

Abstract
The PAX3-FOXO1 fusion gene is generated by a 2;13 chromosomal translocation and is a characteristic feature of an aggressive subset of rhabdomyosarcoma (RMS). To dissect the mechanism of oncogene action during RMS tumourigenesis and progression, doxycycline-inducible PAX3-FOXO1 and constitutive MYCN expression constructs were introduced into immortalised human myoblasts. Though myoblasts expressing PAX3-FOXO1 or MYCN alone were not transformed in focus formation assays, combined PAX3-FOXO1 and MYCN expression resulted in transformation. Following intramuscular injection into immunodeficient mice, myoblasts expressing PAX3-FOXO1 and MYCN formed rapidly growing RMS tumours whereas myoblasts expressing only PAX3-FOXO1 formed tumours after a longer latency period. Doxycycline withdrawal in myoblasts expressing inducible PAX3-FOXO1 and constitutive MYCN following tumour formation in vivo or focus formation in vitro resulted in tumour regression or smaller foci associated with myogenic differentiation and cell death. Following regression, most tumours recurred in the absence of doxycycline. Analysis of recurrent tumours revealed a subset without PAX3-FOXO1 expression, and cell lines derived from these recurrent tumours demonstrated transformation in the absence of doxycycline. The doxycycline-independent oncogenicity in these recurrent tumour-derived lines persisted even after PAX3-FOXO1 was inactivated by a CRISPR-Cas9 editing strategy. Whereas cell lines derived from primary tumours were dependent on PAX3-FOXO1 and differentiated following doxycycline withdrawal, recurrent tumour-derived cells without PAX3-FOXO1 expression did not differentiate under these conditions. These findings indicate that PAX3-FOXO1 collaborates with MYCN during early RMS tumourigenesis to dysregulate proliferation and inhibit myogenic differentiation and cell death. Although most cells in the primary tumours are dependent on PAX3-FOXO1, recurrent tumours can develop by a PAX3-FOXO1-independent mechanism, in which rare cells are postulated to acquire secondary transforming events that were activated or selected by initial PAX3-FOXO1 expression.
Keywords: rhabdomyosarcoma, PAX3-FOXO1, myoblast, differentiation, recurrence
Introduction
Rhabdomyosarcoma (RMS) is a common paediatric soft tissue tumour that is associated with the skeletal muscle lineage. RMS comprises two major histopathological subtypes: alveolar (ARMS) and embryonal. ARMS is the more aggressive subtype and is characterized by a t(2;13)(q35; q14) or t(1;13)(p36; q14) chromosomal translocation, which generates a PAX3-FOXO1 or PAX7-FOXO1 fusion gene [1–4]. These fusion genes combine functional domains from the corresponding wild-type proteins to produce novel chimaeric transcription factors with increased activation of PAX3/PAX7 target genes and associated oncogenic function [5–8].
Earlier studies identified myoblasts as a potential cell of origin for RMS. Ectopic expression of PAX3-FOXO1 in primary human skeletal muscle myoblasts confers a proliferative advantage by bypassing cellular senescence; this bypass is associated with loss of CDKN2A expression [9]. PAX3-FOXO1 alone is not sufficient for tumourigenesis but requires additional changes such as increased MYCN and TERT expression [10,11]. In our previous study of immortalized human myoblasts, PAX3-FOXO1 alone did not transform myoblasts in culture and induced slow-growing tumours in immunocompromised mice. In contrast, the combination of PAX3-FOXO1 and MYCN transformed the myoblasts and induced rapid tumourigenesis [12].
An important limitation of studies with constitutive PAX3-FOXO1 expression is that expression cannot be manipulated to analyse the temporal and quantitative requirements for PAX3-FOXO1. To overcome this limitation, we developed a doxycycline-inducible PAX3-FOXO1 expression system in immortalised human myoblasts. Using this system, expression of PAX3-FOXO1 and its downstream targets can be regulated by doxycycline addition or withdrawal to study the requirements for PAX3-FOXO1. We provide evidence that PAX3-FOXO1 in combination with MYCN contributes to myoblast transformation and tumour formation by stimulating proliferation and inhibiting myogenic differentiation and apoptosis. After tumour formation, cessation of PAX3-FOXO1 expression results in tumour regression. Following regression, recurrent tumours eventually form in which PAX3-FOXO1 is not required to maintain the tumourigenic phenotype. These studies further our understanding of PAX3-FOXO1 function in ARMS tumourigenesis and recurrence, and provide an important model to investigate the effect of therapies directed against the fusion protein.
Materials and Methods
Cell culture
An immortalized human Duchenne muscular dystrophy myoblast cell line (Dbt) and human RMS cell lines were cultured as described [12]. Verification of cell line identity was performed by short tandem repeat genotyping analysis using the AmpFLSTR profiler plus PCR amplification kit (Applied Biosystems).
Lentiviral transduction
The PAX3-FOXO1 fusion cDNA was subcloned into the lentiviral doxycycline-inducible pINDUCER10 vector (provided by Dr. J. Luo). A single-guide RNA (sgRNA) targeting the 5′ PAX3 region was designed (Table S1) and cloned into the lentiCRISPR v2 plasmid (provided by Dr. J. Luo) [13–15]. Oligonucleotides were purchased from Integrated DNA Technologies. Lentiviral particles were prepared in HEK293T cells with the HIV-based pPACK-H1 Packaging Plasmid Mix (System Biosciences).
Establishment of stable cell lines
Dbt cells were transduced with the empty retroviral pK1 vector or a MYCN-expressing construct and selected in hygromycin (120 μg/ml) as described previously [12]. These cells were further transduced with pINDUCER10 containing firefly luciferase or PAX3-FOXO1 and selected with puromycin (1.5 μg/ml) to prepare Dbt-indP3F and Dbt-MYCN/indP3F parental cell populations, respectively (“parental” is specifically used to refer to these starting transduced populations). Subclones were isolated from parental populations by limiting dilution. Engineered Dbt cells were incubated with doxycycline (Sigma-Aldrich) to induce PAX3-FOXO1 expression. In some cases, engineered Dbt cells were further transduced with a GFP expression construct (Addgene plasmid # 17445) [16].
Expression assays
Western blotting of cell and tumour lysates was performed as described previously [17] using antibodies against FOXO1 (1:1000, # 2880; Cell Signaling Technology) and GAPDH (1:1000, #sc-25778; Santa Cruz Biotechnology). Total RNA was extracted and quantitative reverse transcription-PCR (qRT-PCR) was performed as described previously [17]. Taqman gene expression assays (Life Technologies) were used to quantify PAX3-FOXO1 (Hs03024825), MYCN (Hs00232074), and GAPDH (Hs02758991) by standard cycling conditions. To specifically measure endogenous MYCN expression, a qRT-PCR assay was developed for the MYCN 3′ UTR region (Supplementary Table S1). The ΔΔCt method was used to assess the gene expression fold change.
