

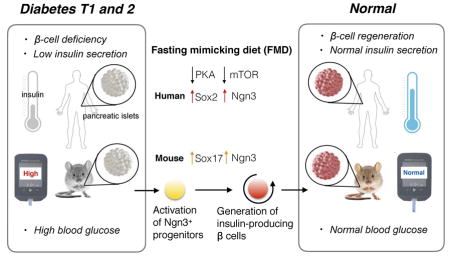
Summary
Stem cell-based therapies can potentially reverse organ dysfunction and diseases but the removal of impaired tissue and reactivation of the program leading to organ regeneration pose major challenges. In mice, a four-day fasting mimicking diet (FMD) induces a step-wise expression of Sox17 and Pdx-1, resembling that observed during pancreatic development, followed by Ngn3-driven generation of insulin-producing β-cells. FMD cycles restore insulin secretion and glucose homeostasis in both a type 2 and type 1 diabetes mouse models. In human type 1 diabetes pancreatic islets, fasting conditions reduce PKA and mTOR activity and induce Sox2 and Ngn3 expression and insulin production. The effects of the FMD are reversed by IGF-1 treatment and recapitulated by PKA and mTOR inhibition. These results indicate that a FMD promotes the reprogramming of pancreatic cells to restore insulin generation in islets from T1D patients and reverse both T1D and T2D phenotypes in mouse models.
Graphical Abstract
A short term diet that mimics periodic fasting modulates beta cell number and promotes inuslin secretion and glucose homeostasis with implications for both type 1 and type 2 diabetes.
Introduction
The ability of animals to survive food deprivation is an adaptive response accompanied by the atrophy of many tissues and organs to minimize energy expenditure. This atrophy and its reversal following the return to a normal diet involves stem cell-based regeneration in the hematopoietic and nervous systems (Brandhorst et al., 2015; Cheng et al., 2014). However, whether prolonged fasting and refeeding can also cause pancreatic regeneration and/or cellular reprogramming leading to functional lineage development is unknown. β cells residing in pancreatic islets are among the most sensitive to nutrient availability. Whereas Type 1 and type 2 diabetes (T1D and T2D) are characterized by β–cell dedifferentiation and trans-differentiation (Cnop et al., 2005; Dor and Glaser, 2013; Piran et al., 2014; Talchai et al., 2012; Wang et al.), β-cell reprogramming, proliferation and/or stepwise re-differentiation from pluripotent cells are proposed as therapeutic interventions (Baeyens et al., 2014; Chera et al., 2014; Maehr et al., 2009; Pagliuca et al., 2014; Sneddon et al., 2012; Zhou et al., 2008), suggesting that lineage-conversion is critical in both diabetes pathogenesis and therapy (Weir et al., 2013).
Although the dietary intervention with the potential to ameliorate insulin resistance and Type II diabetes have been studied extensively for decades, whether these have the potential to promote a lineage-reprogramming reminiscent of that achieved by iPSCs-based engineering remains unknown. We previously showed that cycles of prolonged fasting (2–3 days) can protect mice and humans from toxicity associated with chemotherapy and promote hematopoietic stem cell-dependent regeneration (Cheng et al., 2014; Laviano and Rossi Fanelli; Piran et al., 2014; Raffaghello et al., 2008). In consideration of the challenges and side effects associated with prolonged fasting in humans, we developed a low-calorie, low-protein and low-carbohydrate but high-fat 4-day fasting mimicking diet (FMD) which causes changes in the levels of specific growth factors, glucose and ketone bodies similar to those caused by fasting (Brandhorst et al., 2015)(see also Figure S1 for metabolic cage studies). Here we examine whether cycles of the FMD are able to promote the generation of functional, stable, and insulin-producing β cells and investigate the mechanisms responsible for these effects.
Results
Cycles of a fasting mimicking diet (FMD) rescue mice from late stage T2D
As a consequence of insulin resistance, the decrease in the number of functional insulin-producing β-cells contributes to the pathophysiology of T2D by eventually leading to insulin deficiency (Cnop et al., 2005; Dor and Glaser, 2013). Previously, we showed that a 4-day fasting mimicking diet (FMD) could induce metabolic changes similar to those caused by prolonged fasting and reduce insulin and glucose levels while increasing ketone bodies and igfbp1 (Brandhorst et al., 2015) (Figure S1). Although the role of periodic fasting and fasting mimicking diets on insulin function is unknown, the effects of intermittent fasting and chronic calorie restrictions (CR) on insulin sensitivity have been previously reported (Barnosky et al., 2014). Here our focus is on the putative effects of the FMD in promoting β-cells regeneration, although we have also investigated the effects of the FMD on insulin resistance.
Given that β-cells replicate at an extremely low rate in the adult pancreas (Meier et al., 2008; Teta et al., 2005) and that β-cell neogenesis occurs rarely (Xiao et al., 2013), depletion of β cells and the consequent loss of insulin secretion during late stage diabetes have often been considered conditions whose reversals require islet and stem cells transplantation (Fiorina et al., 2008; Kroon et al., 2008; Milanesi et al., 2012; Pipeleers et al., 2002; Villani et al., 2014). To determine whether the FMD could affect the β-cell deficiency associated with T2D, we studied its effect on mice with a point mutation in the leptin receptor gene (Leprdb/db), which causes insulin resistance and late stage loss of insulin secretion. As reported by others, db/db mice developed hyperglycemia at 10 weeks of age, which we refer to as baseline (BL)(Figure 1A). This was accompanied by high insulin levels initially compensating for insulin resistance followed by the development of severe hyperglycemia at 12 weeks of age caused by the decline in insulin secretion (Figure 1B to 1C)(Arakawa et al., 2001). As a result, db/db mice began to die at around 4 months of age. We attempted to reverse these late-stage T2D phenotypes by treating 12-week-old mice (14 days after the hyperglycemia stabilized, baseline) with weekly cycles of the 4-day FMD (Figure 1A). FMD cycles caused a major reduction and return to nearly normal levels of blood glucose in db/db mice by d60, (Figure 1B). The FMD cycles also reversed the decline in insulin secretion at d30 and improved plasma insulin levels at day 90 (Figure 1C). A Homeostasis Model Assessment (HOMA) was performed to estimate steady state β-cell function (%B) and insulin sensitivity (%S), as previously described (Hsu et al., 2013; Matthews et al., 1985). The results indicate that the reversal of hyperglycemia was mainly caused by an induction of steady state β-cell function (%B) in addition to the minor alleviation of insulin resistance (HOMA-IR) (Figure 1D). Nevertheless, mice receiving the FMD showed improved glucose tolerance and insulin tolerance compared to the ad libitum (AL) fed controls (Figure 1E), due in part to the enhanced insulin secretion functions (Figure S2F). Notably, although db/db mice on the FMD diet gained less weight compared to those on the regular diet, they maintained a weight that was approximately 22% higher than that of their healthy wild-type (WT) littermates during the entire experiment (Figure S2C). Altogether, these results indicate that FMD cycles rescued mice from late stage T2D by restoring insulin secretion and reducing insulin resistance, leading to a major improvement in survival (Figure 1F, *p<0.05, Log-rank test for trend).
Figure 1. FMD cycles promote β-cell regeneration and reverse β-cell failure in T2D.

(A) Experimental scheme to determine effects of the periodic FMD on T2D in the leptin receptor deficient (Leprdb/db) mice. Mice were monitored for hyperglycemia and insulinemia for two weeks (baseline, BL) and then assigned to the dietary groups. Each FMD cycle entails 4-day FMD and up to 10 days of refeeding (RF). During refeeding, mice received a regular chow identical to that given prior to the FMD and that given to the ad libitum (AL) controls.
(B) Plasma glucose levels and (C) Plasma insulin levels; vertical dashed lines indicate each cycle of the FMD and horizontal lines indicate the range of glucose levels (mean ± s.e.m) in age-matched healthy wild-type littermates. Blood samples were collected at the last refeeding day/1st day of the indicated cycles. Mice were fasted for 6 hours (morning fasting) for blood glucose measurements.
(D) Homeostatic model assessment (HOMA) of insulin resistance (IR) and steady state β-cell function (%B) at indicated time points. HOMA-B = (20 x Fasting Insulin)/(Fasting Glucose-3.5) %.
(E) Glucose tolerance test and Insulin tolerance test at day 60.
(F) Survival curve. mean ± s.e.m,*p<0.05, Log-rank (Mantel-Cox) Test for trend.
n ≥ 16 mice per group; for (B–E), each point represents the mean ± s.e.m; *p<0.05, ** p<0.01, ***p<0.005, two-way ANOVA.
(G) Proportion of β cells per islet
(H) Proliferative proportion of β cells per islet.
For (G to I), mean ± s.e.m,*p<0.05, ** p<0.01, ***p<0.005, one-way ANOVA.
(I) Immunostaining of pancreatic sections from Leprdb/db mice and their wild-type littermates at the indicated time points. Arrow in the 8x-enlarged example image indicates a typical proliferative β cell (PCNA+Insulin+). Scale bar represents 50um.
(J) Representative images for size-matched islets isolated from AL-dbdb and FMD-dbdb mice and results of glucose-stimulated insulin secretion (GSIS) test in islets isolated from Leprdb/db mice on FMD or fed ad libitum. Scale bar represents 50um.
Mice are of the C57BL/6J background, of the age indicated. In (a) mice received no additional treatments other than the indicated diet. For (F–J), each point represents the mean ± s.e.m; *p<0.05, ** p<0.01, ***p<0.005, two-way ANOVA. n ≥ 6 mice per group, n ≥ 15 islets per sample;
Cycles of FMD reverse β-cell failure in T2D
Dedifferentiation of β cells, which results in increased non-hormone producing cells within pancreatic islets, is a feature of diabetic β-cell failure (Dor and Glaser, 2013; Kim-Muller et al., 2014; Talchai et al., 2012). Similar to what was previously reported by others, we found an increase in cells producing neither insulin nor glucagon (i.e. non-α/β) and a decrease in β-cell number in pancreatic islets of late stage T2D mice but not in those of WT/AL healthy controls (Figures 1G and S2, db/db BL compared to WT/AL). We also found that β-cell proliferation was low in the late stage of the disease (Figure 1H, AL day 60 compared to BL). Whereas db/db mice fed ad libitum (db/db:AL) showed a 60–80% reduction in β-cell count at day 60, db/db mice receiving FMD cycles (db/db:FMD) displayed a major improvement in the number and proliferation of insulin-generating β cells (comparing db/db BL, Figure 1G–I), but not in the number of non-α/β cells (Figure S2, day 60). Pancreatic islets collected from db/db mice treated with FMD cycles (day 60) displayed increased glucose stimulated insulin secretion (GSIS), compared to that of islets from db/db:AL mice (Figure 1J). We also determined that a longer exposure time (time point 120) was necessary to distinguish between the functionality of islets from db/db:AL and db/db:FMD group mice (Figure 1J). Overall, these results suggest that, in addition to improving insulin sensitivity, FMD induced β-cell regeneration to reverse β-cell loss, which may alleviate late stage T2D symptoms and mortality.
