Sharma HS, Sharma A. Neurotoxicity of engineered nanoparticles from metals. CNS Neurol Disord Drug Targets. 2012;11(1):65–80. doi: 10.2174/187152712799960817. [DOI] [PubMed] [Google Scholar]
- 29.Jose S, Sowwmya S, Cinu TA, Aleykutty NA, Thomas S, Souto EB. Surface modified PLGA nanoparticles for brain targeting of bacoside-A. Eur J Pharm Sci. 2014;63:29–35. doi: 10.1016/j.ejps.2014.06.024. [DOI] [PubMed] [Google Scholar]
- 30.Fonseca C, Simoes S, Gaspar R. Paclitaxel-loaded PLGA nanoparticles: preparation, physicochemical characterization and in vitro anti-tumoral activity. J Control Release. 2002;83(2):273–286. doi: 10.1016/s0168-3659(02)00212-2. [DOI] [PubMed] [Google Scholar]
- 31.Salvador-Morales C, Zhang L, Langer R, Farokhzad OC. Immunocompatibility properties of lipid-polymer hybrid nanoparticles with heterogeneous surface functional groups. Biomaterials. 2009;30(12):2231–2240. doi: 10.1016/j.biomaterials.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Müller RH, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy: rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001;47(1):3–19. doi: 10.1016/s0169-409x(00)00118-6. [DOI] [PubMed] [Google Scholar]
- 33.Bawarski N, Chidlowsky E, Bharali DJ, Mousa SA. Emerging nanopharmaceuticals. Nanomedicine. 2008;4(4):273–282. doi: 10.1016/j.nano.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 34.Lu XF, Shi YF, Lv HL, Fu YY, Ma D, Xue W. Preparation and characterization of molecularly imprinted poly(hydroxyethyl methacrylate) microspheres for sustained release of gatifloxacin. J Mater Sci Mater Med. 2014;25(6):1461–1469. doi: 10.1007/s10856-014-5191-7. [DOI] [PubMed] [Google Scholar]