

Abstract
Age–related macular degeneration (AMD) is a complex and progressive degenerative eye disease resulting in severe loss of central vision. Recent evidence indicates that immune system dysregulation could contribute to the development of AMD. We hypothesize that defective lysosome-mediated clearance causes accumulation of waste products in the retinal pigmented epithelium (RPE), activating the immune system and leading to retinal tissue injury and AMD. We have generated unique genetically engineered mice in which lysosome-mediated clearance (both by phagocytosis and autophagy) in RPE cells is compromised, causing development of features of early AMD. Our recent data indicate a link between Lipocalin-2 (LCN-2) and the inflammatory responses induced in this mouse model. We show that NFκB and STAT-1 may function as a complex in our animal model system, together controlling the up-regulation of LCN-2 expression in the retina and stimulating an inflammatory response. This study revealed increased infiltration of LCN-2 positive neutrophils in the choroid and retina of early AMD patients as compared to age-matched controls. Our results demonstrate that both in our animal model and in human AMD the AKT2/NFκB/LCN-2 signalling axis is involved in activating the inflammatory response, making this pathway a potential target for AMD treatment.
Keywords: age-related macular degeneration, AKT2/NFκB/Lipocalin-2 signaling axis, βA3/A1-crystallin, inflammation, lysosomes
INTRODUCTION
Inflammation plays a key role in the pathogenesis of various age-related diseases, including AMD [1]. Deregulation of the innate immune system is thought to be critical for the onset of AMD; complement has been implicated, and genetic studies show a strong association between AMD and polymorphisms in complement pathway genes [2, 3]. Further, the activation of various cytokines/chemokines [4] and of the NLRP3 inflammasome have been invoked as central to AMD pathogenesis [5].
We have recently shown elevated levels of LCN-2 in RPE of aged mice lacking a lysosomal lumenal protein, βA3/A1-crystallin. Coupled with the associated increases in chemokine (C-C-motif) ligand 2 (CCL2) expression, reactive gliosis, and immune cell infiltration, it seems likely that LCN-2 plays a pivotal role in activating an inflammatory response in AMD [6]. A recent study showed LCN-2 to be a molecular target for reducing cellular stress during retinal degeneration [7]. Here, we provide novel evidence that the activation of inflammation in AMD is mediated through the AKT2/nuclear factor kappa-light-chain enhancer of the activated B cells (NFκB)/LCN-2 signalling axis.
MATERIALS AND METHODS
The detailed methods are given in the supplementary material available online.
Statistics
Statistical analysis was performed using Microsoft Excel Graph Pad Prism 6 software for Windows using one-way analysis of variance (ANOVA). Group means were compared using Tukey’s post hoc test, where significance was set at p < 0.05. The analyses were done on triplicate technical replicates for at least three independent experiments. Results are presented as mean ± SD [8].
Study Approval
All procedures in this study involving human tissue were in accordance with the tenets of the Declaration of Helsinki and were approved by the Institutional Review Boards of the Johns Hopkins University and University of Minnesota.
All animal studies were conducted in accordance with the Guide for the Care and Use of Animals (National Academy Press) and were approved by the Animal Care and Use Committees of the Johns Hopkins University and Noble Life Sciences, Inc.
RESULTS AND DISCUSSION
Pro-inflammatory preference in an animal model of AMD
Lack of a comprehensive animal model of AMD limits our understanding of cellular mechanisms in the critical early stages of the disease. We have recently developed genetically engineered mouse models that exhibit a slowly progressive AMD-like pathology associated with inefficient lysosomal clearance [8]. Altered lysosomal clearance can potentiate an inflammatory response and contribute to pathogenesis in various diseases [9].
Lysosomal function declines with age, the rate varying amongst individuals. Individuals with greater loss of lysosomal clearance functions in RPE during aging may mount inflammatory responses to particular factors/triggers in the retina, leading to AMD. One such trigger could be LCN-2, an adipokine that is elevated in many inflammation-associated diseases, and which may contribute to induction of inflammation in our model [6]. To test this hypothesis, we injected 4 month old Cryba1 (gene encoding βA3/A1-crystallin) knockout (KO) and Cryba1 floxed control mice with lipopolysaccharide (LPS), a regimen followed previously for induction of inflammation [10]. Both retina and RPE from these animals were subsequently screened for pro-inflammatory changes.