FMD cycles restore insulin-dependent glucose homeostasis in STZ-induced T1D
To examine further the role of FMD cycles in stimulating β-cell regeneration, we applied FMD cycles on a T1D model, in which high-dose streptozotocin (STZ) treatment causes the depletion of insulin-secreting β cells (Wu and Huan, 2008; Yin et al., 2006). Starting 5 days after STZ treatment, which we refer to as baseline (STZ BL), hyperglycemia (>300 mg/dl) was observed in both mice fed AL and those subjected to multiple cycles of the 4-day FMD every 7 days (4 days of FMD followed by 3 days of re-feeding, every 7 days per cycle) (Figures 2A and 2B). Levels of blood glucose continued to increase in STZ-treated mice receiving the AL diet, and reached levels above 450 mg/dl at both days 30 and 50. On the other hand, in mice receiving FMD cycles, hyperglycemia and insulin deficiency were both significantly alleviated on day 30. (Figure 2B, sample size indicated in parentheses). More remarkably, the levels of these physiological parameters returned to nearly normal range by day 50–60 after cycles of FMD (Figures 2B and 2C, sample size indicated in parentheses). Intraperitoneal glucose tolerance tests (IPGTTs) at day 50 confirmed that STZ-treated mice undergoing the FMD cycles have improved capacity to clear exogenous blood glucose, compared to mice on the regular chow (AL)(Figure 2D).
Figure 2. FMD cycles reverse STZ-induced β-cell depletion and restore glucose homeostasis.

(A) Experimental scheme of the periodic FMD’s effects on STZ-induced T1D; Baseline measurements (BL) were performed at day 5 after STZ treatment.
(B) Fasting glucose levels and (C) Plasma insulin levels during and 55 days after the FMD cycles (d5 to d35); vertical dashed lines indicate each cycle of FMD; horizontal lines (125±12mg/dl) indicate levels of blood glucose in the naïve control mice.
(D) Glucose tolerance test at d50.
(E) Cytokine profile of mice treated with STZ or STZ+FMD at d30, compared to that in naïve controls.
Pancreatic samples collected at indicated time points were analyzed for:
(F) Proliferative proportion in β cells.
(G) Proportion of insulin-producing β cells per islet and (H) Representative micrographs with immunostaining of insulin, glucagon and DAPI on pancreas sections of mice treated with STZ or STZ + FMD at the indicated time points. Scale bar represents 50um.
Mice of the C57BL/6J background, age 3–6 months, received STZ treatments (150 mg/kg) as indicated in (A). For (B–G), each point represents the mean ± s.e.m and sample size (n) is indicated in parentheses and for (F and G), *p<0.05, **p<0.01, ***p<0.005, one-way ANOVA. Ctrl, STZ-untreated control; STZ BL, baseline level of STZ treated mice at day 5. n≥6 mice per group per time point, n≥15 islets per mouse.
Levels of certain circulating cytokines have been used as indicators to determine islet pathological status in patients with recent-onset T1D (Baeyens et al., 2014; Grunnet et al., 2009; Lebastchi and Herold, 2012). We performed a 23-plex immunoassay to determine the effects of the FMD on inflammatory markers. We found that FMD cycles not only suppressed the circulating cytokines associated with β-cell damage (e.g. TNFalpha and IL-12) but also increased circulating cytokines associated with β-cell regeneration (e.g. IL-2 and IL-10)(Figure 2E, day 30)(Dirice et al., 2014; Rabinovitch, 1998; Zhernakova et al., 2006).
Taken together these results indicate that FMD cycles reduce inflammation and promote changes in the levels of cytokines and other proteins, which may be beneficial for the restoration of insulin secretion and the reversal of hyperglycemia.
FMD cycles reverse STZ-induced β-cell depletion
The characterization of pancreatic islet cells indicate a strong association between restored glucose homeostasis and the replenishment of pancreatic β cells in animals undergoing FMD cycles. STZ treatments resulted in an increase of non-α/β cells (Figure S3) and an about 85% depletion of insulin-secreting β cells (Figure 2F, STZ BL). The transient increase of non-α/β cells was reversed by d30 in both groups (Figures S2D and S2E). Mice receiving weekly cycles of the FMD showed a major increase in proliferative β cells followed by a return to a nearly normal level of insulin generating β cells by d50 (Figures 2F to 2H). In contrast, mice received ad libitum access to regular chow (AL) remained depleted of β-cells for more than 50 days (Figures 2F and 2H). Overall, the increase of non-α/β prior to β-cell proliferation raises the possibility that weekly cycle of the FMD might mediate the fate conversion of non-α/β cells to β cells to reverse the STZ-induced β-cell depletion, although other scenarios are possible.
FMD and post-FMD re-feeding promote β-cell regeneration in non-diabetic mice
We investigated whether and how the FMD and the post-FMD re-feeding period could regulate the cell populations within the islets to promote β-cell regeneration independently of diabetes, with a focus on the non-α/β cells and proliferative β cells. To characterize cellular and hormonal changes, pancreatic samples and peripheral blood of wild-type C57Bl6 mice fed with the FMD for 4 days were collected before the diet (BL), at the end of the diet (d4) and 1 day or 3 days after mice returned to the normal diet (RF1d or RF3d). The FMD caused a trend of decrease in the number and size of pancreatic islets (Figure 3A) and reduced the proportion of β cells by 35% (Figures 3B and 3C, see also Figure S4 for absolute numbers). These effects were reversed within three days of re-feeding (Figures 3A and 3C). Non-α/β cells begun to proliferate at the end of the FMD and this proliferation persisted until 1 day after re-feeding (RF1d), leading to a 2.5-fold increase in non-α/β cells (proportion per islet) at RF1d (Figure S4). By RF3d, the number of non-α/β cells had dropped and that of β cells returned to basal levels (BL), although β cells remained in a much more proliferative state in the FMD group (Figure 3B and 3C). The expression of proliferation marker PCNA was elevated in β cells but not α cells after re-feeding post FMD (Figures 3B, 3C and S4). Despite number of α cells per islet remain the same, the transitional α-to-β or β-to-α cells that co-express both α (i.e. glucagon) and β cell (i.e. Pdx-1 or insulin) markers were increased in mice that received the FMD (Figure 3D). In summary, the FMD promotes a decrease in the numbers of differentiated or committed cells, followed by the induction of transitional cells leading to major increases in the proliferation and number of insulin-generating β cells (Figure 3E).
Figure 3. FMD and post-FMD refeeding promote β-cell proliferation and regeneration.

(A) Size and number of pancreatic islets per pancreatic section.
(B) Proliferative proportion of β cells and proportion of β cells per islet.
(C) Representative images of pancreatic islets with Insulin, glucagon and PCNA immuno-staining.
(D) Transitional cell population co-expressing both the markers of α and β cells: Proportion of α cells and Pdx1+α cells. Arrows in the images with split channels indicating Pdx1+Gluc+ and Insulin+Glucagon+ cells.
(E) Schematic of FMD- and post-FMD refeeding induced cellular changes in pancreatic islets.
Mice of the C57BL/6J background, at age 3–6 months, received no additional treatments other than the indicated diet. Pancreatic samples were collected from mice fed ad libitum (AL) or the fasting mimicking diet (FMD) at indicated time points: the end of the 4d FMD (FMD), 1 day after re-feeding (RF1d) and 3 days after re-feeding (RF3d); for immunohistochemical and morphometric analysis (A to E): n ≥ 6 mice per group, n ≥ 30 islets per staining per time point. mean ± s.e.m,*p<0.05, ** p<0.01, ***p<0.005, one-way ANOVA.†p<0.05, t-test.
FMD promotes a gene expression profile in adult mice similar to that observed during embryonic and fetal development
To identify the genes that may mediate the FMD-induced pancreatic regeneration we measured gene expression in pancreatic islets at the end of the FMD and post-FMD re-feeding. At both time points, we observed a transient up-regulation of Foxo1 (6.9 fold at FMD, 5.3 fold at RF1d, *p<0.05 comparing to AL) and of a set of genes that have been previously identified as dual regulators for both fat-metabolism and fate-determination in mammalian cells (Cook et al., 2015; Haeusler et al., 2014; Johnson et al., 2004; Kim-Muller et al., 2014; Mu et al., 2006; Stanger, 2008; Talchai et al., 2012; Talchai and Accili, 2015; Tonne et al.)(Figure 4A), in agreement with the metabolic changes found in mice receiving the FMD (Figure S1). We further examined whether the metabolic reprogramming caused by the FMD affects lineage determination in pancreatic islets. In Figure 4B, the expression of lineage markers was determined by the mRNA expression of purified islets from mice fed ad libitum (AL) or the FMD. Results from the qPCR array indicate that up-regulation of the following genes was statistically significant (*p<0.05 comparing to AL, Figure 4B; see also Figure S5): i) pluripotency markers (e.g. Lefty1, 3.0 fold during FMD, 7.0 fold at RF1d; Podx1, 3.9 fold during FMD, 9.8 fold at RF1d; Nanog, 2.6 fold during FMD and 5.4 fold RF1d and Dnmt3b, 31.6 fold during FMD and 18.3 fold RF1d), ii) embryonic development markers (e.g. Sox17, 3.4 fold during FMD and Gata6 3.1 fold during FMD and 2.7 fold at RF1d), iii) pancreatic fetal stage markers and iv) β-cell reprogramming markers (e.g. Mafa, 4.7 fold at RF1d; Pdx-1 3 fold during FMD, 5.07 fold at RF1d; and Ngn3, 21.5 fold during FMD, 45.6 fold at RF1d.) (Figure 4B)(Zhou et al., 2008). These changes in gene expression suggest that the FMD either causes: i) a de-differentiation of pancreatic cells towards pluripotency at the end of the diet followed by re-differentiation to pancreatic β-cell lineage during early re-feeding (RF1d) or ii) recruitment of cells with these features from outside of the pancreatic islets. The assessment of protein expression of cells within the islets was also carried out by immunostaining for key proteins associated with pancreatic development (Figure 4C and 4D). In agreement with the results of qPCR array (Figure 4B), we found that protein levels of Sox17, as the early lineage marker, were elevated at the end of the FMD (FMD-4d) and protein levels of Ngn3, a marker for endocrine progenitors, were transiently up-regulated during early re-feeding (FMD-4d to RF1d)(Figure 4C).