Oncogenicity assays
Oncogenic transformation was measured by focus formation assays as described previously [18]. Engineered Dbt myoblasts (106 cells) were injected into the left gastrocnemius muscle of 4-week-old female NOD-SCID mice (Frederick National Laboratory for Cancer Research). Mice were fed either a regular or doxycycline-containing (625 mg/kg) diet (Harlan laboratories) beginning 3 days prior to injection; the feed was replenished twice per week. The mice were observed for tumour formation for 90 days. Tumour volume was calculated according to the formula (length × width2)/2), where length represents the largest tumour diameter and width represents the perpendicular tumour diameter. Cell lines were derived from xenograft tumours as described previously [12] and injected into mice as described above. All experimental procedures were approved by the NIH Animal Care and Use Committee.
Immunohistochemistry (IHC)
Tissue sections from tumour specimens were immunostained as described previously [19]. Primary rabbit and mouse antibodies were applied at the following dilutions: PAX3-FOXO1 fusion-specific antibody [20] at 1:20, Ki67 (#M7240; Dako Cytomation) at 1:300, cleaved- caspase-3 (#9661; Cell Signaling Technology) at 1:750 and Myf4/myogenin (#NCL-L-Myf-4; Leica Biosystems) at 1:80. Concurrent positive and negative controls were stained.
Results
Doxycycline-inducible PAX3-FOXO1 expression system
To investigate the role of PAX3-FOXO1 in RMS tumourigenesis, we transduced immortalized human myoblasts with doxycycline-inducible PAX3-FOXO1 and constitutive MYCN expression constructs to prepare mixed transduced cell populations. In a time course experiment, PAX3-FOXO1 mRNA and protein were expressed by 24 hours following doxycycline treatment in Dbt-indP3F cells (containing inducible PAX3-FOXO1 alone) and Dbt-MYCN/indP3F cells (containing constitutive MYCN and inducible PAX3-FOXO1) (Figure 1A). Doxycycline treatment resulted in a dose-dependent increase in expression of PAX3-FOXO1 (Figures 1B, S1A) and FGFR4, a downstream target of PAX3-FOXO1 (Figure S1B). In contrast, there was constitutive high MYCN expression in Dbt-MYCN/indP3F cells and no detectable MYCN expression in Dbt-indP3F cells (Figure S1C). Using a qRT-PCR assay specific for the MYCN 3′ UTR, there was no evidence of endogenous MYCN expression induced by PAX3-FOXO1 in these cells (Figure S1D).
Figure 1. Generation of doxycycline-inducible PAX3-FOXO1 expression system in human myoblasts.
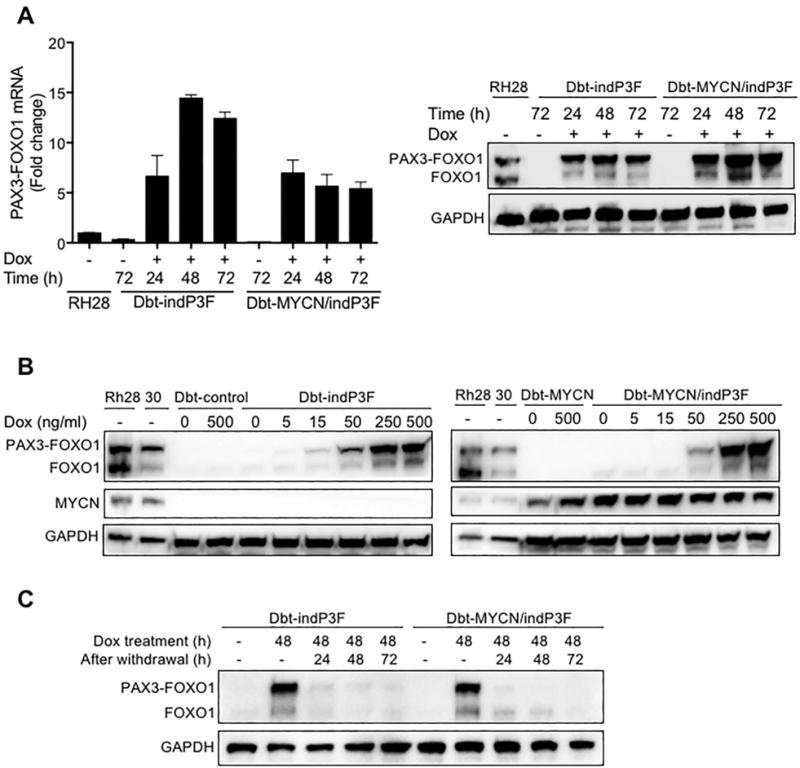
(A) qRT-PCR (left) and Western blot (right) analyses of PAX3-FOXO1 mRNA and protein expression in Dbt-indP3F and Dbt-MYCN/indP3F cells grown in the presence of doxycycline (500 ng/ml) for 24, 48 and 72 h. In qRT-PCR studies, the PAX3-FOXO1 mRNA level was normalized for GAPDH expression, and expressed as mean ± standard deviation of triplicate technical replicate measurements. GAPDH was used as a loading control for Western blot analyses of whole cell lysates. (B) Western blot analysis of doxycycline dose-dependent expression of PAX3-FOXO1 protein and its effect on MYCN protein expression. Dbt-indP3F (left) and Dbt-MYCN/indP3F (right) cells were treated with indicated concentrations of doxycycline for 72 h. RH28 and RH30 ARMS cells were used as positive controls for PAX3-FOXO1 and MYCN expression. (C) Western blot analysis of PAX3-FOXO1 protein expression in Dbt-indP3F and Dbt-MYCN/indP3F cells treated with doxycycline (500 ng/ml) for 48 h and then incubated for 24, 48, or 72 h in fresh medium without doxycycline.
We next investigated the effects of doxycycline withdrawal to determine the reversibility of this inducible system. Engineered Dbt cells were cultured for 48 hours in the presence of doxycycline and then replenished with fresh medium lacking doxycycline. PAX3-FOXO1 mRNA and protein expression decreased to almost undetectable levels as early as 24 hours following doxycycline removal (Figure 1C, S1E). Therefore, this doxycycline-inducible PAX3-FOXO1 system in human myoblasts provides reversible, quantitative and reproducible control of PAX3-FOXO1 expression to study the oncogenic effects of this fusion protein.
Effects of PAX3-FOXO1 on oncogenic transformation
To explore the phenotypic role of PAX3-FOXO1 alone or in combination with MYCN, we assessed oncogenic transformation by focus formation assays. Dbt cells containing the empty vector (Dbt-control), MYCN alone (Dbt-MYCN) or PAX3-FOXO1 alone did not form foci in the absence or presence of doxycycline (Figure 2A). In contrast, Dbt-MYCN/indP3F cells were transformed in the presence of doxycycline. These results corroborated previous findings that multiple genetic changes are required to transform human cells [10,11].
Figure 2. Effect of PAX3-FOXO1 and MYCN on oncogenic transformation.