Using a mouse immune array, we found clear pro-inflammatory responses in both retina and RPE extracts from Cryba1 KO mice treated with LPS, compared to floxed control and untreated Cryba1 KO groups (Supplementary Fig 1A, B). Western blots showed increased expression of pro-inflammatory mediators IL-12 and iNOS and decreased expression of anti-inflammatory mediators IL-10 and Arg-1 (Fig 1A) in retinas from Cryba1 KO mice treated with LPS, as compared to the control and untreated Cryba1 KO groups. The heightened inflammatory state in the retinas of Cryba1 KO mice treated with LPS was further evidenced by increased microglial cell activation and polarization (Fig 1B and C). Microglial activation in the retina occurred concomitantly with activation of Müller cells (Fig 1C), a condition associated with reactive gliosis and an inflammatory response. The combination of increased pro-inflammatory mediators and microglial activation/polarization is critical for the onset of inflammatory processes in retinal diseases [11]. Our data indicated an inflammatory response that was most prominent in the retina of the LPS-treated KO animals. We have previously shown that LCN-2 stimulates inflammation in the retina, particularly when the lysosomal-mediated clearance in RPE is compromised [6]. Loss of Cryba1 diminishes lysosomal-mediated clearance in RPE, resulting in increased expression of LCN-2 [6, 8].
Figure 1. LPS potentiates inflammatory responses in Cryba1 KO mouse retina.
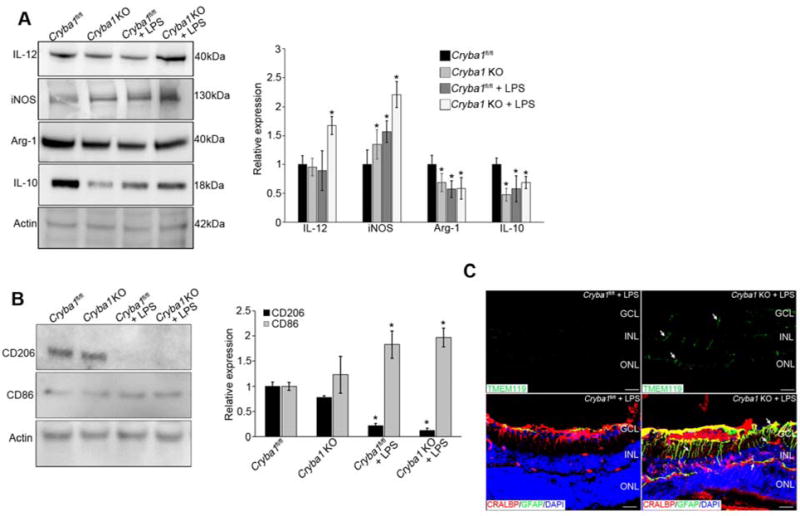
(A) Immunoblot and summary of densitometry show increased expression of pro-(IL12 and iNOS) and decreased expression of anti- inflammatory (Arg-1 and IL-10) mediators in Cryba1 KO+LPS. Error bars represent s.d.; * P<0.05 relative to Cryba1fl/fl. (B) Immunoblot and summary of densitometry show increased expression (activation) of CD86 (M1 polarization) versus CD206 (M2 polarization) in the retina of Cryba1 KO+LPS, indicating transition of the glial cell population to a more M1 phenotype. Error bars indicate s.d.; *P<0.05. (C) Top panel shows Tmem119-positive microglia activation (white arrows) in LPS-treated Cryba1 KO (right panel), but not in treated Cryba1fl/fl retinal flatmounts (left panel). In the bottom panel, immunostaining of Cryba1 KO+LPS retina with GFAP (green) or CRALBP (red) antibodies shows extensive staining of the Müller glia processes (green indicating activation, white arrows) which is not seen in the Cryba1fl/fl retinas treated with LPS. The Müller cell bodies in LPS-treated Cryba1 KO mice express both CRALBP and GFAP (merged-yellow). Bar=30μm. n=6 (4 month old) mice/group. GCL: ganglion cell layer, INL: inner nuclear layer, ONL: outer nuclear layer.