Figure 4. Fasting mimicking diet (FMD) initiates metabolic reprogramming in pancreatic islets and stepwise redirect development of β cells in adult mice.

(A) mRNA expression profile indicating changes of metabolic genes in pancreatic islets and (B) mRNA expression profile indicating changes in lineage markers in pancreatic islets, at the end of 4d FMD (FMD) and 1d after refeeding (RF1d), comparing the ad libitum (AL) control; *p<0.05. t-test. Heat-map generated by Qiagen RT2 PCR array indicating a fold-regulation ranging from 77 (max, red) to −4 (min, green).
(C) Quantification of protein expressing cells of lineage markers in pancreatic islets, from mice fed AL or on FMD at indicated time points. Protein expression was defined as a marker+ area/total islet area. See also Figure S5B.
(D) Representative images of immunofluorescent staining indicating stepwise transition of Sox17/Pdx1 and Pdx1/Ngn3. Scale bar represents 50um.
Mice of the C57Bl6J background, at age 3–6 months, received no additional treatments other than the indicated diet. Pancreatic samples were collected from mice fed ad libitum (AL) or on the fasting mimicking diet (FMD) at indicated time points: the end of 4d FMD (FMD), 1 day after re-feeding (RF1d) and 3 days after re-feeding (RF3d); n=6 mice per group, ≥ 30 islets per marker.
To determine whether step-wise β-cell conversion from the dedifferentiated cells occurs during early refeeding, we performed double-staining for the targeted developmental markers (i.e. Sox17, Pdx-1, Ngn3), across the time points of FMD. We also measured the expression of Oct4 (Pou5f1), which has been previously reported to be expressed in the nucleus of adult pancreatic islets in association with Foxo-1 related diabetic β-cell dedifferentiation (Talchai et al., 2012; Xiao et al., 2013). Oct4 (Pou5f1) mRNA expression showed a trend for an increase in mice on the FMD, which is not significant (Figure 4C, p>0.05). Results of immunostaining indicate that Oct4 (Pou5f1) and Sox17 may only be co-expressed very transiently after overnight re-feeding (Figure S5B, RF12hr) followed by robust expansion of Sox17+Pdx1+ and then Pdx1+Ngn3+ cells at RF1d (Figure 4D and see also Figure S5B for all time points). Although Ngn3+ cells were also detectable in AL mice, they were mainly located outside or on the edge of the islets, in agreement with what was reported in previous studies (Baeyens et al., 2014; Gomez et al., 2015) (Figure 4D). The number of Ngn3+ cells was increased both inside and outside the islets during the FMD and re-feeding (Figure 4D).
These results suggest that as a result of the FMD and re-feeding cycle, the pancreatic islets contain an elevated number of cells with features of progenitor cells, which may differentiate and generate insulin-producing cells.
FMD induces Ngn3 expression to generate insulin-producing β cells
Ngn3+ cells within the pancreatic islets have been previously described as progenitor cells able to generate all lineages of endocrine cells including the insulin-producing β cells, although the role of Ngn3 in adult β-cell regeneration remains unclear (Baeyens et al., 2014; Van de Casteele et al., 2013; Xu et al., 2008). To investigate whether the FMD causes de novo expression of Ngn3 and whether Ngn3+ cells may contribute to FMD-induced β-cell regeneration, we generated Ngn3-CreER;tdTomatoLSL-reporter mice to trace the lineage of putative Ngn3-expressing cells and their progeny in the adult mice treated with the FMD (Figure 5A). To initiate the loxP recombination for lineage tracing, low-dose tamoxifen injections (2mg per day, for 3 days) inducing the recombination (maximized at 48hr and minimized within a week) were given to mice before or after the FMD and to mice fed ad libitum (AL control) (Figure 5A). Tissue collection time points are relative to the time of injection and to that of FMD treatments (Figure 5A). Results indicate that the FMD induced the expansion of the Ngn3-derived lineages (Figure 5B and 5C). Characterization of tdTomato+ cells by immunostaining indicates that tdTomato+ cells contributed 50.8 ± 8.3% of the overall beta cell pool following the FMD (Figure 5C, group C).
Figure 5. FMD promotes Ngn3-dependent lineage reprogramming to generate insulin-producing β cells.

(A) Genetic strategy used to perform lineage tracing (tdTomato) of NGN3-expressing cells in pancreas and schematic time line of tamoxifen (TAM) treatments for lineage tracing experiments. A, mice fed ad libitum were treated with TAM; B, mice receiving FMD 3 days after TAM injection; C, mice receiving TAM and FMD concurrently and D, mice receiving FMD and vehicle (corn oil) concurrently. Pancreatic tissues were collected 11 days after TAM injection to analyze the effects of FMD on Ngn3-lineage generation. Tdtomato+ cells (red, arrows) are Ngn3-derived cells; n=6 for each group.
(B) Representative images of the labeled Ngn3-lineage cells (red, tdTomato) and Insulin-producing β cells (green, Ins) at the indicated time-points in pancreatic islets. Left panel, scale bar represents 150um; right panel, scale bar represents 50um.
(C) Quantification of total tdTomato labeled Ngn3-lineage cells per islet (top) and proportion of labeled insulin-producing β cells (ins+tdTomato+) (bottom). mean ± s.e.m, **p<0.01, ***p<0.005, t-test.
(D) Genetic strategy used to perform diphtheria toxin gene A chain (DTA)-mediated Ngn3-lineage ablation in pancreas and schematic time line of tamoxifen (TAM) treatments for lineage ablation experiments (left) and results of glucose homeostasis (right). Mice were injected with TAM prior to and after the FMD, to ablate Ngn3 lineage developed and/or expanded during FMD and early refeeding (RF3d). Alternatively, mice were given additional STZ injection and then assigned to the indicated dietary groups (i.e. AL+STZ or FMD+STZ), to analyze the contribution of FMD-induced β-cell conversion to glucose homeostasis.
(E) Representative images of pancreatic islets with insulin and Pdx1 immunostaining for β cells, DAPI for nuclei. See also Figure S5 for the images of vehicle controls.
(F) Quantification of insulin-producing β cells from Ngn3-lineage ablated mice of indicated groups. mean ± s.e.m,*p<0.05, **p<0.01 t-test, (top) paired t-test (bottom). n=6 for TAM and STZ, n=3 for vehicle controls. Scale bar represents 50um.
(G) Glucose levels in homeostasis and intraperitoneal glucose tolerance tests (IPGTTs) for the indicated groups. mean ± s.e.m,**p<0.01 t-test, (top) paired t-test (bottom). n=6 for TAM and STZ, n=3 for vehicle controls.
For (A) to (C), mice of ICR and B6;129S6 mixed background, at age 3–6 months, received the diet and/or STZ treatments indicated in (B). For (D) to (G), mice of ICR and B6;129S6 mixed background, at age 3–6 months, received the diet and/or STZ treatments indicated in (F).
To confirm the contribution of FMD-induced Ngn3 lineages in reconstituting insulin-secreting beta cells, we generated another mouse model (Ngn3-CreER/LSL-R26RDTA) and performed lineage-ablation experiments in both wild-type non-diabetic mice and STZ-treated mice (Figure 5D). The results indicate that ablation of Ngn3+ lineage reversed FMD-induced β-cell regeneration and its effects on fasting glucose levels in the naïve wild-type mice (Figures 5E, 5F and S5) and that on glucose clearance capacities (IPGTT assay) in STZ-treated diabetic mice (Figure 5G), confirming the FMD-induced generation of an Ngn3-lineage and suggesting a critical role for this Ngn3-dependent β-cell neogenesis in glucose homeostasis.
Fasting conditions or inhibition of nutrient signaling pathways promote Ngn3 expression and insulin production in human pancreatic cells
In both mouse and humans, Ngn3 expression occurs right before and during endocrine cell generation. Ngn3 mRNA expression in the developing mouse pancreas peaks around E15.5, which is roughly equivalent to week 7–8 (Carnegie Stages 21–22) in human development. Expression of Ngn3 in adult mouse islets, although rare, has been demonstrated by rigorous lineage reporter analysis (Wang et al., 2009). In agreement with results from others, our data (Figure 5 and S5) indicate that Ngn3+ cells in adult pancreas islets are important for beta-cell regeneration in mice. On the other hand, the role of Ngn3 in human islet development and β-cell regeneration in adulthood remains poorly understood (McKnight et al., 2010).
To investigate how the fasting mimicking conditions affect Ngn3 expression and β-cell functions in human pancreatic cells, we performed ex vivo experiments using primary human pancreatic islets (Figure 6A). Briefly, the pancreatic islets from healthy and T1D subjects (HI and T1DI, respectively) were cultured according to the manufacturer’s instructions. The cultured islets were then treated with serum from subjects enrolled in a clinical trial testing the effects of a low protein and low calorie FMD lasting 5 days (NCT02158897) 17. Serum samples were collected at baseline and at day 5 of the fasting mimicking diet in 5 subjects. We then measured IGF-1, glucose and ketone bodies and treated human pancreatic islets with the subject-derived serum (Figure 6B, Table S1). In both healthy islets and T1D islets exposed to the serum of FMD-treated subjects, we observed a trend for glucose-dependent induction in the expression of Sox2 and Ngn3 (Figure S6A).
Figure 6. Ngn3 expression and insulin producing function of human pancreatic islets in response to fasting conditions.

(A) Experimental scheme for fasting conditioning treatments on human pancreatic islet. Pancreatic islets from healthy human subjects (HI) or from T1D subjects (T1DI) were cultured separately based on manufacturer’s instructions and then exposed to fasting conditions (i.e. STS media, mTOR and PKA inhibitors and PKA siRNA) or control media for 36hr.