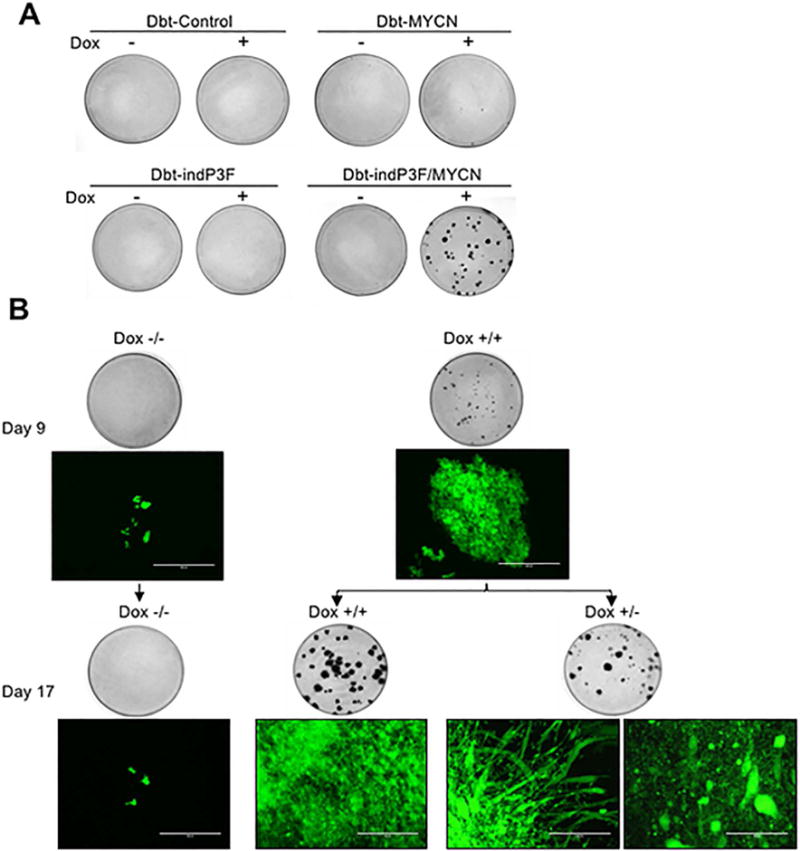
(A) Focus formation was assayed in engineered Dbt myoblasts (103 cells) co-cultured with NIH 3T3 fibroblasts (106 cells) in 10 cm dishes in a medium consisting of 50% F-10 and 50% DMEM. The medium and doxycycline were replaced every 48–72 h, plates were fixed with methanol and stained with Giemsa on day 14. (B) The effect of PAX3-FOXO1 depletion in Dbt-MYCN/indP3F cells (also transduced with GFP) was evaluated in the focus formation assay by withdrawing doxycycline at day 9 from one group of plates (+/−) while another group was continuously treated with doxycycline (+/+). Representative images are shown of fluorescent microscopy on day 9 and day 17 as well as whole plates fixed and stained on day 9 and 17. Scale bar = 400 μm.
We then analysed the requirement for continued PAX3-FOXO1 expression in transformed foci arising in the focus formation assay. We initially performed the assay with Dbt-MYCN/indP3F cells in the presence of doxycycline as described above. After small foci appeared at day 9, doxycycline treatment was discontinued in half of the plates. In plates with continued doxycycline treatment, foci continued to grow during the following 8 days (Figure 2B). Microscopy revealed that these large foci contained tightly packed small undifferentiated cells. In contrast, in plates in which doxycycline treatment was discontinued for 8 days, foci were smaller and showed prominent myotube formation consistent with myogenic differentiation. In other areas, there was bleb formation and cell swelling indicative of cell injury and death. These results indicate that PAX3-FOXO1 inhibits myogenic differentiation and cell death, and maintains cells in the proliferative state to exert transforming effects.
Effect of PAX3-FOXO1 on tumour formation
To assess the oncogenic role of PAX3-FOXO1 in vivo and determine whether MYCN modifies this phenotype, we performed intramuscular injections of the mixed Dbt-control, Dbt-MYCN, Dbt-indP3F and Dbt-MYCN/indP3F parental cells in NOD-SCID mice fed a standard or doxycycline-supplemented diet. We observed that only Dbt-indP3F and Dbt-MYCN/indP3F cells formed tumours during the 90-day observation period when PAX3-FOXO1 expression was induced with doxycycline (Table S2). The combination of constitutive MYCN and inducible PAX3-FOXO1 expression resulted in rapid tumour formation (4–5 weeks) in all mice (Figure 3A). In contrast, PAX3-FOXO1 expression alone caused tumours after a much longer latency period (9–10 weeks). RT-PCR, Western and IHC assays confirmed that doxycycline induced high PAX3-FOXO1 expression in tumour cells (Figures 3B, 3C, S2A, S2B). The IHC assay identified a heterogeneous PAX3-FOXO1 expression pattern, which may be due to cell cycle differences, microenvironmental effects or clonality heterogeneity. In addition, Western and qRT-PCR studies detected expression of endogenous MYCN mRNA and protein in these tumours (Figures 3C, S2A) in contrast to the findings described above in parental cell lines.
Figure 3. Tumourigenic potential of Dbt cells expressing inducible PAX3-FOXO1 with or without constitutive MYCN.
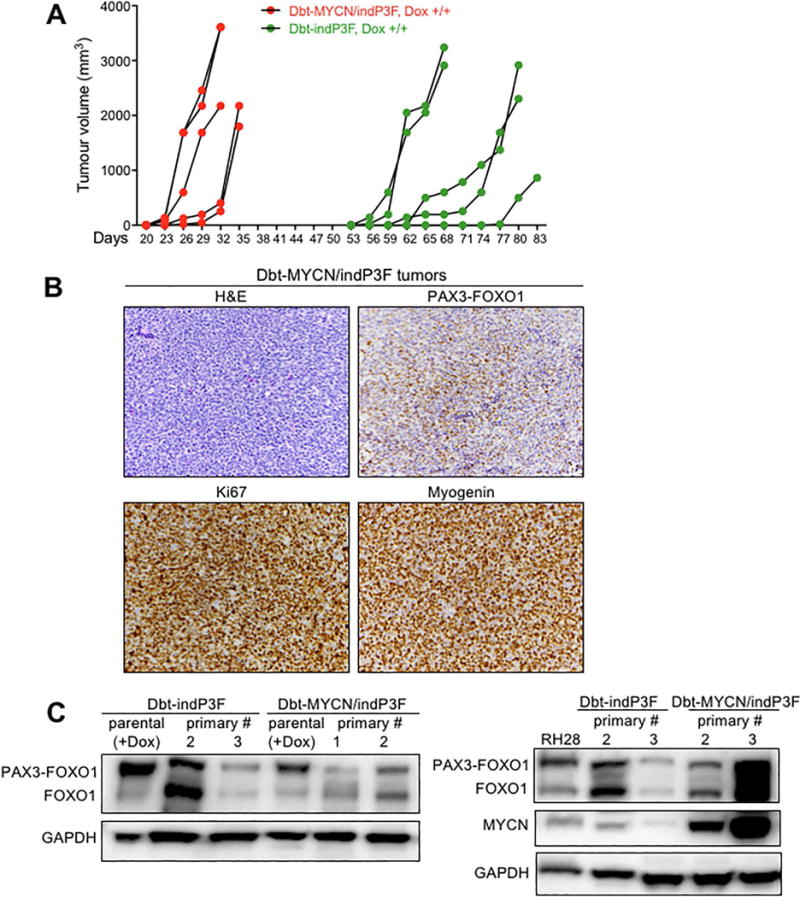
(A) Tumour growth curves in NOD-SCID mice (n=5/group) injected with Dbt-indP3F and Dbt-MYCN/indP3F cells and fed doxycycline-containing diet. Engineered Dbt cells (106) were suspended in 50 μl Hank’s balanced salt solution (HBSS) and injected into the left hind leg of mice. (B) Histopathological (H&E) and IHC (PAX3-FOXO1, Ki67, Myogenin) analyses of tumours from mice injected with Dbt-MYCN/indP3F myoblasts and fed doxycycline-containing diet (original magnification, X200). (C) Western blot analyses of PAX3-FOXO1 and MYCN protein expression in primary tumours formed from Dbt-indP3F and Dbt-MYCN/indP3F cells. In contrast to Dbt-MYCN/indP3F tumours, which express exogenous MYCN, the Dbt-indP3F tumours only express endogenous MYCN.