Lipocalin-2 levels increase in human AMD patients
We next asked if LCN-2 is also activated in human AMD patients. We assessed LCN-2 expression in human donor eyes: 5 age-matched normal control eyes and 9 AMD eyes graded for disease severity according to the Minnesota Grading System (MGS) (Supplementary Table 1) [12]. Western data (Fig 2A) confirmed that LCN-2 expression increased significantly in early AMD (MGS2) compared to age-matched controls (MGS1). Moreover, expression remained high in more advanced AMD (MGS3) and in donors with late AMD exhibiting central geographic atrophy (MGS4). These studies suggest that LCN-2 could be a novel indicator of early AMD. Further, immunostaining of human AMD tissue sections revealed LCN-2 positive infiltrating neutrophils in the retina and sub-macular choroid (Fig 2B). Neutrophils contribute to various inflammatory diseases and higher levels of circulating neutrophils have been associated with AMD [13]. These data indicate a role for LCN-2 in activating inflammation in both human AMD and in our animal model, prompting us to evaluate how LCN-2 might induce inflammatory pathways in the pathogenesis of AMD.
Figure 2. Increased LCN-2 in human AMD and Binding of NFkB to the LCN-2 promoter.
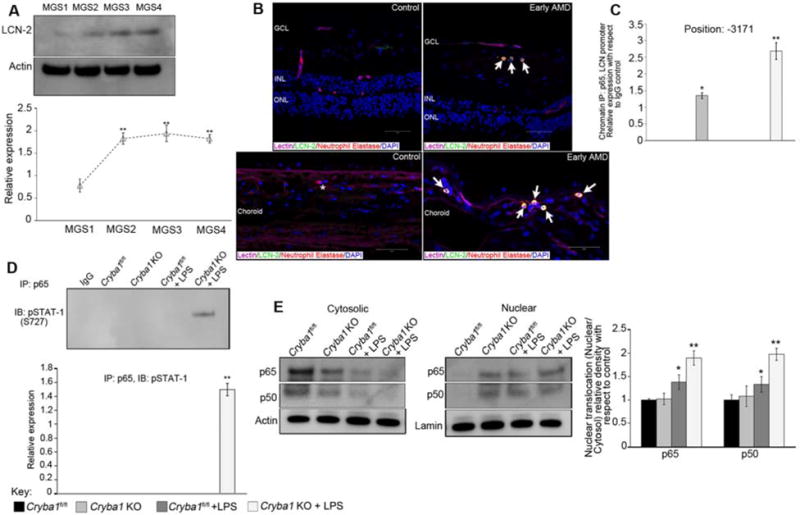
(A) In human samples (see methods section for grading system of human AMD), data from immunoblots shows a significant increase in LCN-2 expression in early AMD (MGS2) compared to age-matched controls (MGS1), which persisted in the later stages of the disease (n=5 control donors/and n=3 donors/disease stage). (B) Immunofluorescence demonstrates LCN-2 expressing infiltrating cells (Neutrophils stained with anti-neutrophil elastase) in the sub-macular choroid and retina of an early AMD patient (arrows). In age-matched control samples, many fewer neutrophils are detected (asterisk) and they are not positive for LCN-2. Bars= 50 μm. GCL: ganglion cell layer, INL: inner nuclear layer, ONL: outer nuclear layer. (C) ChIP analysis of LCN-2 promoter-binding activity for NFκB p65 subunit in retinal cells from Cryba1 KO mice (+/−LPS) showing association of NFκB in the promoter region (−3171) of the LCN-2 gene, but not in floxed controls. (D) Reverse ChIP analysis followed by western blotting indicated association between NFκB and STAT1 in the same region as described in (C). (E) Immunoblot shows significantly higher nuclear expression of NFκB-p65 and p50 subunits in Cryba1 KO+LPS retinal cells, as compared to floxed control.