(B) Levels of hIGF-1, glucose, insulin and ketone bodies in the serum from human subjects, prior (baseline) and after receiving the FMD (FMD). n=5 per group.
(C) Insulin secretion capacity of HI and T1DI pre-treated with short-term starvation (STS) conditioned medium (2% FBS and 0.5 g/L glucose) and then induced with 25mM glucose, compared to that of islets cultured in standard medium (STD). n=3 per group.
(D) Sox2 and (E) Ngn3 expression in HI and T1DI pre-treated with STS-conditioned medium with or without administration of IGF-1 (40ng/ml). n=6 per group.
(F) Immunostaining for Ngn3 protein expression in HI and T1DI. n=5 per group. Scale bar represents 100um.
(G) Insulin gene expression, (H) PKA activity and (I) mTOR activity in HI and T1DI pretreated with STS-conditioned medium with or without administration of IGF-1 (40ng/ml); phosphorylated versus total p70S6K ratio was used as an indicator of mTOR activity, which was normalized to the levels of STD (standard medium); n=6 per group.
(J and K) expression of lineage markers (Sox2 and Ngn3) in HI and T1DI treated with inhibitors dampening IGF-1 signaling; rapamycin, mTOR inhibitor; H89, PKA inhibitor and PKA siRNA. mean ± s.e.m,*p<0.05, ** p<0.01, ***p<0.005, unpaired t-test.
We then applied the low-glucose and low-serum fasting mimicking medium (STS) to the cultured pancreatic islets, and found that it significantly stimulated the secretion of insulin in both HI and T1DI (Figure 6C). We further investigated the expression of lineage reprogramming markers, which we found to be up-regulated in mice as a result of the FMD-treatment (i.e. Nanog, Sox17, Sox2, Ngn3 and Ins). The results indicate that the fasting mimicking conditions had strong effects in inducing the expression of Sox2, Ngn3 and insulin in human pancreatic islets from healthy (healthy islets, HI) and T1D subjects (T1D islets, T1DI) (Figure 6D–F). In cells from normal human subjects, these effects were reversed by IGF-1 treatment (Figure 6G). Notably, in human T1D cells, IGF-1 reversed the increased insulin and Sox 2 gene expression but not that of Ngn3 expression caused by the STS medium (Figure 6G vs. Figure 6D and 6E). Future studies are warranted to further investigate the role of circulating IGF-1 in the expression of lineage reprogramming markers and pancreatic islet cells regeneration in vivo.
In both healthy and T1D human islets, STS medium significantly reduced the activity of PKA, an effect reversed by IGF-1 treatment (Figure 6H). It also dampened the activity of mTOR, which is a key mediator of amino acid signaling (Figure 6I). To further investigate the role of these nutrient sensing signaling pathways in regulating the expression of lineage markers (i.e. Sox2 and Ngn3), we tested the role of the Tor-S6K and PKA pathways, which function downstream of IGF-1, in the reprogramming of pancreatic cells. Human pancreatic islets cultured in standard medium were treated with rapamycin, which inhibits mTOR, and H89, which inhibits PKA. mTOR and PKA were implicated by our group and others in the regeneration of other cell types (Cheng et al., 2014; Yilmaz et al., 2012). We found that in human islets from T1D subjects (T1DI), expression of the essential lineage markers Sox2 and Ngn3 was not induced by inhibition of either mTOR or PKA, but was significantly induced when both mTOR and PKA were inhibited (Figure 6J and 6K). Interestingly, the constitutive mTOR, but not PKA, activity is trending higher in HI compared to T1D1 cells (Figure 6I, lane 1 for both sets for mTOR activity and Figure 6H for PKA activity), which may explain the overall differences between HI and T1DI in Sox2 and Ngn3 expression shown in Figure 6J. Taken together, these results indicate that fasting may be effective in promoting lineage reprogramming and insulin generation in pancreatic islet cells, in part by reducing IGF-1 and inhibiting both Tor and PKA signaling. Pancreatic cells from T1D subjects displayed constitutively elevated activity of Tor-S6K and PKA, which points to the potential for inhibitors of both pathways in the induction of Ngn3-mediated lineage reprogramming. These results raise the possibility that the effect of the FMD on pancreatic regeneration in T1D subjects could be mimicked or enhanced by pharmacological inhibition of these pathways.
Discussion
During mouse development, at embryonic day E8.5, pancreatic progenitor cells co-express the SRY-Related HMG-Box transcription factors Sox17 and homeodomain transcription factor Pdx1. These multipotent pancreatic progenitors are then converted into bipotent epithelial cells that generate duct cells or a transient population of endocrine precursor cells expressing the bHLH factor Neurogenin3 (Ngn3). Ngn3+ endocrine precursors give rise to all the principal islet endocrine cells including glucagon+ α cells and insulin+ β cells (Arnes et al., 2012). In mice, expression of Ngn3 in the developing pancreas is transient, detectable between E11.5 and E18 (Arnes et al., 2012). Whether developmental genes including Sox17, Pdx-1 and Ngn3 could be activated to generate functional β-cells in adults was previously unknown.
Both cell-based therapy and the use of cytokines and hormones that stimulate β-cell self-replication have the potential to restore insulin-producing β cells in diabetic patients (Dirice et al., 2014; Yi et al., 2013). However, despite some success with transplantation-based therapy, the short supply of donor pancreata plus the inefficient conversion of stem cells into specialized derivatives have represented obstacles for clinical application, suggesting that a successful β-cell regeneration might depend on the coordinated activation and re-programming of endogenous progenitors (Blum et al., 2014; Sneddon et al., 2012; Wang et al.; Xiao et al., 2013). Recently, this in vivo lineage reprogramming or trans-differentiation, has become an emerging strategy to regenerate β cells (Cohen and Melton, 2011; Heinrich et al., 2015) (Abad et al., 2013; Xu et al., 2015).
In this study, we discovered that a low protein and low sugar fasting mimicking diet (FMD) causes a temporary reduction in β-cell number followed by its return to normal levels after re-feeding, suggesting an in vivo lineage reprogramming. We show that the severe hyperglycemia and insulinemia in both the late stage Leprdb/db T2 and the STZ-treated T1 mouse diabetes models were associated with severe β-cell deficiency in pancreatic islets. 6-to-8 cycles of the FMD and re-feeding were required to restore the β-cell mass, insulin secretion function and to return the 6hr-fasting blood glucose to nearly normal levels. In non-diabetic wild-type mice, the portion of beta cells per islet, as well as the total number of β cells per pancreas was reduced by about 60% at the end of a 4-day FMD but their normal level was completely restored within 3-to-5 days post re-feeding. Also, insulin and blood glucose levels were reduced by 70% or more at the end of the FMD period but returned to normal levels within 24-to-36 hours of re-feeding. Interestingly, in diabetic mice the majority of cells residing in the islets expressed neither insulin nor glucagon (i.e. non-α/β). This phenotype was also found in non-diabetic wild-type mice during the FMD and was accompanied by an increase of other transitional cell types (i.e. Pdx1+Glucagon+ cells and Insulin+glucagon+) followed by significant β-cell regeneration upon re-feeding. This suggests that the FMD alters the gene expression profile that normally suppresses the generation of β cells. More importantly, these results suggest that dietary-induced lineage-conversion occurring prior to the β-cell proliferation may play an important role in beta-cell regeneration across the diabetic and non-diabetic mouse models. One possibility is that glucagon and insulin expression are transiently suppressed in α and β cells during the FMD, followed by lineage reprograming in committed cells. Another possibility is that the FMD may cause cell death and then stimulate progenitor or other cells to regenerate β cells.
The FMD reversed the dedifferentiated expression profile for a number of genes associated with maturity-onset diabetes of the young (MODY) and regulated by Foxo1 (Kim-Muller et al., 2014). The FMD appears to cause pancreatic islets to first increase the expression of Foxo1 and its transcriptional targets, then induce transitionally the expression of the progenitor cell marker Ngn3+ upon re-feeding, leading to β-cell regeneration. We conclude that together with the changes in a wide range of cytokines associated with β-cell regeneration, FMD and post-FMD re-feeding generate the complex and highly coordinated conditions that promote the generation of stable insulin producing β-cells to reverse severe β-cell depletion. The key changes priming pancreatic islet cells for regeneration during the FMD, appear to be the reduction of IGF-1 levels, and the consequent down-regulation of PKA and mTor activity, in agreement with the role for these pathways in hematopoietic (Cheng et al., 2014) and intestinal stem cell self-renewal (Yilmaz et al., 2012). It was proposed that transient dedifferentiation of β-cells may play a role in their in vivo dynamics (Kim-Muller et al., 2014; Weinberg et al., 2007). The capacity of these de-differentiated cells to re-differentiate fundamentally changes the therapeutic potential of existing cells in promoting β-cell regeneration and reverse T1D symptoms (Blum et al., 2014; Wang et al., 2014). Thus, our study provides an example of a potent and coordinated dietary regulation of cell fate-determination with the potential to serve as a therapeutic intervention to treat diabetes and other degenerative diseases. Our preliminary results from a pilot clinical trial also indicate that the use of periodic cycles of a prolonged FMD is feasible and ready to be tested in large randomized clinical trials for effects on both insulin resistance and pancreatic β-cell regeneration for the treatment of both T1D and T2D.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Valter D. Longo (vlongo@usc.edu).
Experimental Model and Subject Details
Mouse models
All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Southern California (USC). All mice were maintained in a pathogen-free environment and housed in clear shoebox cages in groups of five animals per cage with constant temperature and humidity and 12 hr/12 hr light/dark cycle. Prior to supplying the FMD diet, animals were transferred into fresh cages to avoid feeding on residual chow and coprophagy. All animals had access to water at all times. Unless otherwise on experimental diets, mice were fed ad libitum with regular chow (e.g. AIN93G).