Microscopic examination of H&E stained slides revealed that tumours derived from Dbt-indP3F and Dbt-MYCN/indP3F myoblasts showed a similar histologic pattern resembling human ARMS (Figures 3B, S2B). In particular, the tumours consisted of a dense and often sheet-like population of small to medium round cells. Tumour giant cells were present at varying numbers and were particularly frequent in tumours formed from Dbt-indP3F cells (Figure S2B).
To further characterise these tumours, IHC was performed to detect proliferative and myogenic markers (Figures 3B, S2B). Staining for Ki67 demonstrated strong and diffuse reactivity, confirming the presence of a high proliferative rate in tumours cells. Most tumour cells also stained for the myogenic marker myogenin, consistent with the diagnosis of RMS. These IHC findings were similar in tumours expressing PAX3-FOXO1 with or without MYCN.
Tumour regression following PAX3-FOXO1 depletion
We next investigated whether continuous PAX3-FOXO1 expression was required in later stages of tumourigenesis by downregulating PAX3-FOXO1 expression in these tumours. In particular, mice were switched to a standard diet without doxycycline supplementation after palpable tumours formed (Figures 4A, S3A, S3B). Shortly after discontinuing doxycycline, all tumours formed from Dbt-indP3F or Dbt-MYCN/indP3F cells regressed, became undetectable within a week, and remained in this state during the next 3 weeks of observation.
Figure 4. Effect of PAX3-FOXO1 depletion on tumour growth.
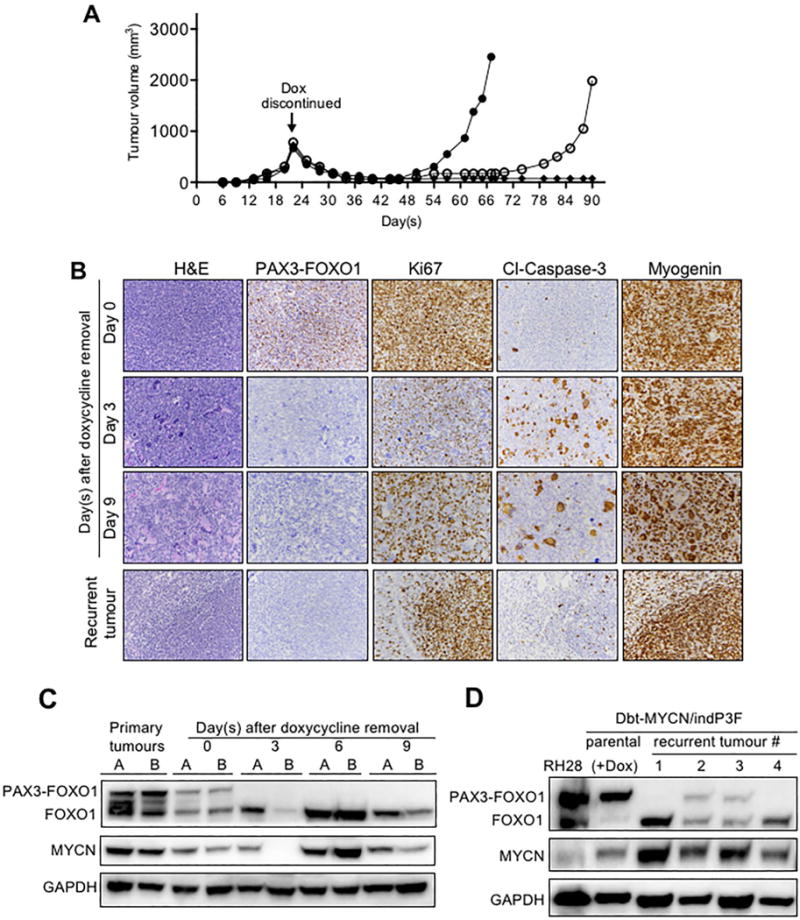
(A) Tumour regression and recurrence in Dbt-MYCN/indP3F-injected mice after doxycycline withdrawal. The doxycycline-containing diet was replaced with regular base diet when tumours were palpable around day 22, and three injected mice from one representative experiment are shown (by open circles, closed circles and diamonds). (B) Histopathological (H&E) and IHC (PAX3-FOXO1, Ki67, cleaved-caspase-3, and myogenin) analyses of tumours from mice injected with Dbt-MYCN/indP3F cells and harvested on 0, 3, and 9 days after doxycycline removal (original magnification, X200). A representative fully developed recurrent tumour is also shown; this specific tumour was harvested 42 days after doxycycline withdrawal, which corresponds to 64 days after injection. (C) Western blot analyses of PAX3-FOXO1 and MYCN protein expression in tumours from mice injected with Dbt-MYCN/indP3F cells and harvested 0, 3, 6, and 9 days after doxycycline withdrawal. Two primary tumours harvested 28 days after mice were injected with Dbt-MYCN/indP3F cells and fed a continuous doxycycline-supplemented diet were used as positive controls. (D) Western blot analysis of PAX3-FOXO1 and MYCN protein expression in recurrent tumours forming after doxycycline withdrawal in mice injected with Dbt-MYCN/indP3F cells. RH28 ARMS cells and Dbt-MYCN/indP3F parental cells cultured in the presence of doxycycline (500 ng/ml) were used as positive controls. GAPDH was used as a loading control.
The events occurring during tumour regression were examined in a time-course experiment in which tumours were harvested 0, 3, 6 and 9 days after doxycycline withdrawal. RT-PCR, Western and IHC assays revealed that PAX3-FOXO1 mRNA and protein expression decreased to near undetectable levels by 3 days after doxycycline withdrawal and remained at this level through day 9 (Figure 4B, 4C, S3C, S3D). This observation confirms that high PAX3-FOXO1 expression is crucial to maintain the tumourigenic phenotype.
Microscopic examination of the regressing lesions further elucidates pathways involved in maintaining the tumourigenic phenotype. By 3 days following doxycycline withdrawal, cell proliferation, as assessed by Ki67 expression, was almost completely shut down (Figures 4B, S3D). In addition, there was widespread formation of multinucleated giant cells expressing high myogenin levels, indicative of cell fusion and myogenic differentiation. In addition, many cells showed hyperchromasia and cleaved-caspase-3 expression, consistent with superimposed cell death. Therefore, as found in our in vitro assays, PAX3-FOXO1 is needed in vivo to stimulate proliferation and inhibit differentiation and cell death of tumour cells. By 9 days after doxycycline withdrawal, the giant cells were very prominent, both in number and size, consistent with continued differentiation. Expression of cleaved-caspase-3 decreased and hyperchromasia was no longer detectable, indicating that a wave of cell death was ending. Furthermore, there was increased Ki67 expression in mononuclear tumour cells, indicating restored proliferation of cells that did not undergo differentiation or cell death. These findings suggest that a subpopulation of proliferative cells is able to recover after loss of PAX3-FOXO1 expression.