The activation of the AKT2/NFκB/LCN2 signalling axis in AMD
The LCN-2 promoter has binding sites for several transcription factors [14]. It has been reported that NFκB regulates LCN-2 by binding to specific promoter sites during inflammatory stress [15]. This information prompted us to perform a Chromatin immunoprecipitation (ChIP) study to identify NFκB binding sites in the LCN-2 promoter region in our mouse model. Interestingly, we found that in retinas from Cryba1 KO mice (+/−LPS), the NFκB-p65 subunit was associated with a binding motif in the promoter region (−3171) of the LCN-2 gene (Table 1 and Fig 2C). This was not true in Cryba1 floxed retinas (+/− LPS). This suggests that NFκB may regulate LCN-2 gene expression in our mouse model, thereby inducing inflammation. However, a previous study reported that STAT-1 binds to the -3171 promoter region of the LCN-2 gene in adipocytes [15]. Further investigation using reverse ChIP analysis followed by immunoblotting with pSTAT-1 (S727) revealed an association between NFκB and STAT-1 at the -3171 promoter binding site of the LCN-2 gene in the LPS-treated Cryba1 KO retina (Fig 2D). Our studies provide novel evidence that NFκB and STAT-1 may function as a complex, thereby controlling the upregulation of LCN-2 in the retina and stimulating an inflammatory response. Such dual stimulation has been previously shown in the inflammatory responses of CXCL1 and CXCL2 to both NFκB and STAT-1 [16]. We also observed increased nuclear translocation of NFκB-p65/p50 subunits (Fig 2E), specifically as a heterodimer (Supplementary Fig 2), in the retinas of LPS treated Cryba1 KO mice, as compared to the control and untreated Cryba1 KO animals. A previous study [15] showed that IFN-γ and TNF-α induce STAT-1 and NFκB respectively, and that these transcription factors are required for LCN-2 activation. Our array data also show an increase in both IFN-γ and TNF-α in Cryba1 KO mice treated with LPS (Supplementary Fig 1).
Table 1.
Represents binding affinity for NFkB-p65 subunit on the LCN-2 promoter region in the retina from all the experimental groups.
| Gene | Position | Groups | ChIP (IP:p65; LPS responsiveness) |
|---|---|---|---|
| LCN-2 | −266 | Cryba1fl/fl | No |
| Cryba1 KO | No | ||
| Cryba1fl/fl +LPS | No | ||
| Cryba1 KO +LPS | No | ||
| LCN-2 | −619 | Cryba1fl/fl | No |
| Cryba1 KO | No | ||
| Cryba1fl/fl +LPS | No | ||
| Cryba1 KO +LPS | No | ||
| LCN-2 | −676 | Cryba1fl/fl | No |
| Cryba1 KO | No | ||
| Cryba1fl/fl +LPS | No | ||
| Cryba1 KO +LPS | No | ||
| LCN-2 | −1014 | Cryba1fl/fl | No |
| Cryba1 KO | No | ||
| Cryba1fl/fl +LPS | No | ||
| Cryba1 KO +LPS | No | ||
| LCN-2 | −1822 | Cryba1fl/fl | No |
| Cryba1 KO | No | ||
| Cryba1fl/fl +LPS | No | ||
| Cryba1 KO +LPS | No | ||
| LCN-2 | −3171 | Cryba1fl/fl | No |
| Cryba1 KO | Yes | ||
| Cryba1fl/fl +LPS | No | ||
| Cryba1 KO +LPS | Yes |
To further delineate possible upstream regulators of NFκB, we performed a human proteome high-throughput microarray assay and found that βA3/A1-crystallin interacts with AKT2, a serine/threonine kinase. Through mass spectrometry analysis, we observed that pAKT2 is associated with βB2-crystallin in the mouse retina (Supplementary Fig 3). βB2-crystallin was the strongest binding partner of βA3/A1-crystallin in the same array. In the lens, heterodimer formation between βA3- and βB2-crystallin is energetically favoured and important to transparency [17]. In retina, the association between βB2-crystallin and βA3/A1-crystallin may regulate AKT2 phosphorylation upon inflammatory stimuli. Lack of βA3/A1-crystallin in the Cryba1 KO mice would disrupt this particular association. AKT2 is directly associated with the phosphorylation of IKKα, which is required for activation of NFκB during inflammatory stress [18]. Since we have previously shown activation of LCN-2 in our aging Cryba1 conditional KO mice [6], a mouse model that exhibits a slowly progressing form of AMD-like pathology, we determined if AKT2 and NFκB are also activated. There was increased phosphorylation of AKT2, as well as increased levels of NFκB and LCN-2 in retinas from 1 year old Cryba1 cKO mice, as compared to floxed controls (Fig 3A–B). These results suggest that activation of the AKT2-mediated NFκB signalling axis exerts a pro-inflammatory bias in the retina of our mouse model by upregulating expression of LCN-2. To confirm this, Triciribine, a potent and selective inhibitor of AKT2 phosphorylation, was injected intravitreally in Cryba1 cKO mice. Not only was pAKT2 reduced in the retina, but activation of NFκB and LCN-2 was also diminished (Fig 3A–B). Further, in human retina specimens, those from AMD patients showed increased expression of pAKT2 and NFκB compared to age-matched controls (Fig 3C). Taken together, our findings suggest that the AKT2/NFκB/LCN-2 signaling axis represents a potential therapeutic target for AMD and that LCN-2 may be a novel indicator of early disease.