T1D: High-dose streptozotocin (STZ) injection (180 mg/kg) for inducing T1D was performed to cause β-cell depletion (>70% loss) and hyperglycemia (>250dl/ml)(Wu and Huan, 2008). Male C57Bl6 mice (16 weeks, n=18) were weighed and fasted 4 to 6 hours prior to STZ injection. Briefly, STZ was dissolved in sodium citrate buffer (pH 4.5, freshly prepared) at a concentration of 10 mg/ml immediately prior to use and STZ solution was administered intraperitoneally (IP) within 5 minutes from preparation according to mice weight. An equal volume of citrate buffer was injected into male and female C57Bl6 mice (4- to 8-months-old, n=18) that were used as healthy controls. Mice were returned to their cage and food was replaced and 10% sucrose water was provided to prevent hypoglycemia in the days immediately following STZ injection. T2D: Ten-week-old male BKS.Cg-+Leprdb/+Leprdb/Jcl (db/db) mice and BKS.Cg-m+/m+/Jcl (m/m) control mice (n=15 for db/db and n=7 for control) were purchased from The Jackson Laboratory. Age-matched wild-type littermates were used as the healthy controls. Unless otherwise specified, mice were not fasted at the time of tissue collection. All experiments and procedures were performed according to an approved protocol by the Institutional Animal Care and Use Committee at University of Southern California.
Lineage tracing and Lineage ablation
Hemizygous transgenic Ngn3-CreER mice were bred with R26RCAG-LSL-tdTomato reporter mice for lineage tracing experiments, or bred with R26RDTA mice for lineage ablation experiments (Jackson Laboratory). Tamoxifen (TAM) injections were performed as previously described (Madisen et al., 2010). Briefly, 75 mg/kg TAM was injected i.p. at the interested time points (as indicated in Figure 5B and Figure 5F). Both sex mice (12–16 weeks old) were used n=6 for each group, n=3 for vehicle control.
Human pancreatic islets
Human Pancreatic Type 1 Langerhans Cells (#35001-03) and non-diabetic control (#35002-04) were purchased from a commercial source Celprogen (Torrance, CA). The cells were seeded at 80% of confluence and cultured with standard medium: Celprogen, M35002-04S for healthy islets and M35001-03S for T1D islets. The cells were plated at 105 per ml density and incubated in the appropriate size flask at 37°C in a 5% CO2 humidified incubator. Medium were changed every 48h. When reached 65–75% of confluence, cells were passaged with Celprogen Trypsin-EDTA (T1509-014) and subcultured for expansion.
Human subjects (FMD)
Study subjects were recruited under protocols approved by the IRB (approval # HS-12-00391) of the University of Southern California (USC) based on established inclusion and exclusion criteria. Informed consent was obtained from all subjects. Examination and tests included, but were not limited to, blood draws through venipuncture before (AL) and after the 1st FMD cycle (FMD) and after 5–8 days after completion of the 3rd dietary intervention cycle (RF). Presented are preliminary results for a set of 6 to 8 healthy subjects (see Table S1 for the profiles of these individuals).
Criteria for inclusion of human subjects
Generally healthy adult volunteers, subjects of 18–70 years of age, body mass index: 18.5 and up, ability and willingness to provide written informed consent, ability and willingness to undergo multiple cycles of a 5-day dietary regimen, ability and willingness to provide blood samples via venipuncture. Exclusion criteria: Any major medical condition and chronic diseases, mental illness including severe depression and dementia, drug dependency, hormone replacement therapy (DHEA, estrogen, thyroid, testosterone), severe hypertension (systolic BP > 200 mm Hg and/or diastolic BP > 105 mm Hg), underweight (BMI < 18.5 kg/m2), females who are pregnant or nursing, special dietary requirements incompatible with the study interventions or food allergies, alcohol dependency). Profile: Subjects in this study are at age 49.1±6.9, with heights of 166.7±4.7cm, with body weights of 63.32±4.8 kg at baseline and 61.36±4.3 at the end of FMD.
Method Details
Mouse fasting mimicking diet (FMD)
The mouse version of the FMD is a 4-day regimen using ingredients similar to those used for the human FMD (see Table S2) (Brandhorst et al., 2015).. All diet ingredients were thoroughly mixed and then blended together with heated hydrogel (ClearH2O, Maine). Mice on the FMD diet were fed 50% of the standard daily calorie intake on day 1 and 10% of normal daily calorie intake on days 2 to 4.All mice were supplied with fresh food during the morning hours (8am–10am). FMD mice generally consumed the supplied food within the first few hours of the light cycle. Control-fed animals usually consumed the supplied food during the dark hours. Post-FMD refeeding: After the end of the day 2–4 diet, mice were fed ad libitum regular chow for up to 10 days to regain body weight before the next FMD cycle.
Blood glucose and insulin measurement
Unless otherwise specified, mice were fasted for 6 hours (morning fasting) before blood glucose measurements. Blood glucose was measured through tail vein bleeding with the use of the OneTouch UltraMini Blood Glucose Monitoring System (Lifescan, Milpitas, CA, USA). Plasma insulin levels were determined by mouse enzyme-linked immunosorbent assay (Mercodia, Winston-Salem, NC, USA), according to manufacturer’ s instructions. Whole blood was withdrawn from the facial vein, and plasma was separated by centrifugation at 14,000 rpm for 3 min in plasma separator tubes with lithium heparin (BD, Franklin Lakes, NJ, USA). Briefly, 10 mL of plasma samples and calibrators were aliquoted in a mouse monoclonal anti-insulin antibody-coated 96-well plate. Working solution containing peroxidase-conjugated anti-insulin antibody was then added to each well (100 m L per well), and the plate was put under shaking conditions at room temperature (RT) for 2 h. After six washes with washing buffer, 200 mL of 3,3′,5,5′-Tetramethylbenzidine substrate was added to each well and allowed to incubate for 15 min at RT. The reaction was stopped with 0.5 mol/L H2SO4 and optical density (OD) was read at 450 nm with the use of the Victor3 multilabel plate reader (PerkinElmer, Waltham, MA, USA). A standard curve was drawn by plotting OD450 and insulin concentration of calibrators with the use of Sigmaplot 11 data analysis software (Systat Software Inc, San Jose, CA, USA) and used as reference to extrapolate sample values for insulin concentrations (Milanesi et al., 2012; Villani et al., 2014). The homeostatic model assessment (HOMA) was the method used to quantify insulin resistance (HOMA-IR) and beta-cell function (%B). HOMA-IR was calculated using the following formula: HOMA-B = (Fasting glucose x Insulin)/22.5. HOMA %B was calculated using the following formula: HOMA-B = (20 x Fasting Insulin)/(Fasting Glucose-3.5) % (Matthews, 2001).
Intraperitoneal glucose tolerance testing (IPGTT) and insulin tolerance testing
Mice were single-caged and fasted for 4 hours prior to the injections. Glucose (1.5 mg/g) or Insulin (0.75 unit/kg) was injected intraperitoneally. Blood glucose levels were measured at 5, 15, 30, 60, and 90 min after the injection.
Immunofluorescence Analysis
Briefly, the pancreatic samples were collected without saline perfusion and immediately processed for paraffin embedding and histology as previously described (Milanesi et al., 2012; Villani et al., 2014). Tissues were fixed in 10% neutral phosphate buffer formalin (Polysciences Inc, Warrington, PA, USA) for 1 h at 4 °C and stored in 70% ethanol overnight at 4 °C. Tissues were subsequently dehydrated through graded ethanol, toluene and finally embedded in paraffin (TissuePrep, Fisher Scientific, Pittsburgh, PA, USA). Pancreatic sections (5 mm) were sliced with the use of a Leica RM2235 rotary microtome and dried ON. The following day sections were deparaffinized in Histochoice clearing agent (Sigma-Aldrich) and rehydrated through graded ethanol series (100%, 90%, 70%, 50% and 30%) followed by rinsing in distilled water before use. Heat-mediated antigen retrieval with antigen unmasking solution (H-3300, Vector Laboratories) was performed and sections were blocked with 2% bovine serum albumin (BSA) solution for 30 min at RT. The following primary antibodies were used: anti-insulin (Abcam, ab8304), anti-glucagon (Abcam, ab8055), anti-PCNA (Abcam, ab2426), anti-PDX1 (Abcam, ab47383). Additional antibodies used for developmental markers are Anti-Oct4 antibody - ChIP Grade (Abcam, #ab19857) and anti-Oct-4A (C30A3) rabbit mAb (Cell Signaling, 2840P), Anti-Sox17 (Santa Cruz Biotechnology #sc-17355) and Anti-NGN3 (LifeSpan Biosciences, #LS-C97692). Sections were incubated with primary antibodies in a humidified chamber for 1 hour at RT. After PBS washing, sections were incubated for 30 min at RT with secondary antibodies, respectively, Alexa-fluor 555 donkey anti-mouse immunoglobulin (Ig)G, Alexa-fluor 488 donkey anti-rabbit IgG, Alexa-fluor anti-goat 647 (dilution 1/500). All secondary fluorochrome-conjugated anitbodies were purchased from Life Technologies. Sections were mounted with Vectashield mounting medium with 40, 6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA, USA). The images of the stained sections were captured using a 10x, 20x or a 40x objective as indicated in figure legends with a Leica AF6000 fluorescent microscope. Double staining for Pdx1+Glucagon+ cells, Insulin+Glucagon+ cells and Ngn3-tdTomato+glucagon+ cells was verified by confocal imaging (Zeiss LSM 710). Numbers of cells or areas of interest were measured from 3–5 mice per group, 6–8 pancreas sections per mouse for 20–30 islets per condition per time point. “Islet area” was measured as previously described (Brereton et al., 2014). Basically, we measure total islet cross-sectional area per 2.5mm2 pancreas section (%). Please also see Figures S2–4 for the absolute cell counts per islet, the numbers and/or area of islets per pancreas section.
Pancreatic Islet isolation
Pancreatic islets were isolated form mice as previously described (Li et al., 2009). In brief, mice were sacrificed by CO2 inhalation and immediately processed for pancreatic perfusion. The pancreas was perfused with 3 ml of collagenase P (Roche Diagnostics Corp, 1–1.5 mg/ml, ca 1,000 U/mL) dissolved in HBSS. The collagenase solution was injected by inserting a 30G1/2-G needle into the common bile duct through the joint site of the hepatic duct under a dissecting microscope. After full pancreas distension the tissue was digested by incubation at 37°C for 30 min in a tota l of 2 ml of digestion solution. Digested tissue was vigorously shaken and subsequently washed twice in HBSS containing CaCl2. Islets were purified using Falcon cell strainers (Fisher Scientific) and hand-picked under a dissecting microscope. Islets were washed with PBS and used to extract total RNA for qPCR analysis (> 40 uM in size) or incubated in HBSS solutions for GSIS assay (average sized between 40 and 70 uM) in a humidified incubator (95% air, 5% CO2) at 37°C.