Recurrence of myoblast-derived tumours
Although regressed tumours from Dbt-MYCN/indP3F myoblasts were undetectable for 3 weeks following doxycycline withdrawal, tumour cells eventually grew in the absence of doxycycline and formed recurrent tumours (Figure 4A; this recurrence phenomenon was not studied in tumours derived from Dbt-indP3F cells because of the late occurrence of the primary tumours). Microscopic analysis showed that the recurrent tumours consisted of one or more prominent nodules or multiple smaller foci containing densely packed small round cells (Figures 4B, S3D). Surrounding these nodules and foci, there were extensively differentiated areas consisting of numerous giant cells and myotubes. The proliferative status of the nodules and foci was demonstrated by high Ki67 levels, which contrasted with the relatively low expression in surrounding differentiated cells. Cells in the proliferating areas also stained with myogenin, confirming the myogenic nature of the recurrences. Myogenin was also expressed in differentiated areas but appeared less prominent.
We next assessed PAX3-FOXO1 expression in these recurrent tumours. Sixty percent of recurrences showed no detectable PAX3-FOXO1 expression by IHC and Western blotting (Figures 4B, 4D, S3D), consistent with a PAX3-FOXO1-independent recurrence mechanism. In another subset of recurrent tumours, PAX3-FOXO1 was expressed in the absence of doxycycline (Figure 4D); these latter changes are considered experimental artefacts that uncouple PAX3-FOXO1 from the doxycycline-inducible system.
Characterisation of tumour-derived cell lines
Cell lines were generated from primary and recurrent tumours arising in these experiments. Western blotting showed that all examined primary and recurrent tumour-derived cell lines only expressed PAX3-FOXO1 protein in the presence of doxycycline except for one recurrent tumour-derived line that expressed PAX3-FOXO1 with or without doxycycline (Figure 5A). The expression findings in the tumour-derived lines are in agreement with findings in the corresponding tumours, except for most recurrent tumours expressing PAX3-FOXO1 in the absence of doxycycline. The discordance in PAX3-FOXO1 expression without doxycycline between these recurrent tumours and associated cell lines suggests that the mechanism for doxycycline-independent PAX3-FOXO1 expression in these tumours reflects environmental specificity or cell line-independent heterogeneity. MYCN expression in these tumour-derived lines resembled the corresponding parental lines. In particular, the increased endogenous MYCN expression found in tumours was not maintained in the tumour-derived lines (Figure S4A), suggesting that MYCN is only induced in vivo by PAX3-FOXO1 in this experimental system.
Figure 5. Characterization of primary and recurrent tumour-derived cell lines.
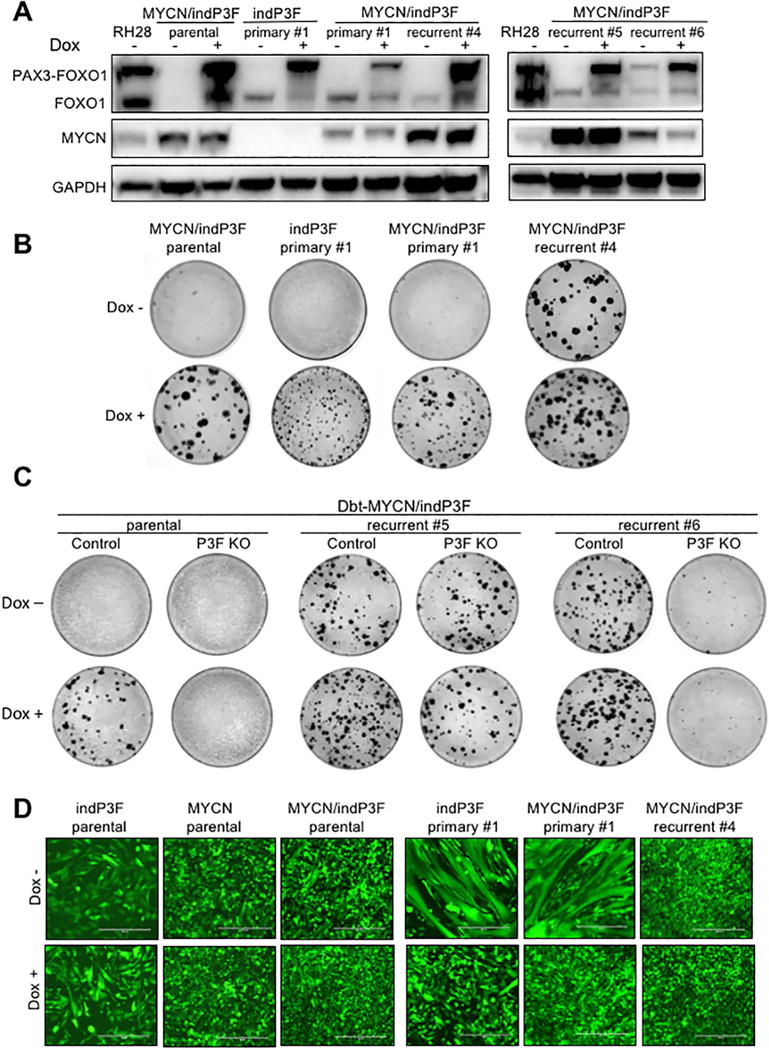
(A) Western blot analysis of PAX3-FOXO1 and MYCN in parental and representative primary (Dbt-indP3F and Dbt-MYCN/indP3F) and recurrent (Dbt-MYCN/indP3F) tumour-derived cell lines. All cells were grown in standard culture medium with and without doxycycline (500 ng/ml) for 72 h. (B) Focus formation assay of parental and representative tumour-derived cell lines in the presence or absence of doxycycline. Plates were fixed with methanol and stained with Giemsa on day 14. (C) Focus formation assay of empty lentiCRISPR (Control) or 5′ PAX3 sgRNA-expressing lentiCRISPR (P3F KO) transduced cells in the presence or absence of doxycycline. (D) Spontaneous myogenic differentiation in parental and representative tumour-derived cell lines (transduced with GFP) in the presence or absence of doxycycline. Cell lines (3×105/6 cm dish) were cultured in medium consisting of 50% F-10 and 50% DMEM. Medium and doxycycline (500 ng/ml) were replenished every 2–3 days and incubated for up to 6 days. Scale bar = 400 μm.
The transformation potential of these tumour-derived cell lines was assessed by focus formation assays. All primary tumour-derived lines were transformed in the presence of PAX3-FOXO1 induction (Figure 5B). Of note, tumour-derived cells expressing PAX3-FOXO1 alone formed foci whereas the parental Dbt-indP3F myoblasts did not. This discordance may be explained by in vivo selection of transformed clones with additional molecular changes during tumour formation and/or changes caused by the tumour microenvironment. All lines derived from recurrent tumours were also transformed in the presence of doxycycline, indicating that the fusion protein is still functional when induced. In addition, most lines derived from recurrent tumours that did not express PAX3-FOXO1 formed foci in the absence of PAX3-FOXO1 induction, supporting the premise that many recurrent tumours develop a PAX3-FOXO1-independent mechanism of transformation and tumourigenesis.