Figure 3. Activation of LCN-2 through the AKT2/NFκB axis.
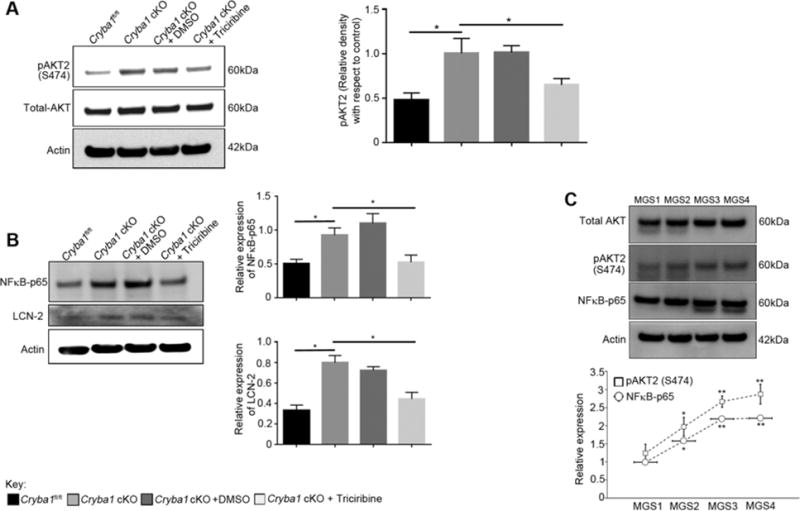
(A) Immunoblot and summary of densitometry showing a significant increase in the phosphorylation of AKT2 in retinas from 1 year old Cryba1 cKO mice. Treatment with Triciribine, a potent inhibitor of AKT2 significantly decreased the levels of pAKT2 in the Cryba1 cKO retinas. DMSO had no effect. Additionally, levels of total AKT did not change in the samples. (B) Immunoblot and summary of densitometry showing increased expression of both NFκB-p65 subunit and LCN-2 in the retinas from 1 year old Cryba1 cKO mice. After intravitreal injections of Triciribine in Cryba1 cKO mice, the activation of both NFκB and LCN-2 was significantly reduced. (C) Immunoblot data showing significant increase in expression of pAKT2 (S474) and NFκB-p65 subunit in retinas of early AMD subjects compared to age-matched controls, which increased with disease severity (n=3 donors/stage). Error bars indicate s.d.; *P<0.05. **P<0.01 relative to Cryba1fl/fl.
Supplementary Material
Acknowledgments
We thank Drs. Morton Goldberg and Eric Wawrousek for critical reading and discussions regarding this manuscript and the personnel at the Minnesota Lions Eye Bank for assistance in procuring and processing donor eyes. We thank Drs. Mariko Bennett and Ben Barres for the Tmem119 antibody. SG is recipient of the Fulbright-Nehru Doctoral Research Fellowship. DS is a recipient of the Carolyn K. McGillvray Memorial Award for Macular Degeneration Research from BrightFocus Foundation and the Sybil B. Harrington Special Scholar Award for Macular Degeneration from Research to Prevent Blindness. This research was supported by BrightFocus Foundation (DS), Research to Prevent Blindness (an unrestricted grant to the Wilmer Eye Institute and Department of Ophthalmology and Visual Sciences, University of Minnesota), National Eye Institute: EY019037-S (DS), EY01765 (Wilmer Imaging Core), the Arnold and Mabel and Beckman Foundation (DF) and an anonymous benefactor for AMD research (DF).
Footnotes
AUTHOR CONTRIBUTIONS:
DS designed the study. SG, PS, MY, TL conducted the experiments. SG, DS, JQ, SH, SZ analyzed the data. SC generated the inflammatory model. DF, SM, IB, GL contributed to the human studies. SG, SH, SZ, DS wrote the paper. All authors have approved the final manuscript.
CONFLICT OF INTEREST STATEMENT:
DS has research funding from Bayer Healthcare, Germany and F. Hoffmann-La Roche, Switzerland.