Cell Lineage Identification qPCR Array
Total RNA was extracted from purified islets by column method using the RNeasy Mini kit (Qiagen, Valencia, CA) according to manufacturer’s instructions and quantified with the Nanodrop system (Thermo Scientific, Waltham, MA). cDNA was obtained from the extracted RNA by retrotranscription with the RT2 First Strand Kit (SABiosciences, Qiagen, Valencia, CA) following manufacturer’s instructions. The cDNA of each sample was then added to the RT2 SYBR Green qPCR Master Mix and aliquoted for gene expression analysis on specific arrays for the mouse cell lineage identification (PAMM-508Z, SABiosciences). Gene analysis, including significant values and fold changes, was performed with the online tool provided by SABiosciences (http://www.sabiosciences.com/pcrarraydataanalysis.php).
Glucose stimulated insulin secretion (GSIS) Assay
Isolated purified islets were incubated in RPMI1640 medium (containing 11 mmol/l glucose and supplemented with 100 μg/ml streptomycin, 100 μg/ml penicillin, 2 mmol/l glutamine and 10% FBS) overnight at 37°C (5% CO2) and then hand-picked into the following medium for GSIS assay: glucose-starved preparation in HEPES buffer (pH 7.4, containing 125 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2 and 1.28 mM CaCl2, 1 mg/ml BSA) for 45min and then transferred to the basal glucose conditioned medium (2.5G)(10 islets per well into 500ul assay medium containing 2.5mM glucose) for 60min. Islets were then washed in glucose starvation medium for 5 min and then transferred to glucose stimulation medium (25G) containing 25mM glucose for 60 min. Medium was collected at the indicated time points (Figure 1K). The GSIS experiment was performed at least three times and in triplicate in 24-well tissue culture plates with 10 equally-sized islets per well. Washes in between treatments were performed under a dissecting microscope to preserve the number of islet throughout the assay.
Cytokines profiling
Mouse cytokine 23-plex immunoassay (BIO-RAD, USA) was used to perform simultaneous quantitative detection of multiple analytes from a single serum sample.
Human pancreatic islet treatments and qPCR
Cells were treated for 36h with: Standard medium: Celprogen, M35002-04S for healthy islets and M35001-03S for T1D islets). STS condition (0.5g/liter of Glucose; 2% FBS), H89 (20uM) (Sigma-Aldrich), Rapamycin (RAPA) (20nM) (LC Laboratories, Woburn, MA, USA), IGF1 (40ng/ml) (ProSpec, East Brunswick, NJ, USA) and PKA siRNA (Cell signaling, 6574S). For PKA silencing, cells were seeded at 80% of confluence, after 24h they were transfected for 48h with 100nM PKA siRNA (Cell signaling, 6574S) using Lipofectamine RNAiMAx Reagent (ThermoFisher) according to manufacturer instructions. Total RNA was extracted using RNeasy Mini kit (Qiagen) according to the manufacturer’s instructions and quantified with the use of the Nanodrop system (Thermo Scientific, Waltham, MA, USA). Briefly, 1ug of total RNA was extracted using RNeasy Mini kit and reverse transcribed with SuperScript II Reverse transcriptase (Invitrogen) and the detection of the different transcripts (Ngn3, Sox2, Insulin, and 18S as a reference gene) was carried out using the Kapa sybr fast qPCR kit (KAPA BIOSYSTEMS) according to the instructions of the manufacturer. The expression level were analyzed by quantitative real-time polymerase chain reaction (PCR) on a Roche LightCycler 480II. Gene expression was quantified using the comparative Ct method.
Western Blotting
Total cell lysates were prepared using the M-Per Protein Extraction Reagent (ThermoScientific, Rockford, IL, USA) according to manufacturer instructions. Protein concentration was measured with BCA assay (Thermo Scientific). Equal amounts of protein (60 μg) were heat-denatured in lane marker reducing sample buffer (Thermo Scientific), resolved by SDS-PAGE using Novex 4–20% Tris-Glycine MiniProtein Gels (Thermo Scientific), and transferred to PVDF membranes (Millipore,Darmstadt, Germany).The filters were blocked in 5% BSA for 1h at room temperature and then incubated overnight at 4°C with primary antibody directed against p70 S6 Kinase and phospho-p70 S6 Kinase(Thr389) (Cell Signaling rabbit mAb #2708, rabbit mAb #9234) and antibody against beta-tubulin (Millipore, #05-661). Peroxidase conjugated IgG (Santa Cruz, CA, USA) was used as secondary antibody. Membrane-bound immune complexes were detected by ultra-sensitive enhanced chemiluminescence system (Thermo Scientific) on a photon-sensitive film (Hyperfilm ECL, GE Healthcare, Milano, Italy). Quantification was performed by densitometric analysis using ImageJ64 software.
PKA activity
PKA activity was measured using the PKA kinase activity kit (Enzo Life Science, Inc., Farmingdale, NY, USA) according to manufacturer instructions.
Human fasting mimicking diet
The human version of the FMD is a propriety formulation belonging to L-Nutra (http://l-nutra.com/prolon/). It is a plant-based diet designed to attain fasting-like effects on the serum levels of IGF-I, IGFBP1, glucose and ketone bodies while providing both macro- and micronutrients to minimize the burden of fasting and adverse effects (Brandhorst et al., 2015). Day 1 of the FMD supplies ~4600 kJ (11% protein, 46% fat, 43% carbohydrate), whereas days 2–5 provide ~3000 kJ (9% protein, 44% fat, 47% carbohydrate) per day. The FMD comprises proprietary formulations of vegetable-based soups, energy bars, energy drinks, chip snacks, tea, and a supplement providing high levels of minerals, vitamins and essential fatty acids (Fig. S3). All items to be consumed per day were individually boxed to allow the subjects to choose when to eat while avoiding accidentally consuming components of the following day. For the human subjects, a suggested FMD meal plan was provided that distributes the study foods to be consumed as breakfast, lunch, snacks, and dinner. See lists below for ingredients and supplements:
Ingredients
Mushroom Soup
Rice flour, Carrot powder, Dried onion, Champignon mushroom powder, Inulin (chicory fiber), Dried champignon mushroom, Salt, Yeast extract, Potato starch, Olive oil, Dried parsley, Natural flavor.
Vegetable Soup
Rice flour, Dried onion, Inulin(chicory fiber), Dried tomato, Dried carrot, Salt, Dried red pepper, Dried leek, Potato starch, Olive oil, Freeze dried basil, Spinach powder, Dried parsley, Natural flavor.
Tomato Soup
Rice flour, Dried tomato powder, Dried onion, dried tomato pieces, dried carrot, chicory fiber, potato starch, olive oil, Salt, Yeast extract, Dried basil, Dried parsley, Natural flavor.
Pumpkin Soup
Pumpkin powder, rice flour, inulin (chicory fiber), dried carrot, salt, yeast extract, potato starch, olive oil, dried onion, dried parsley, natural flavor.
Energy Bar
Almond meal, Macadamia nut butter, Honey, Pecan, Coconut, Flaxseed meal, Coconut oil, Vanilla extract, Sea salt.
Kale Crackers
kale, golden flax seeds, sunflower seeds, cashews, Sesame seeds, nutritional yeast, apple cider vinegar, hemp seeds, pumpkin seeds, sea salt, onion powder, dill week, black pepper
Algal Oil Capsule
Algal oil, Gelatin, Glycerin, Purified water, Turmeric, Annatto extract
Teas
chamomile, spearmint, or Lemon
Energy drink
Purified water, natural vegetable glycerin, polylysine.
Supplements
Vitamin A (as Beta Carotene), Vitamin C (Ascorbic Acid), Vitamin D (as Cholecalciferol), Vitamin E (as DL-Alpha Tocopherol Acetate), Vitamin K (as Phytonadione), Thiamine (as Thiamine Mononitrate), Riboflavin, Niacin (as Niacinamide), Vitamin B6 (as Pyridoxine HCI), Folic Acid, Vitamin B12 (as Cyanocobalamin), Biotin, Pantothenic Acid (as Calcium-D-Pantothenate), Calcium (as Calcium Carbonate and Tribasic Calcium Phosphate), Iron (as Ferrous Fumarate), Phosphorous (as Tribasic Calcium Phosphate), Iodine (as Potassium Iodine), Magnesium (as Magnesium Oxide), Zinc (Zinc Oxide), Selenium (as Sodium Selenate), Copper (as Cupric Sulfate), Manganese (as Manganese Sulfate), Chromium (as Chromium Picolinate), Molybdenum (as Sodium Molybdate). Proprietary Blend: Beet Root Powder, Spinach Leaf Powder, Tomato Fruit Powder, Carrot Root Powder, Collards Greens Powder, Collards (Kale) Leaf Powder. Other Ingredients: Stearic Acid, Microcrystalline Cellulose, Dicalcium Phosphate, Croscarmellose Sodium, Magnesium Stearate, Silicon Dioxide, Pharmaceutical Glaze.
Metabolic cages
Whole-body fat and lean mass was noninvasively measured using LF90 time domain nuclear magnetic resonance scanner (Bruker Optics, Inc). Indirect calorimetric and energy balance parameters including food/water intake, energy expenditure, respiratory exchange ratio (RER), and physical activity were noninvasively assessed for 10d using metabolic cages (TSE Systems Inc., Chesterfield, MO, USA).
Quantification and Statistical Analysis
All experiments reported in Figs 1 to 4 were repeated at least three independent times and those in Figs 5 and 6 were repeated twice. All samples represent biological replicates. Unless otherwise specified in figure legends, all center values shown in graphs refer to the mean. For statistical significance of the differences between the means of two groups, we used two-tailed Student’s t-tests. Statistical significance of differences among multiple groups (≥3) was calculated by performing ANOVA multiple comparisons of the means for each group. No samples or animals were excluded from analysis, and sample size estimates were not used. Animals were randomly assigned to groups. Studies were not conducted blinded, with the exception of all histological analyses.
Supplementary Material
metabolic effects of FMD and short-term starvation (STS) on (A) body weights with lean- and fat-mass ratio prior to, after STS or FMD and 3 days after refeeding. (B) Water intake, food intake (kcal/day), Total movement and VCO2/VO2 before, during and after STS and (C) after FMD. (D) Levels of circulating insulin and ketone body (β-HB) in mice on FMD and post-FMD refeeding, comparing to that of mice under prolonged fasting (24, 36 and 60 hr). *p<0.05, **p<0.01 and ***p<0.005, t-test.