To provide further evidence that many recurrent tumours do not require PAX3-FOXO1 expression, we used the CRISPR/Cas9 system to knock down PAX3-FOXO1 in Dbt-MYCN/indP3F parental cells and representative recurrent tumour-derived cells. Following transduction of a CRISPR construct targeting the 5′ PAX3 region, PAX3-FOXO1 protein expression was knocked down to nearly undetectable levels in all these lines in the presence of doxycycline (Figure S4B). In oncogenicity assays, parental cells transduced with the 5′ PAX3-specific CRISPR construct failed to form foci when treated with doxycycline whereas control transduced cells formed numerous foci as expected (Figure 5C). In contrast, the PAX3-specific CRISPR did not affect the ability of the postulated PAX3-FOXO1-independent recurrent tumour-derived lines to form foci in the absence or presence of doxycycline, confirming that the oncogenicity of these cells does not require fusion protein expression. However, in a recurrent tumour-derived line expressing PAX3-FOXO1 in a doxycycline-independent fashion, transforming activity (with or without doxycycline) was abolished by the PAX3-specific CRISPR construct, indicating that PAX3-FOXO1 expression is essential for transformation in this second category of recurrent tumours.
There was also a striking distinction in differentiation status between primary and recurrent tumour-derived cells. In standard growth-promoting medium, Dbt-indP3F, Dbt-MYCN and Dbt-MYCN/indP3F parental cells proliferate without any differentiation in the absence or presence of doxycycline. In contrast, cells derived from primary tumours expressing PAX3-FOXO1 with or without MYCN showed morphologic evidence of differentiation when PAX3-FOXO1 expression was turned off by doxycycline withdrawal (Figure 5D). In the presence of doxycycline (and thus PAX3-FOXO1 expression), the primary tumour-derived cells proliferate with significantly less or no differentiation, confirming the necessity of PAX3-FOXO1 for inhibiting differentiation and maintaining the proliferative phenotype. Furthermore, recurrent tumour-derived cells did not differentiate in the absence or presence of PAX3-FOXO1 induction, indicating that these cells developed a PAX3-FOXO1-independent differentiation block as part of the recurrence process. Similar findings were noted in focus formation assays in which doxycycline treatment was either discontinued or maintained after small foci formed (Figure S4C). In the continued presence of doxycycline, foci formed from all primary and recurrent tumour-derived lines were composed of tightly packed small round undifferentiated cells. However, when doxycycline was withdrawn after small foci formed, there was widespread differentiation in foci formed from primary tumour-derived cells but no differentiation was evident in foci formed from recurrent tumour-derived cells, corroborating the PAX3-FOXO1-independent differentiation block.
Tumourigenicity of tumour-derived cell lines
We finally compared in vivo tumourigenesis of the parental Dbt-indP3F and Dbt-MYCN/indP3F mixed populations with cell lines derived from primary and recurrent tumours. The primary tumour-derived cell lines from Dbt-indP3F myoblasts formed tumours more rapidly (3–4 weeks) than the parental Dbt-indP3F population (9–10 weeks) (Figure 6A). This finding further confirms the premise that tumourigenic cells with additional genetic changes were selected during tumourigenesis. In contrast, cells derived from primary tumours expressing MYCN and PAX3-FOXO1 formed tumours at nearly the same rate as the parental population, indicating that many (and perhaps most) cells in the parental population have transforming and tumourigenic potential (Figure 6B). This premise is confirmed by studies of subclones prepared from the parental population; all three tested subclones showed relatively rapid tumour formation with some heterogeneity in growth rates (Figure 6C).
Figure 6. Tumourigenic potential of primary and recurrent tumour-derived cell lines in comparison to parental cells and subclones.
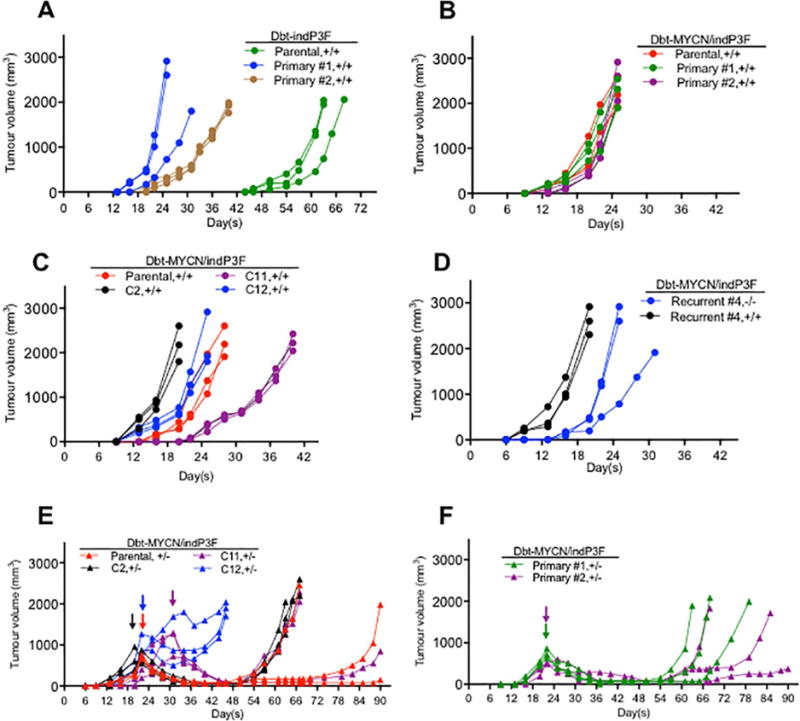
(A) Tumour growth of parental Dbt-indP3F cells and associated primary tumour-derived cell lines in the presence of doxycycline. (B) Tumour growth of parental Dbt-MYCN/indP3F cells and associated primary tumour derived cell lines in the presence of doxycycline. (C) Tumour growth of parental Dbt-MYCN/indP3F cells and three subclones (C2, C11, C12) in the presence of doxycycline. (D) Tumour growth of a Dbt-MYCN/indP3F recurrent tumour-derived cell line in the presence or absence of doxycycline. (E) Regression and recurrent tumour formation of parental Dbt-MYCN/indP3F cells and subclones C2, C11, and C12 after doxycycline withdrawal. (F) Regression and recurrent tumour formation of Dbt-MYCN/indP3F primary tumour-derived cell lines after doxycycline withdrawal. In parts E and F, doxycycline was withdrawn after the tumours were palpable. Arrows indicate time points of doxycycline withdrawal. Symbols: +/+, continuous doxycycline treatment; −/−, no doxycycline treatment; +/−, doxycycline treatment followed by doxycycline withdrawal.
The recurrent tumour-derived cells were also assayed for tumourigenesis with or without doxycycline. A recurrent tumour-derived line that did not express PAX3-FOXO1 and was transformed in the absence of doxycycline formed tumours rapidly in the absence of doxycycline. This finding corroborates a PAX3-FOXO1-independent mechanism of transformation and tumourigenesis (Figure 6D). Of note, these cells formed tumours even more rapidly in the presence of doxycycline, suggesting that PAX3-FOXO1-dependent and independent oncogenic mechanisms have additive effects that collaborate during tumourigenesis.