LIST OF SUPPLEMENTARY MATERIAL:
References
- 1.Whitcup SM, Sodhi A, Atkinson JP, et al. The role of the immune response in age-related macular degeneration. Int J Inflam. 2013;348092 doi: 10.1155/2013/348092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sparrow JR, Ueda K, Zhou J. Complement dysregulation in AMD: RPE-Bruch’s membrane-choroid. Molecular Aspects of Medicine. 2012;33:436–445. doi: 10.1016/j.mam.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorin MB. Genetic insights into age-related macular degeneration: controversies addressing risk, causality, and therapeutics. Molecular Aspects of Medicine. 2012;33:467–486. doi: 10.1016/j.mam.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagineni CN, Kommineni VK, Ganjbaksh N, et al. Inflammatory Cytokines Induce Expression of Chemokines by Human Retinal Cells: Role in Chemokine Receptor Mediated Age-related Macular Degeneration. Aging Dis. 2015;6:444–55. doi: 10.14336/AD.2015.0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tarallo V, Hirano Y, Gelfand BD, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. 2012;149:847–59. doi: 10.1016/j.cell.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valapala M, Edwards M, Hose S, et al. Increased Lipocalin-2 in the retinal pigment epithelium of Cryba1 cKO mice is associated with a chronic inflammatory response. Aging Cell. 2014;13:1091–4. doi: 10.1111/acel.12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parmar T, Parmar VM, Arai E, et al. Acute Stress Responses Are Early Molecular Events of Retinal Degeneration in Abca4−/−Rdh8−/− Mice After Light Exposure. Invest Ophthalmol Vis Sci. 2016;57:3257–67. doi: 10.1167/iovs.15-18993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valapala M, Wilson C, Hose S, et al. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/βA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy. 2014;10:480–96. doi: 10.4161/auto.27292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choudhury S, Ghosh S, Gupta P, et al. Inflammation-induced ROS generation causes pancreatic cell death through modulation of Nrf2/NF-κB and SAPK/JNK pathway. Free Radic Res. 2015;49:1371–83. doi: 10.3109/10715762.2015.1075016. [DOI] [PubMed] [Google Scholar]
- 11.Zhu T, Miller AG, Deliyanti D, et al. Prorenin stimulates a pro-angiogenic and pro-inflammatory response in retinal endothelial cells and an M1 phenotype in retinal microglia. Clin Exp Pharmacol Physiol. 2015;42:537–48. doi: 10.1111/1440-1681.12376. [DOI] [PubMed] [Google Scholar]
- 12.Decanini A, Nordgaard CL, Feng X, et al. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am J Ophthalmol. 2007;143:607–615. doi: 10.1016/j.ajo.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lechner J, Chen M, Hogg RE, et al. Alterations in circulating immune cells in neovascular age-related macular degeneration. Sci Rep. 2015;2015:16754. doi: 10.1038/srep16754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao P, Elks CM, Stephens JM. The induction of lipocalin-2 protein expression in vivo and in vitro. J Biol Chem. 2014;289:5960–9. doi: 10.1074/jbc.M113.532234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao P, Stephens JM. STAT1, NF-κB and ERKs play a role in the induction of lipocalin-2 expression in adipocytes. Mol Metab. 2013;2:161–70. doi: 10.1016/j.molmet.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burke SJ, Lu D, Sparer TE, et al. NF-κB and STAT-1 control CXCL1 and CXCL2 gene transcription. Am J Physiol Endocrinol Metab. 2014;306:E131–E149. doi: 10.1152/ajpendo.00347.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sergeev YV, Hejtmancik JF, Wingfield PT. Energetics of domain-domain interactions and entropy driven association of beta-crystallins. Biochemistry. 2004;43:415–24. doi: 10.1021/bi034617f. [DOI] [PubMed] [Google Scholar]
- 18.Madrid LV, Mayo MW, Reuther JY, et al. Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J Biol Chem. 2001;276:18934–40. doi: 10.1074/jbc.M101103200. [DOI] [PubMed] [Google Scholar]
- 19.Bennett ML, Bennett FC, Liddelow SA, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113:E1738–46. doi: 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Truax AD, Greer SF. ChIP and Re-ChIP assays: investigating interactions between regulatory proteins, histone modifications, and the DNA sequences to which they bind. Methods Mol Biol. 2012;809:175–88. doi: 10.1007/978-1-61779-376-9_12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.