Effects of FMD in Leprdb/db mice. (A) Numbers of indicated cell type per islet, (B) Proliferation frequency of indicated cell type per islet, (C) Body weight and (D) Proliferation frequency and numbers and (E) example image of non-insulin/glucagon producing cells (non-α/b) and Pdx1+α cells. (f) Levels of circulating insulin during IPGTT. (G) illustration of pancreatic islet sampling. *p<0.05, ** p<0.01, ***p<0.005, one-way ANOVA
Effects of FMD cycles on STZ-treated mice. (A) body weight, one cycle of FMD (B) Numbers of indicated cell type per islet, (C) Proliferation frequency of indicated cell type per islet, (D) Proliferation frequency of α cells and number of Pdx1+α cells per islet and (E) Proliferation frequency and numbers of the non-insulin/glucagon producing cells (non-α/b) and (F) Levels of circulating cytokines. *p<0.05, ** p<0.01, ***p<0.005, one-way ANOVA.
Effects of FMD and post-FMD refeeding on non-diabetic wild-type mice. (A) Number and area of pancreatic islets per pancreas section. (B) Numbers of indicated cell type per islet. (C) Proportion and (D) number of Proliferation frequency of indicated cell type per islet. (E) Number of Pdx1+α transitional cells per islet,(F) Representative images of Pdx1+α transitional cells. (G) z-stack confocal microscopy images of Gluc+ Ins+ cells (H) Proliferation frequency and numbers of the non-insulin/glucagon producing cells (non-α/b) in wild-type mice without STZ treatments. *p<0.05, ** p<0.01, ***p<0.005, one-way ANOVA.
Effects of FMD on expression of developmental markers of β cell in adult mice. (A) Fold-regulation of genes significantly (*p<0.05) up- or down-regulated by FMD or RF1d comparing to AL. The p values are calculated based on a student t-test of the replicate 2^(-Delta Ct) values for each gene in the control group and treatment groups. (B) Immunostaining for proteins expression of lineage markers in pancreatic islets. (C) schematic time line and representative images of corn oil (vehicle control) treatments for Ngn3-lineage ablation experiments shown in Figure 5F.
(A) Gene expression and (B) insulin production of healthy pancreatic islets (HI) and T1DI treated with serum form subjects at indicated time points. ****p<0.0001, t-test.
Highlights.
Fasting mimicking diet induces prenatal-development gene expression in adult pancreas
FMD promotes Ngn3 expression to generate insulin-producing β cells
Cycles of FMD reverse β-cell failure and rescue mice from T1D and T2D
Inhibition of PKA or mTOR promote Ngn3-driven β-cell regeneration in human T1D islets
Acknowledgments
We thank Dr. Lora Barsky (USC Flow Cytometry Core Facility) for assistance in the FACS analysis, Dr. Michael Sheard (CHLA Flourescence Activated Cell Sorting Core) for assistance in the cytokines profiling, and Gerrardo Navarrete for preparing the diet. This study was funded in part by NIH/NIA grants AG20642, AG025135 and P01 AG034906 to V.D.L. V.D.L. has equity interest in L-Nutra, a company that develops medical food.
Footnotes
Author contribution
V.D.L conceptualized and supervised the study. C.W.C, V.V. and R.B. designed and preformed experiments, analyzed data, interpreted results and wrote manuscript, with inputs from M.W., S.K., P.C., J.B.S., L.P. and O.H.Y.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abad M, Mosteiro L, Pantoja C, Canamero M, Rayon T, Ors I, Grana O, Megias D, Dominguez O, Martinez D, et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature. 2013;502:340–345. doi: 10.1038/nature12586. [DOI] [PubMed] [Google Scholar]
- Arakawa K, Ishihara T, Oku A, Nawano M, Ueta K, Kitamura K, Matsumoto M, Saito A. Improved diabetic syndrome in C57BL/KsJ-db/db mice by oral administration of the Na(+)-glucose cotransporter inhibitor T-1095. British journal of pharmacology. 2001;132:578–586. doi: 10.1038/sj.bjp.0703829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnes L, Hill JT, Gross S, Magnuson MA, Sussel L. Ghrelin expression in the mouse pancreas defines a unique multipotent progenitor population. PloS one. 2012;7:e52026. doi: 10.1371/journal.pone.0052026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeyens L, Lemper M, Leuckx G, De Groef S, Bonfanti P, Stange G, Shemer R, Nord C, Scheel DW, Pan FC, et al. Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nature biotechnology. 2014;32:76–83. doi: 10.1038/nbt.2747. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Barnosky AR, Hoddy KK, Unterman TG, Varady KA. Intermittent fasting vs daily calorie restriction for type 2 diabetes prevention: a review of human findings. Translational research : the journal of laboratory and clinical medicine. 2014;164:302–311. doi: 10.1016/j.trsl.2014.05.013. [DOI] [PubMed] [Google Scholar]
- Blum B, Roose AN, Barrandon O, Maehr R, Arvanites AC, Davidow LS, Davis JC, Peterson QP, Rubin LL, Melton DA. Reversal of beta cell de-differentiation by a small molecule inhibitor of the TGFbeta pathway. eLife. 2014;3:e02809. doi: 10.7554/eLife.02809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandhorst S, Choi IY, Wei M, Cheng CW, Sedrakyan S, Navarrete G, Dubeau L, Yap LP, Park R, Vinciguerra M, et al. A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab. 2015;22:86–99. doi: 10.1016/j.cmet.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brereton MF, Iberl M, Shimomura K, Zhang Q, Adriaenssens AE, Proks P, Spiliotis II, Dace W, Mattis KK, Ramracheya R, et al. Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nature communications. 2014;5:4639. doi: 10.1038/ncomms5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CW, Adams GB, Perin L, Wei M, Zhou X, Lam BS, Da Sacco S, Mirisola M, Quinn DI, Dorff TB, et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell stem cell. 2014;14:810–823. doi: 10.1016/j.stem.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, Furuyama K, Thorel F, Gribble FM, Reimann F, et al. Diabetes recovery by age-dependent conversion of pancreatic delta-cells into insulin producers. Nature. 2014;514:503–507. doi: 10.1038/nature13633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(Suppl 2):S97–107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- Cohen DE, Melton D. Turning straw into gold: directing cell fate for regenerative medicine. Nature reviews Genetics. 2011;12:243–252. doi: 10.1038/nrg2938. [DOI] [PubMed] [Google Scholar]
- Cook JR, Matsumoto M, Banks AS, Kitamura T, Tsuchiya K, Accili D. A Mutant Allele Encoding DNA-Binding-Deficient Foxo1 Differentially Regulates Hepatic Glucose and Lipid Metabolism. Diabetes. 2015 doi: 10.2337/db14-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirice E, Kahraman S, Jiang W, El Ouaamari A, De Jesus DF, Teo AK, Hu J, Kawamori D, Gaglia JL, Mathis D, et al. Soluble factors secreted by T cells promote beta-cell proliferation. Diabetes. 2014;63:188–202. doi: 10.2337/db13-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor Y, Glaser B. beta-cell dedifferentiation and type 2 diabetes. The New England journal of medicine. 2013;368:572–573. doi: 10.1056/NEJMcibr1214034. [DOI] [PubMed] [Google Scholar]
- Fiorina P, Shapiro AM, Ricordi C, Secchi A. The clinical impact of islet transplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2008;8:1990–1997. doi: 10.1111/j.1600-6143.2008.02353.x. [DOI] [PubMed] [Google Scholar]
- Gomez DL, O’Driscoll M, Sheets TP, Hruban RH, Oberholzer J, McGarrigle JJ, Shamblott MJ. Neurogenin 3 Expressing Cells in the Human Exocrine Pancreas Have the Capacity for Endocrine Cell Fate. PloS one. 2015;10:e0133862. doi: 10.1371/journal.pone.0133862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunnet LG, Aikin R, Tonnesen MF, Paraskevas S, Blaabjerg L, Storling J, Rosenberg L, Billestrup N, Maysinger D, Mandrup-Poulsen T. Proinflammatory cytokines activate the intrinsic apoptotic pathway in beta-cells. Diabetes. 2009;58:1807–1815. doi: 10.2337/db08-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler RA, Hartil K, Vaitheesvaran B, Arrieta-Cruz I, Knight CM, Cook JR, Kammoun HL, Febbraio MA, Gutierrez-Juarez R, Kurland IJ, et al. Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nature communications. 2014;5:5190. doi: 10.1038/ncomms6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich C, Spagnoli FM, Berninger B. In vivo reprogramming for tissue repair. Nature cell biology. 2015;17:204–211. doi: 10.1038/ncb3108. [DOI] [PubMed] [Google Scholar]
- Hsu FL, Huang CF, Chen YW, Yen YP, Wu CT, Uang BJ, Yang RS, Liu SH. Antidiabetic effects of pterosin A, a small-molecular-weight natural product, on diabetic mouse models. Diabetes. 2013;62:628–638. doi: 10.2337/db12-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JD, Han Z, Otani K, Ye H, Zhang Y, Wu H, Horikawa Y, Misler S, Bell GI, Polonsky KS. RyR2 and calpain-10 delineate a novel apoptosis pathway in pancreatic islets. J Biol Chem. 2004;279:24794–24802. doi: 10.1074/jbc.M401216200. [DOI] [PubMed] [Google Scholar]
- Kim-Muller JY, Zhao S, Srivastava S, Mugabo Y, Noh HL, Kim YR, Madiraju SR, Ferrante AW, Skolnik EY, Prentki M, et al. Metabolic inflexibility impairs insulin secretion and results in MODY-like diabetes in triple FoxO-deficient mice. Cell Metab. 2014;20:593–602. doi: 10.1016/j.cmet.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, Young H, Richardson M, Smart NG, Cunningham J, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nature biotechnology. 2008;26:443–452. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- Laviano A, Rossi Fanelli F. Toxicity in chemotherapy--when less is more. The New England journal of medicine. 366:2319–2320. doi: 10.1056/NEJMcibr1202395. [DOI] [PubMed] [Google Scholar]