To further study the generality of these recurrence events, we compared recurrence rates in primary tumour-derived cells, parental Dbt-MYCN/indP3F cells, and subclones. We again withdrew doxycycline after palpable tumours were formed in the presence of doxycycline (Figures 6E and 6F). Except for one subclone (C12), there was a similar pattern of recurrence among all cell lines tested, indicating that recurrent tumours form from rare cells in the population after doxycycline withdrawal. Of note, there was variation in the rate of recurrent tumour formation among the injected replicates of each cell line. For subclone C12, recurrent tumours arose at a uniformly fast rate, suggesting that there is a pre-existing PAX3-FOXO1-independent tumourigenesis mechanism in this clonal population.
Discussion
In this paper, we continued our longstanding studies of the PAX3-FOXO1 oncoprotein in ARMS tumourigenesis. To study the dynamic and temporal relationship of this fusion oncoprotein to the tumourigenesis process, we developed a doxycycline-inducible PAX3-FOXO1 expression system in human myoblasts, postulated to be a cell of origin of RMS. By inducing PAX3-FOXO1 in the presence or absence of constitutive MYCN expression, this system has validated previous findings of collaboration between PAX3-FOXO1 and MYCN in oncogenic transformation and tumourigenesis [12,21]. Furthermore, this inducible expression system permitted us to dissect the role of PAX3-FOXO1 in initiating and maintaining the oncogenic phenotype. Our findings indicate that PAX3-FOXO1 induces transformation while inhibiting differentiation and cell death during the early phases of tumourigenesis. However, these phenotypes may be uncoupled from the fusion protein in later stages of tumour progression.
Our analysis is compatible with an initial phase of oncogene addiction during ARMS tumourigenesis. It is widely known that some tumours exhibit exquisite dependence on a single oncogenic protein (or pathway) for sustaining growth and survival despite the presence of other mutations and aberrant pathways [22]. Such dependence has been demonstrated for oncogenic versions of MYC [23,24] and RAS [25] in tissue culture and transgenic murine systems. In our current study, we also observed that transformed myoblasts expressing PAX3-FOXO1 are dependent on the fusion protein to overcome contact inhibition and induce cell proliferation in vitro and tumourigenesis in vivo. When PAX3-FOXO1 was depleted in these myoblasts in vitro or in vivo, the cells stopped proliferating and showed widespread differentiation and apoptosis. These changes resulted in shrinkage of the tumour to a non-palpable state within a few days. Our findings in this inducible myoblast system are concordant with previous studies of human ARMS cell lines in which knockdown of PAX3-FOXO1 resulted in reduced proliferation along with increased differentiation and cell death [12,26,27], ultimately culminating in decreased tumour volume in vivo [28].
Following regression, most tumours (>80%) in our model system recurred in the absence of doxycycline during the 90-day observation period. Though some of these recurrences resulted from acquisition of mechanisms to express PAX3-FOXO1 without doxycycline induction, many recurrences (60%) were associated with no detectable PAX3-FOXO1 protein expression. For these latter tumours, the initial oncogene-addicted tumour cells acquired mechanisms to bypass the addiction and overcome the detrimental effects of loss of fusion protein expression. Tumour recurrence in the absence of the initiating oncogene was previously described in several inducible tumour mouse models, such as Myc-inducible breast cancer [29]. We propose that such bypass mechanisms were acquired by one or more cells in the initial RMS tumour followed by gradual regrowth until a recurrent RMS tumour was detectable after a latent period of several weeks. The recurrent tumours demonstrated heterogeneity in the length of this latent period, suggesting stochastic differences or differences in the underlying bypass mechanisms, re-growth rate, or number of aberrant cells in the initial tumour. Since primary tumours do not form in the absence of doxycycline, even after extended observation, the appearance of these PAX3-FOXO1-independent tumours requires a prior period of PAX3-FOXO1 expression, indicating that these alternative oncogenic pathways may be initially activated or otherwise enabled by fusion protein expression. Though there may be multiple different molecular mechanisms underlying these recurrences, a common theme is a PAX3-FOXO1-independent block in myogenic differentiation. We propose that cells in which the transcriptional environment favours PAX3-FOXO1-mediated inhibition of myogenic differentiation are selected during ARMS tumourigenesis. Furthermore, many recurrent tumours also possess a PAX3-FOXO1-independent mechanism for oncogenic transformation.
These inducible systems are valuable to model therapy targeted against oncoproteins. In these experimental systems, targeted therapy is simulated by withdrawal of the inducing agent whereas in clinical practice, the targeted therapy is addition of an agent that inactivates oncoprotein function. This targeted therapeutic effect can be bypassed following the development of primary or acquired resistance in a subset of tumour cells. Some resistance mechanisms involve genetic alterations, such as point mutation or amplification in the oncoprotein, whereas other resistance mechanisms involve acquisition of alternative oncogenic pathways in tumour cells [30,31]. In the inducible experimental systems, recurrence is most likely caused by the latter group of mechanisms. In the doxycycline-inducible Myc breast cancer model, recurrence was associated with acquisition of activating point mutations in Ras family genes [32]
In summary, our findings highlight the role of PAX3-FOXO1 in ARMS tumourigenesis and provide evidence for the evolution of fusion-positive RMS to a fusion protein-independent stage. The findings in this model animal system are consistent with our earlier molecular pathology studies of human ARMS tumours that identified cases in which a fusion gene was detectable by in situ hybridisation but the corresponding fusion transcript was not expressed [33]. Due to the paucity of banked recurrent tumour specimens from ARMS patients, the frequency of fusion protein-independent recurrences with the current therapeutic regimens is not known. However, it should be acknowledged that there are currently no therapies targeted against the PAX3-FOXO1 fusion oncoprotein in RMS. We therefore hope that further investigation of this inducible model system will assist in the elucidation of the types of primary and acquired resistance that will be encountered as these targeted therapies are developed in coming years.
Supplementary Material
Figure S1. Efficacy of doxycycline-inducible PAX3-FOXO1 expression system in human myoblasts.
Figure S2. Analysis of tumours from Dbt-indP3F and Dbt-MYCN/indP3F cells.
Figure S3. Effect of PAX3-FOXO1 depletion on tumour growth.
Figure S4. Analyses of primary and recurrent tumour-derived cell lines.
Acknowledgments
This research was supported by the Intramural Research Program of the National Cancer Institute and by the Joanna McAfee Childhood Cancer Foundation. We thank Dr. Zengfeng Wang, Kristofferson Ylaya, and Julia Hisey for their technical assistance and Dr. Ji Luo for providing lentiviral constructs.
Footnotes
Disclosure of Potential Conflicts of Interest: The authors have no potential conflicts of interest to disclose.
Author contribution statement
FGB conceived and supervised the overall study. PRP and FGB designed the experiments. PRP performed the experiments and made the figures. BC and MEO performed additional molecular biology experiments. SMH directed the tissue processing, and both SMH and MMM directed immunohistochemistry studies. JK provided the PAX3-FOXO1-specific antibody and gave advice on the experimental plan. PRP and FGB analysed the data and wrote the manuscript. BC, MEO, JK, MMM and SMH critically reviewed the manuscript. All authors read and gave final approval for the submitted version.