- Lebastchi J, Herold KC. Immunologic and metabolic biomarkers of beta-cell destruction in the diagnosis of type 1 diabetes. Cold Spring Harbor perspectives in medicine. 2012;2:a007708. doi: 10.1101/cshperspect.a007708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DS, Yuan YH, Tu HJ, Liang QL, Dai LJ. A protocol for islet isolation from mouse pancreas. Nature protocols. 2009;4:1649–1652. doi: 10.1038/nprot.2009.150. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature neuroscience. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehr R, Chen S, Snitow M, Ludwig T, Yagasaki L, Goland R, Leibel RL, Melton DA. Generation of pluripotent stem cells from patients with type 1 diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:15768–15773. doi: 10.1073/pnas.0906894106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews DR. Insulin resistance and beta-cell function--a clinical perspective. Diabetes, obesity & metabolism. 2001;3(Suppl 1):S28–33. [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- McKnight KD, Wang P, Kim SK. Deconstructing pancreas development to reconstruct human islets from pluripotent stem cells. Cell stem cell. 2010;6:300–308. doi: 10.1016/j.stem.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57:1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanesi A, Lee JW, Li Z, Da Sacco S, Villani V, Cervantes V, Perin L, Yu JS. beta-Cell regeneration mediated by human bone marrow mesenchymal stem cells. PloS one. 2012;7:e42177. doi: 10.1371/journal.pone.0042177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu J, Woods J, Zhou YP, Roy RS, Li Z, Zycband E, Feng Y, Zhu L, Li C, Howard AD, et al. Chronic inhibition of dipeptidyl peptidase-4 with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes. 2006;55:1695–1704. doi: 10.2337/db05-1602. [DOI] [PubMed] [Google Scholar]
- Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA. Generation of functional human pancreatic beta cells in vitro. Cell. 2014;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipeleers D, Keymeulen B, Chatenoud L, Hendrieckx C, Ling Z, Mathieu C, Roep B, Ysebaert D. A view on beta cell transplantation in diabetes. Ann N Y Acad Sci. 2002;958:69–76. doi: 10.1111/j.1749-6632.2002.tb02948.x. [DOI] [PubMed] [Google Scholar]
- Piran R, Lee SH, Li CR, Charbono A, Bradley LM, Levine F. Pharmacological induction of pancreatic islet cell transdifferentiation: relevance to type I diabetes. Cell death & disease. 2014;5:e1357. doi: 10.1038/cddis.2014.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch A. An update on cytokines in the pathogenesis of insulin-dependent diabetes mellitus. Diabetes/metabolism reviews. 1998;14:129–151. doi: 10.1002/(sici)1099-0895(199806)14:2<129::aid-dmr208>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Raffaghello L, Lee C, Safdie FM, Wei M, Madia F, Bianchi G, Longo VD. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:8215–8220. doi: 10.1073/pnas.0708100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneddon JB, Borowiak M, Melton DA. Self-renewal of embryonic-stem-cell-derived progenitors by organ-matched mesenchyme. Nature. 2012;491:765–768. doi: 10.1038/nature11463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanger BZ. HNF4A and diabetes: injury before insult? Diabetes. 2008;57:1461–1462. doi: 10.2337/db08-0454. [DOI] [PubMed] [Google Scholar]
- Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. 2012;150:1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talchai SC, Accili D. Legacy Effect Of Foxo1 In Pancreatic Endocrine Progenitors On Adult beta-cell Mass And Function. Diabetes. 2015 doi: 10.2337/db14-1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- Tonne JM, Sakuma T, Deeds MC, Munoz-Gomez M, Barry MA, Kudva YC, Ikeda Y. Global gene expression profiling of pancreatic islets in mice during streptozotocin-induced beta-cell damage and pancreatic Glp-1 gene therapy. Dis Model Mech. 6:1236–1245. doi: 10.1242/dmm.012591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Casteele M, Leuckx G, Baeyens L, Cai Y, Yuchi Y, Coppens V, De Groef S, Eriksson M, Svensson C, Ahlgren U, et al. Neurogenin 3+ cells contribute to beta-cell neogenesis and proliferation in injured adult mouse pancreas. Cell death & disease. 2013;4:e523. doi: 10.1038/cddis.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani V, Milanesi A, Sedrakyan S, Da Sacco S, Angelow S, Conconi MT, Di Liddo R, De Filippo R, Perin L. Amniotic fluid stem cells prevent beta-cell injury. Cytotherapy. 2014;16:41–55. doi: 10.1016/j.jcyt.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Jensen JN, Seymour PA, Hsu W, Dor Y, Sander M, Magnuson MA, Serup P, Gu G. Sustained Neurog3 expression in hormone-expressing islet cells is required for endocrine maturation and function. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9715–9720. doi: 10.1073/pnas.0904247106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic beta Cell Dedifferentiation in Diabetes and Redifferentiation following Insulin Therapy. Cell Metab. 19:872–882. doi: 10.1016/j.cmet.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic beta cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014;19:872–882. doi: 10.1016/j.cmet.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg N, Ouziel-Yahalom L, Knoller S, Efrat S, Dor Y. Lineage tracing evidence for in vitro dedifferentiation but rare proliferation of mouse pancreatic beta-cells. Diabetes. 2007;56:1299–1304. doi: 10.2337/db06-1654. [DOI] [PubMed] [Google Scholar]
- Weir GC, Aguayo-Mazzucato C, Bonner-Weir S. beta-cell dedifferentiation in diabetes is important, but what is it? Islets. 2013;5:233–237. doi: 10.4161/isl.27494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KK, Huan Y. Streptozotocin-induced diabetic models in mice and rats. Curr Protoc Pharmacol. 2008;Chapter 5(Unit 5):47. doi: 10.1002/0471141755.ph0547s40. [DOI] [PubMed] [Google Scholar]
- Xiao X, Chen Z, Shiota C, Prasadan K, Guo P, El-Gohary Y, Paredes J, Welsh C, Wiersch J, Gittes GK. No evidence for beta cell neogenesis in murine adult pancreas. The Journal of clinical investigation. 2013;123:2207–2217. doi: 10.1172/JCI66323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Du Y, Deng H. Direct lineage reprogramming: strategies, mechanisms, and applications. Cell stem cell. 2015;16:119–134. doi: 10.1016/j.stem.2015.01.013. [DOI] [PubMed] [Google Scholar]
- Xu X, D’Hoker J, Stange G, Bonne S, De Leu N, Xiao X, Van de Casteele M, Mellitzer G, Ling Z, Pipeleers D, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132:197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- Yi P, Park JS, Melton DA. Betatrophin: a hormone that controls pancreatic beta cell proliferation. Cell. 2013;153:747–758. doi: 10.1016/j.cell.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yilmaz OH, Katajisto P, Lamming DW, Gultekin Y, Bauer-Rowe KE, Sengupta S, Birsoy K, Dursun A, Yilmaz VO, Selig M, et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature. 2012;486:490–495. doi: 10.1038/nature11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin D, Tao J, Lee DD, Shen J, Hara M, Lopez J, Kuznetsov A, Philipson LH, Chong AS. Recovery of islet beta-cell function in streptozotocin-induced diabetic mice: an indirect role for the spleen. Diabetes. 2006;55:3256–3263. doi: 10.2337/db05-1275. [DOI] [PubMed] [Google Scholar]
- Zhernakova A, Alizadeh BZ, Eerligh P, Hanifi-Moghaddam P, Schloot NC, Diosdado B, Wijmenga C, Roep BO, Koeleman BP. Genetic variants of RANTES are associated with serum RANTES level and protection for type 1 diabetes. Genes and immunity. 2006;7:544–549. doi: 10.1038/sj.gene.6364326. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
metabolic effects of FMD and short-term starvation (STS) on (A) body weights with lean- and fat-mass ratio prior to, after STS or FMD and 3 days after refeeding. (B) Water intake, food intake (kcal/day), Total movement and VCO2/VO2 before, during and after STS and (C) after FMD. (D) Levels of circulating insulin and ketone body (β-HB) in mice on FMD and post-FMD refeeding, comparing to that of mice under prolonged fasting (24, 36 and 60 hr). *p<0.05, **p<0.01 and ***p<0.005, t-test.
Effects of FMD in Leprdb/db mice. (A) Numbers of indicated cell type per islet, (B) Proliferation frequency of indicated cell type per islet, (C) Body weight and (D) Proliferation frequency and numbers and (E) example image of non-insulin/glucagon producing cells (non-α/b) and Pdx1+α cells. (f) Levels of circulating insulin during IPGTT. (G) illustration of pancreatic islet sampling. *p<0.05, ** p<0.01, ***p<0.005, one-way ANOVA
Effects of FMD cycles on STZ-treated mice. (A) body weight, one cycle of FMD (B) Numbers of indicated cell type per islet, (C) Proliferation frequency of indicated cell type per islet, (D) Proliferation frequency of α cells and number of Pdx1+α cells per islet and (E) Proliferation frequency and numbers of the non-insulin/glucagon producing cells (non-α/b) and (F) Levels of circulating cytokines. *p<0.05, ** p<0.01, ***p<0.005, one-way ANOVA.
Effects of FMD and post-FMD refeeding on non-diabetic wild-type mice. (A) Number and area of pancreatic islets per pancreas section. (B) Numbers of indicated cell type per islet. (C) Proportion and (D) number of Proliferation frequency of indicated cell type per islet. (E) Number of Pdx1+α transitional cells per islet,(F) Representative images of Pdx1+α transitional cells. (G) z-stack confocal microscopy images of Gluc+ Ins+ cells (H) Proliferation frequency and numbers of the non-insulin/glucagon producing cells (non-α/b) in wild-type mice without STZ treatments. *p<0.05, ** p<0.01, ***p<0.005, one-way ANOVA.
Effects of FMD on expression of developmental markers of β cell in adult mice. (A) Fold-regulation of genes significantly (*p<0.05) up- or down-regulated by FMD or RF1d comparing to AL. The p values are calculated based on a student t-test of the replicate 2^(-Delta Ct) values for each gene in the control group and treatment groups. (B) Immunostaining for proteins expression of lineage markers in pancreatic islets. (C) schematic time line and representative images of corn oil (vehicle control) treatments for Ngn3-lineage ablation experiments shown in Figure 5F.
(A) Gene expression and (B) insulin production of healthy pancreatic islets (HI) and T1DI treated with serum form subjects at indicated time points. ****p<0.0001, t-test.