References
- 1.Olanich ME, Barr FG. A call to ARMS: targeting the PAX3-FOXO1 gene in alveolar rhabdomyosarcoma. Expert Opin Ther Targets. 2013;17:607–623. doi: 10.1517/14728222.2013.772136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barr FG. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene. 2001;20:5736–5746. doi: 10.1038/sj.onc.1204599. [DOI] [PubMed] [Google Scholar]
- 3.Sorensen PH, Lynch JC, Qualman SJ, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol. 2002;20:2672–2679. doi: 10.1200/JCO.2002.03.137. [DOI] [PubMed] [Google Scholar]
- 4.Davis RJ, Barr FG. Fusion genes resulting from alternative chromosomal translocations are overexpressed by gene-specific mechanisms in alveolar rhabdomyosarcoma. Proc Natl Acad Sci USA. 1997;94:8047–8051. doi: 10.1073/pnas.94.15.8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lam PY, Sublett JE, Hollenbach AD, et al. The oncogenic potential of the Pax3-FKHR fusion protein requires the Pax3 homeodomain recognition helix but not the Pax3 paired-box DNA binding domain. Mol Cell Biol. 1999;19:594–601. doi: 10.1128/mcb.19.1.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fredericks WJ, Galili N, Mukhopadhyay S, et al. The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol Cell Biol. 1995;15:1522–1535. doi: 10.1128/mcb.15.3.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia SJ, Barr FG. Analysis of the transforming and growth suppressive activities of the PAX3-FKHR oncoprotein. Oncogene. 2004;23:6864–6871. doi: 10.1038/sj.onc.1207850. [DOI] [PubMed] [Google Scholar]
- 8.Scheidler S, Fredericks WJ, Rauscher FJ, 3rd, et al. The hybrid PAX3-FKHR fusion protein of alveolar rhabdomyosarcoma transforms fibroblasts in culture. Proc Natl Acad Sci USA. 1996;93:9805–9809. doi: 10.1073/pnas.93.18.9805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linardic CM, Naini S, Herndon JE, 2nd, et al. The PAX3-FKHR fusion gene of rhabdomyosarcoma cooperates with loss of p16INK4A to promote bypass of cellular senescence. Cancer Res. 2007;67:6691–6699. doi: 10.1158/0008-5472.CAN-06-3210. [DOI] [PubMed] [Google Scholar]
- 10.Linardic CM, Downie DL, Qualman S, et al. Genetic modeling of human rhabdomyosarcoma. Cancer Res. 2005;65:4490–4495. doi: 10.1158/0008-5472.CAN-04-3194. [DOI] [PubMed] [Google Scholar]
- 11.Naini S, Etheridge KT, Adam SJ, et al. Defining the cooperative genetic changes that temporally drive alveolar rhabdomyosarcoma. Cancer Res. 2008;68:9583–9588. doi: 10.1158/0008-5472.CAN-07-6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xia SJ, Holder DD, Pawel BR, et al. High expression of the PAX3-FKHR oncoprotein is required to promote tumorigenesis of human myoblasts. Am J Pathol. 2009;175:2600–2608. doi: 10.2353/ajpath.2009.090192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheong TC, Compagno M, Chiarle R. Editing of mouse and human immunoglobulin genes by CRISPR-Cas9 system. Nat Commun. 2016;7:10934. doi: 10.1038/ncomms10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shalem O, Sanjana NE, Hartenian E, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campeau E, Ruhl VE, Rodier F, et al. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One. 2009;4:e6529. doi: 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olanich ME, Sun W, Hewitt SM, et al. CDK4 Amplification Reduces Sensitivity to CDK4/6 Inhibition in Fusion-Positive Rhabdomyosarcoma. Clin Cancer Res. 2015;21:4947–4959. doi: 10.1158/1078-0432.CCR-14-2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia SJ, Rajput P, Strzelecki DM, et al. Analysis of genetic events that modulate the oncogenic and growth suppressive activities of the PAX3-FKHR fusion oncoprotein. Lab Invest. 2007;87:318–325. doi: 10.1038/labinvest.3700521. [DOI] [PubMed] [Google Scholar]
- 19.Miettinen M, Wang ZF, Paetau A, et al. ERG transcription factor as an immunohistochemical marker for vascular endothelial tumors and prostatic carcinoma. Amer J Surg Pathol. 2011;35:432–441. doi: 10.1097/PAS.0b013e318206b67b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao L, Yu Y, Bilke S, et al. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 2010;70:6497–6508. doi: 10.1158/0008-5472.CAN-10-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tonelli R, McIntyre A, Camerin C, et al. Antitumor activity of sustained N-myc reduction in rhabdomyosarcomas and transcriptional block by antigene therapy. Clin Cancer Res. 2012;18:796–807. doi: 10.1158/1078-0432.CCR-11-1981. [DOI] [PubMed] [Google Scholar]
- 22.Weinstein IB. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 23.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 24.Felsher DW, Bishop JM. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci USA. 1999;96:3940–3944. doi: 10.1073/pnas.96.7.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chin L, Tam A, Pomerantz J, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 26.Ebauer M, Wachtel M, Niggli FK, et al. Comparative expression profiling identifies an in vivo target gene signature with TFAP2B as a mediator of the survival function of PAX3/FKHR. Oncogene. 2007;26:7267–7281. doi: 10.1038/sj.onc.1210525. [DOI] [PubMed] [Google Scholar]
- 27.Kikuchi K, Tsuchiya K, Otabe O, et al. Effects of PAX3-FKHR on malignant phenotypes in alveolar rhabdomyosarcoma. Biochem Biophys Res Commun. 2008;365:568–574. doi: 10.1016/j.bbrc.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 28.Ayyanathan K, Fredericks WJ, Berking C, et al. Hormone-dependent tumor regression in vivo by an inducible transcriptional repressor directed at the PAX3-FKHR oncogene. Cancer Res. 2000;60:5803–5814. [PubMed] [Google Scholar]
- 29.D’Cruz CM, Gunther EJ, Boxer RB, et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7:235–239. doi: 10.1038/84691. [DOI] [PubMed] [Google Scholar]
- 30.Holohan C, Van Schaeybroeck S, Longley DB, et al. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 31.Zahreddine H, Borden KL. Mechanisms and insights into drug resistance in cancer. Front Pharmacol. 2013;4:28. doi: 10.3389/fphar.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jang JW, Boxer RB, Chodosh LA. Isoform-specific ras activation and oncogene dependence during MYC- and Wnt-induced mammary tumorigenesis. Mol Cell Biol. 2006;26:8109–8121. doi: 10.1128/MCB.00404-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barr FG, Qualman SJ, Macris MH, et al. Genetic heterogeneity in the alveolar rhabdomyosarcoma subset without typical gene fusions. Cancer Res. 2002;62:4704–4710. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Efficacy of doxycycline-inducible PAX3-FOXO1 expression system in human myoblasts.
Figure S2. Analysis of tumours from Dbt-indP3F and Dbt-MYCN/indP3F cells.
Figure S3. Effect of PAX3-FOXO1 depletion on tumour growth.
Figure S4. Analyses of primary and recurrent tumour-derived cell lines.